
Diagnostic Evaluation of Pulmonary Hypertension: A Comprehensive Approach for Primary Care Physicians




Abstract
:1. Introduction
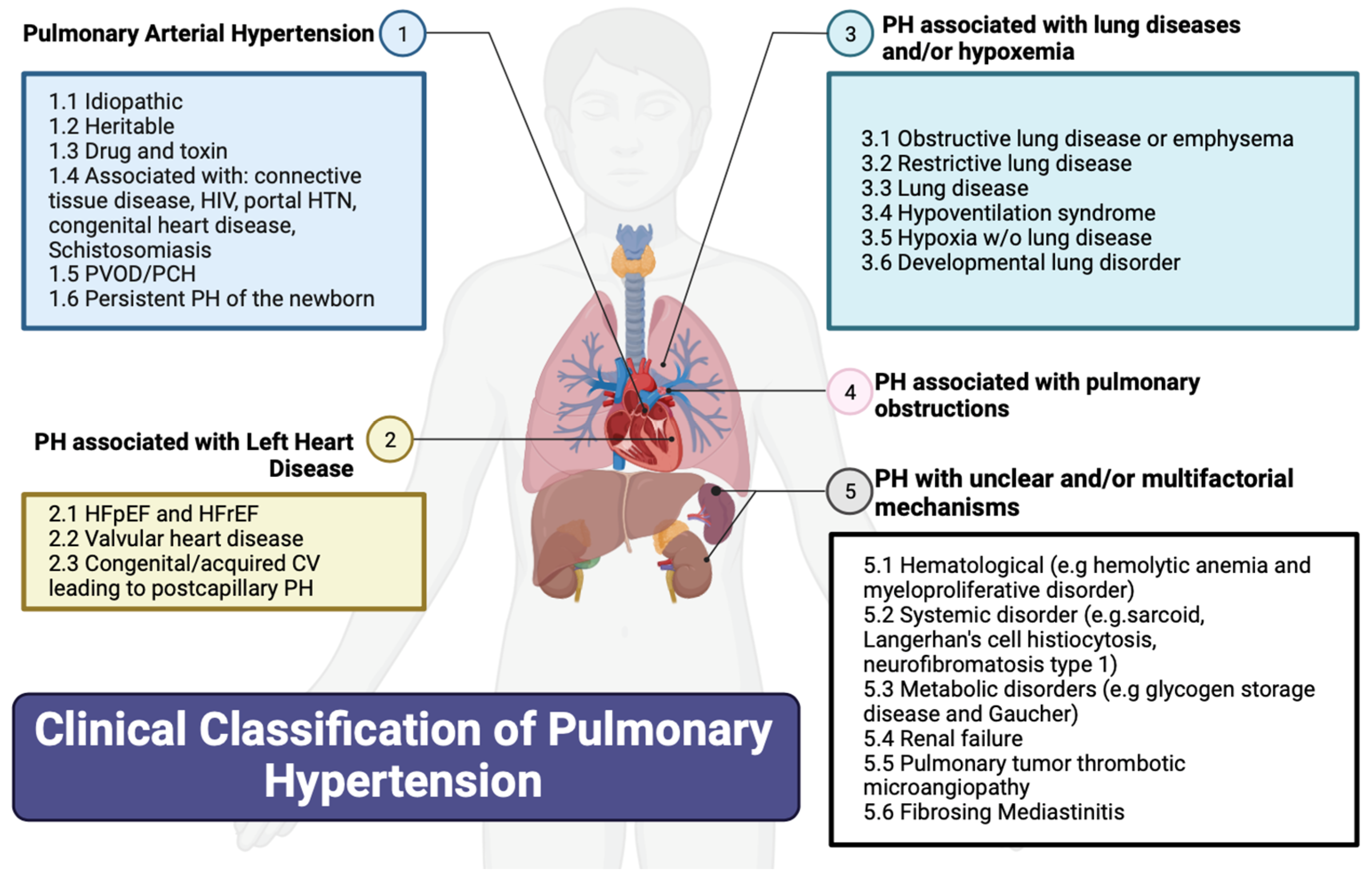
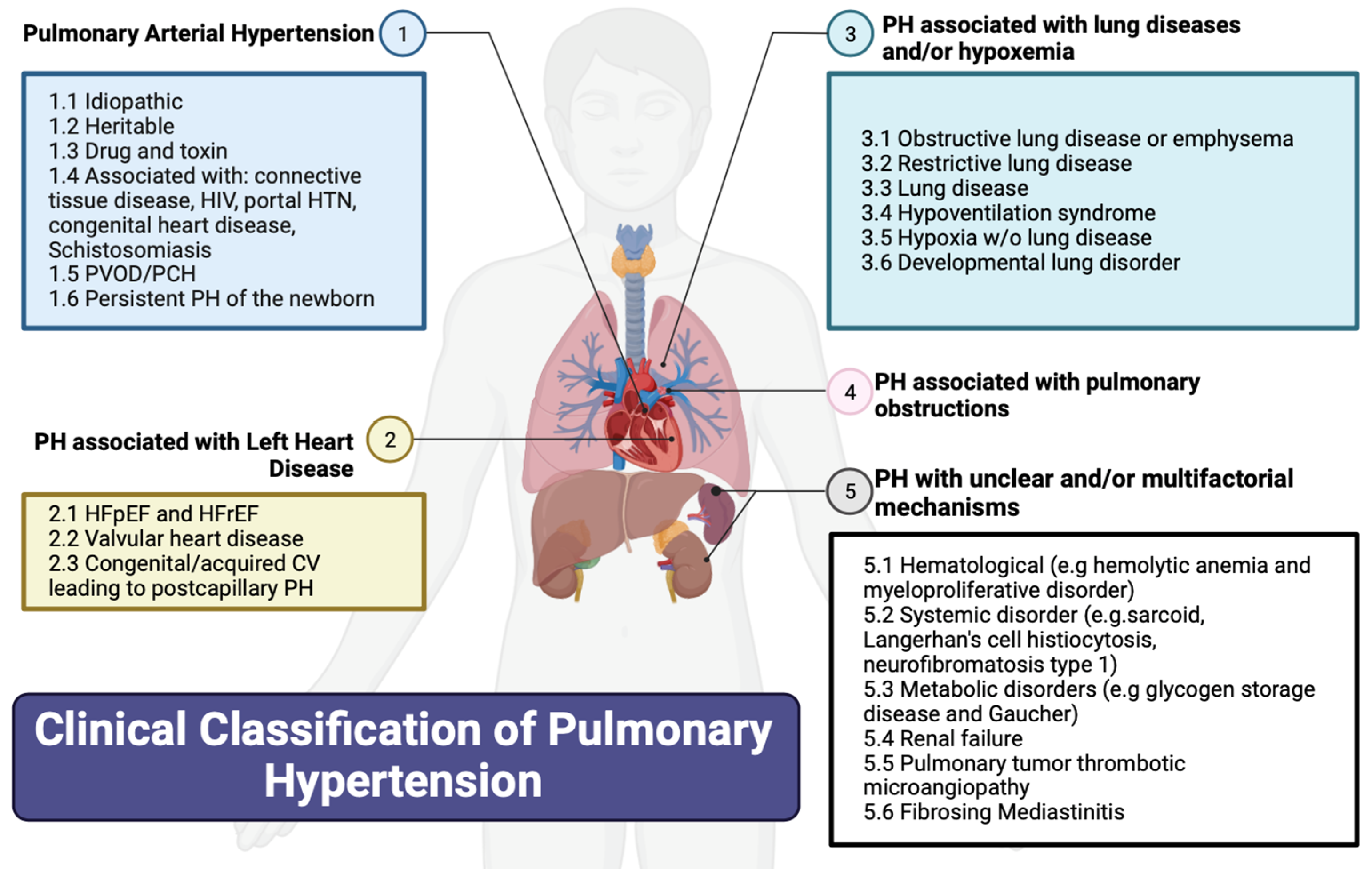
2. Classification of PH
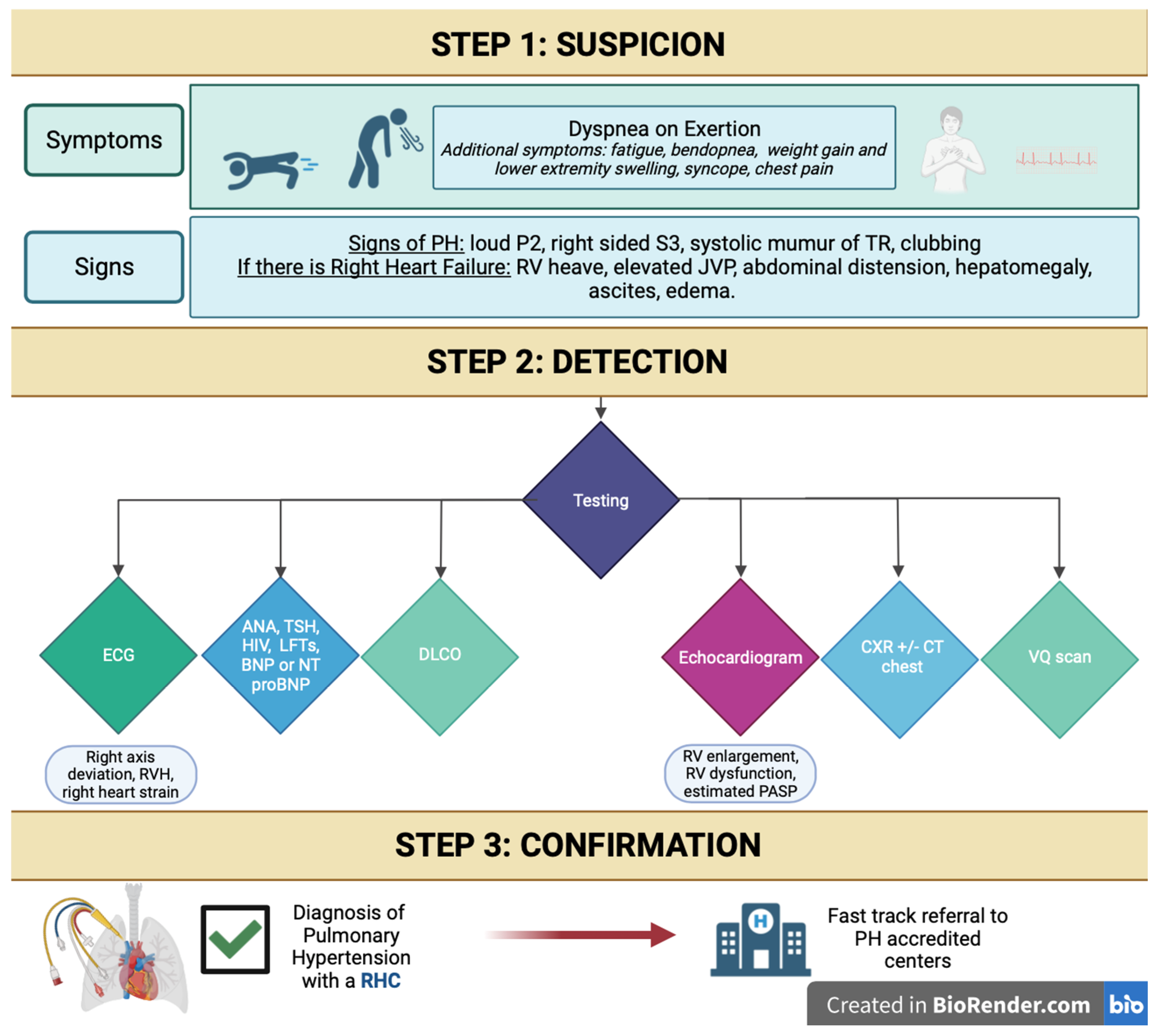
3. History and Physical Exam
4. Risk Factors for Pulmonary Hypertension
5. Diagnostic Tools
5.1. Laboratory Markers
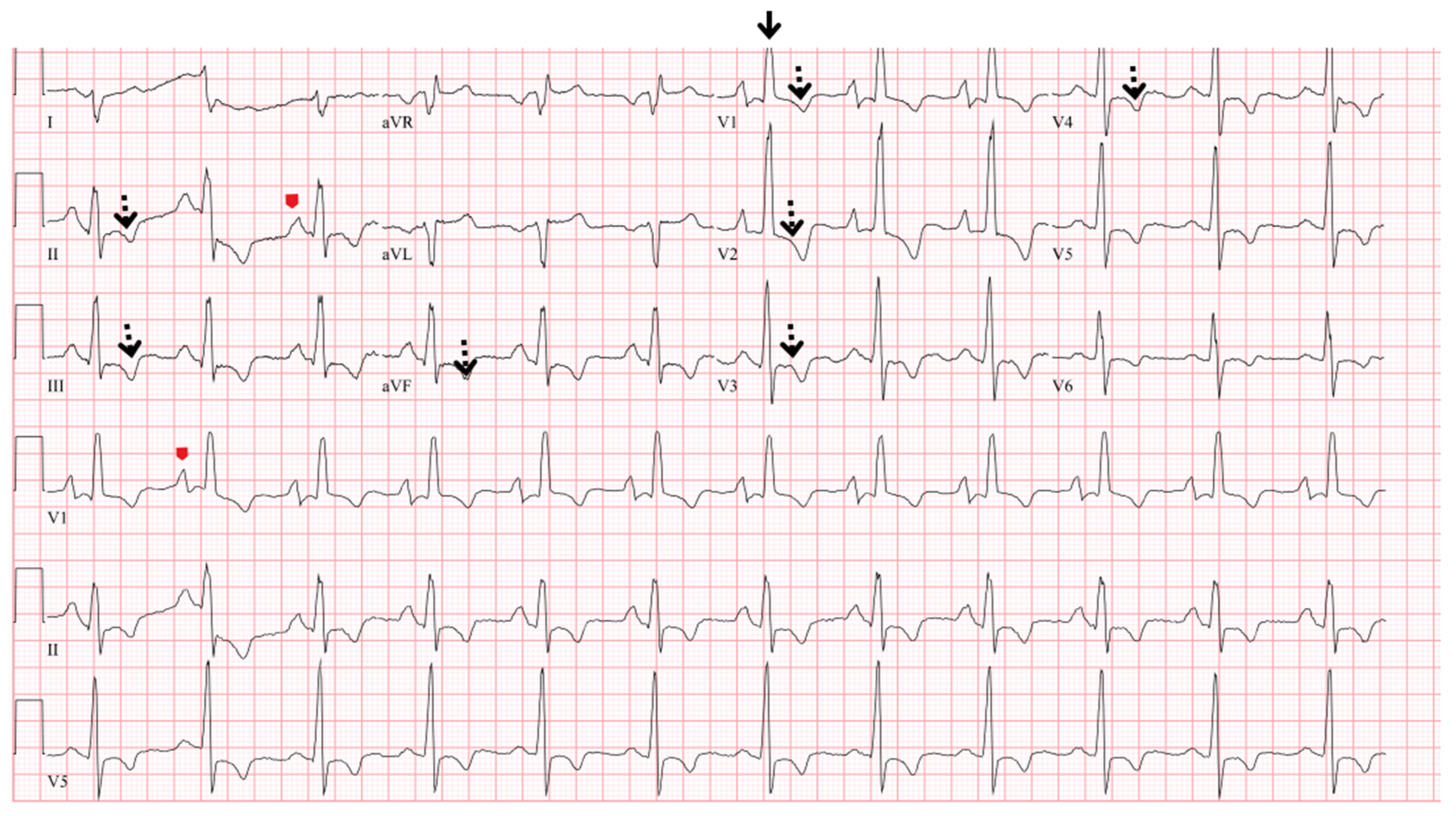
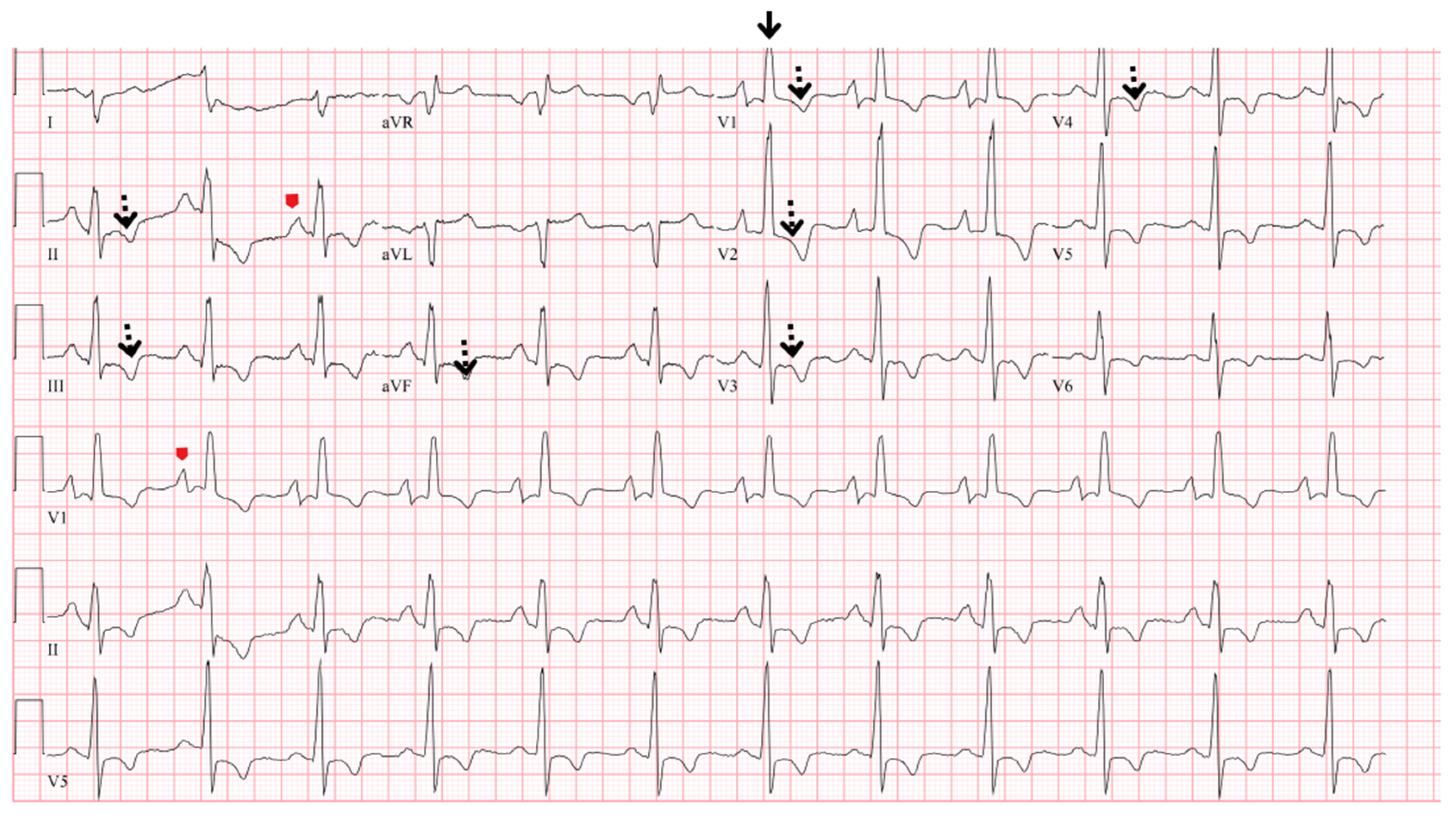
5.2. Electrocardiogram
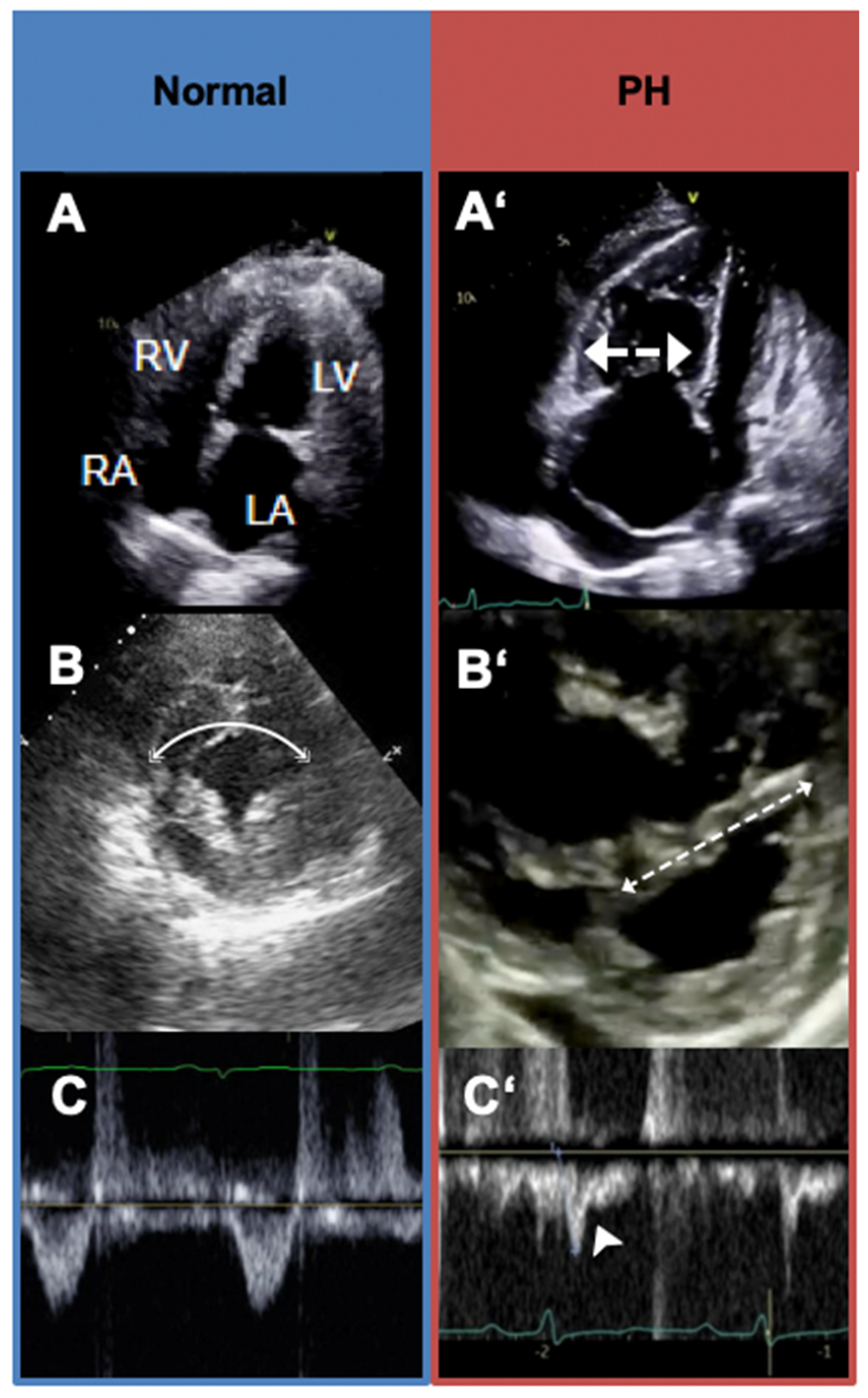
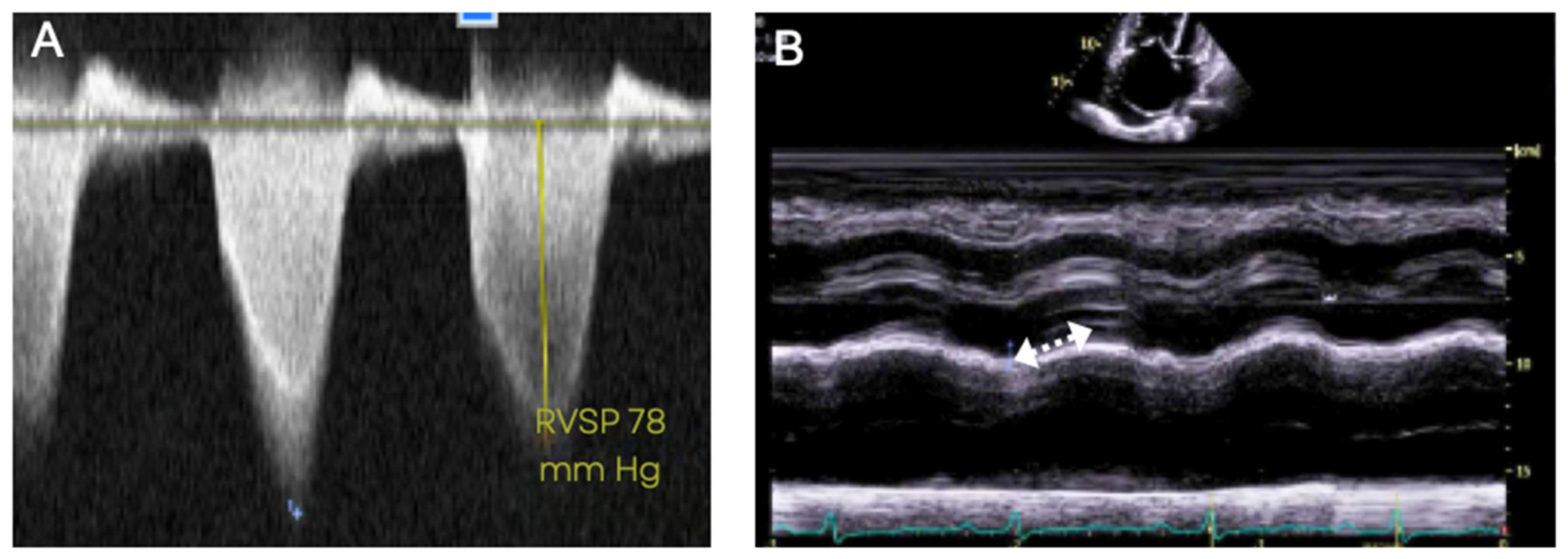
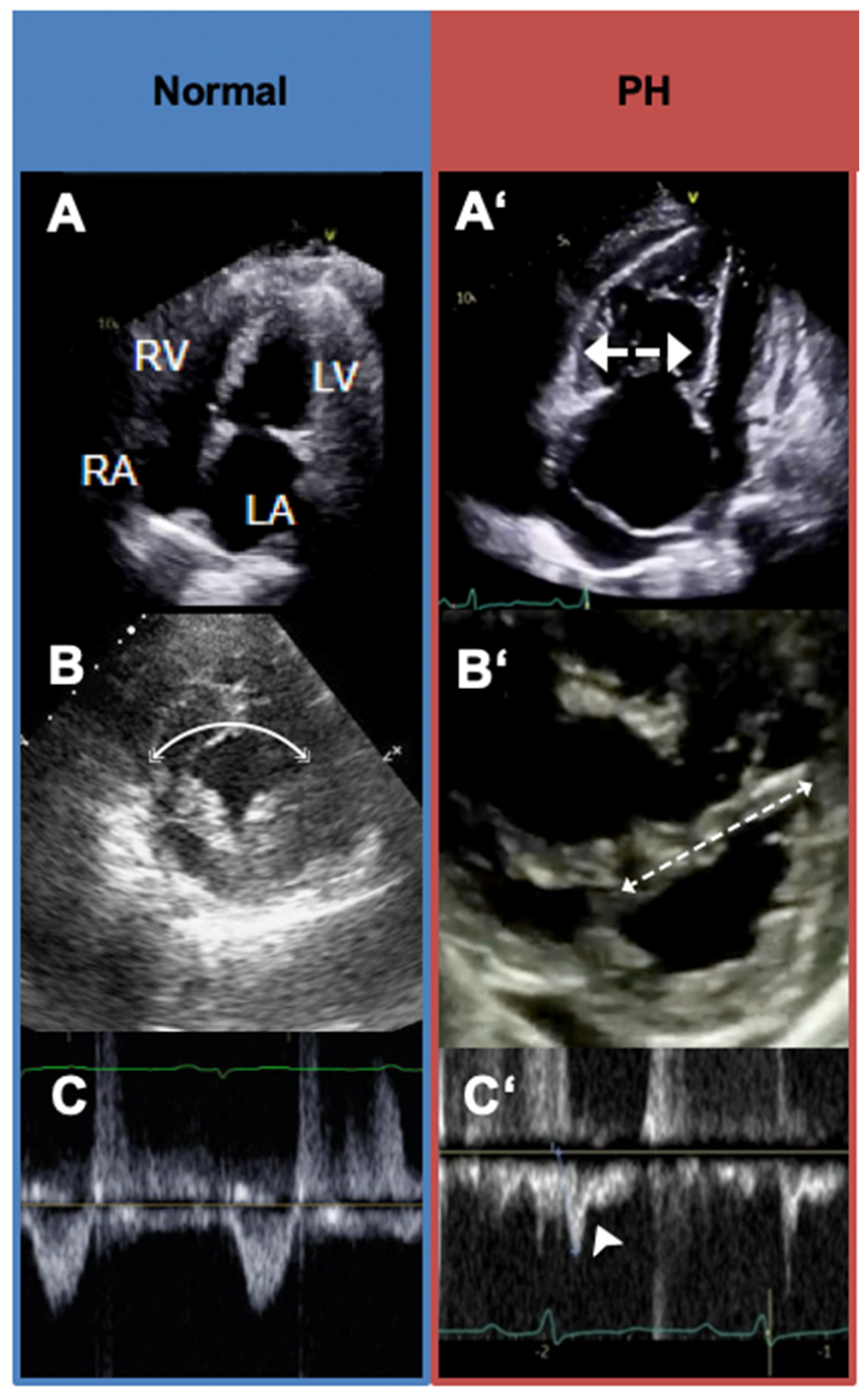
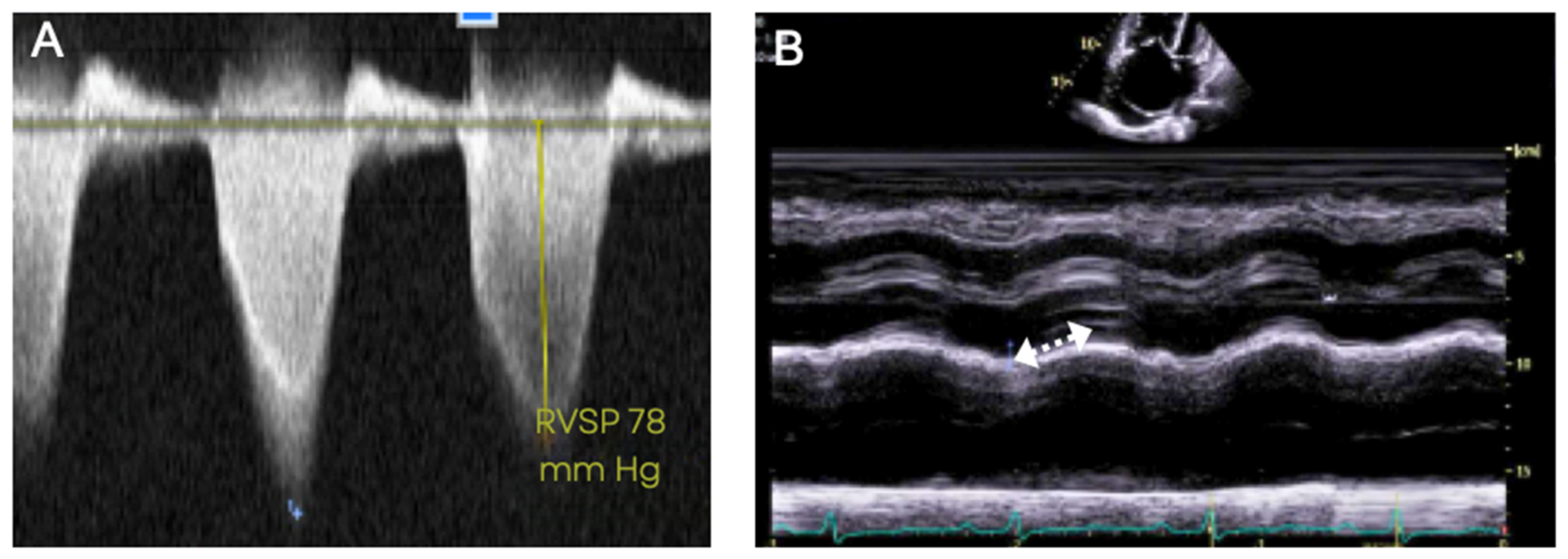
5.3. Transthoracic Echocardiography
5.4. Chest X-ray
5.5. Pulmonary Function Test
5.6. Arterial Blood Gas
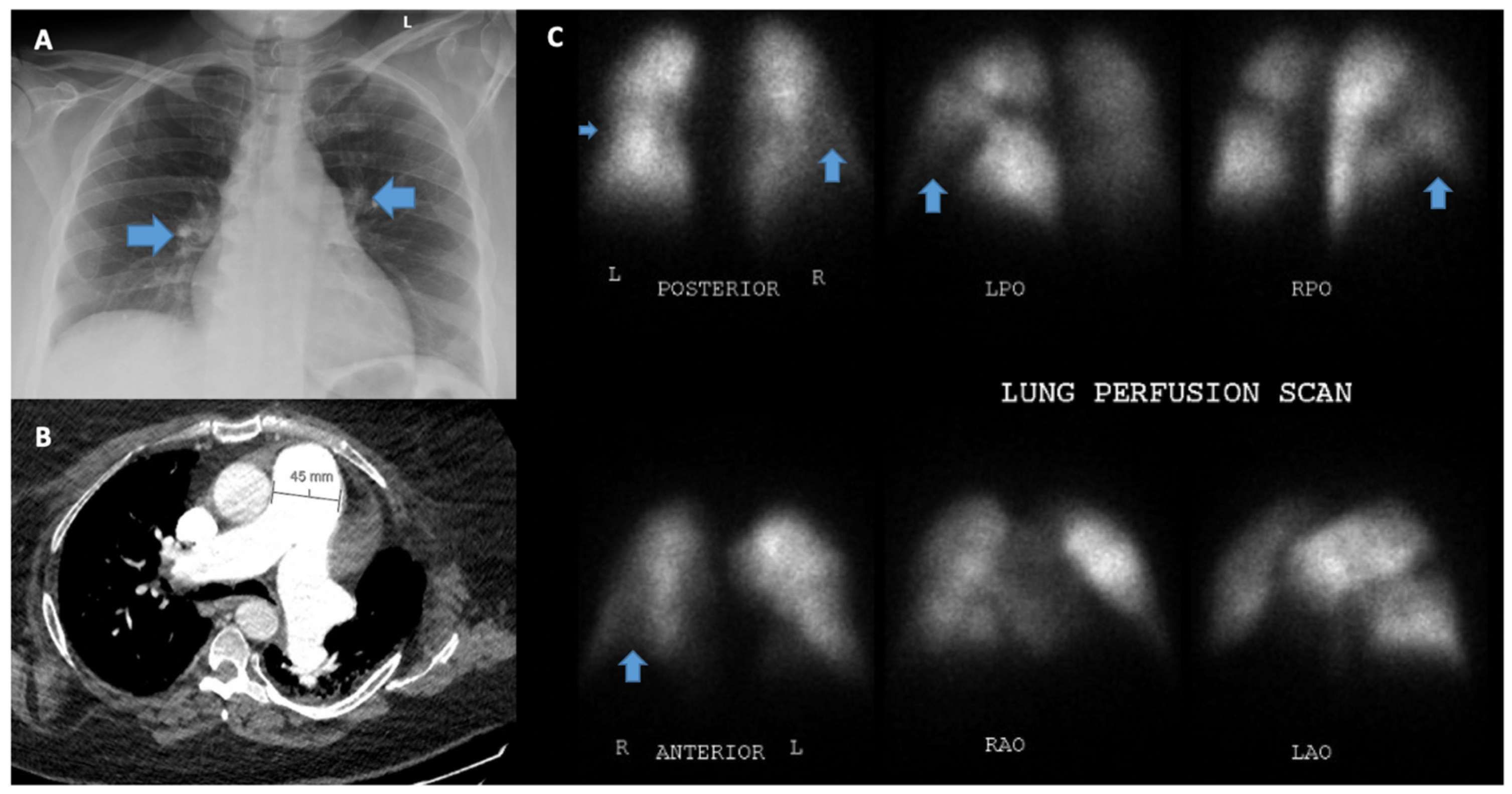
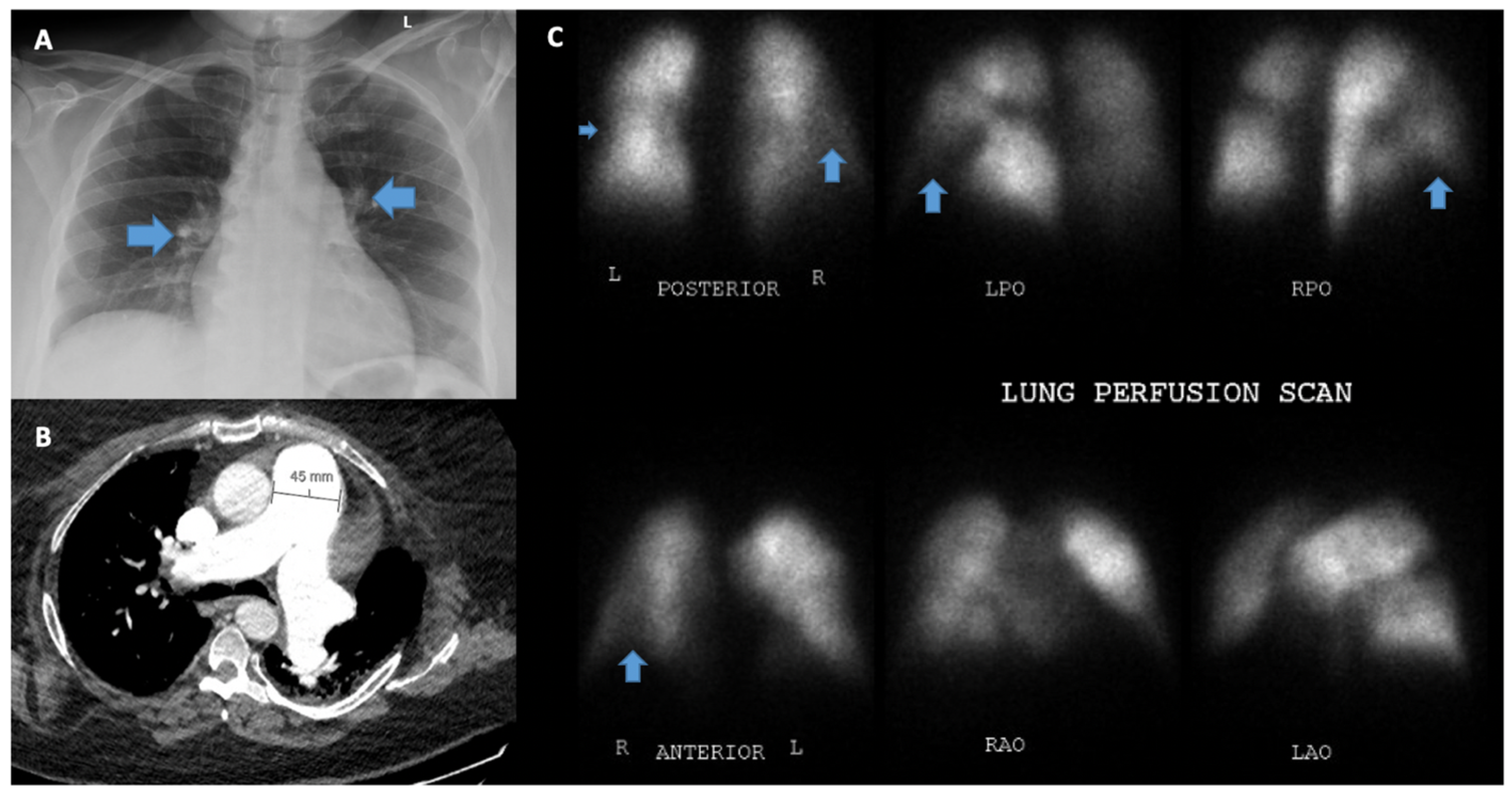
5.7. Computed Tomography of the Chest and Pulmonary Angiography
5.8. Ventilation/Perfusion Scanning (V/Q)
5.9. Cardiac Magnetic Resonance Imaging
5.10. Right Heart Catheterization
5.11. Cardiopulmonary Exercise Testing
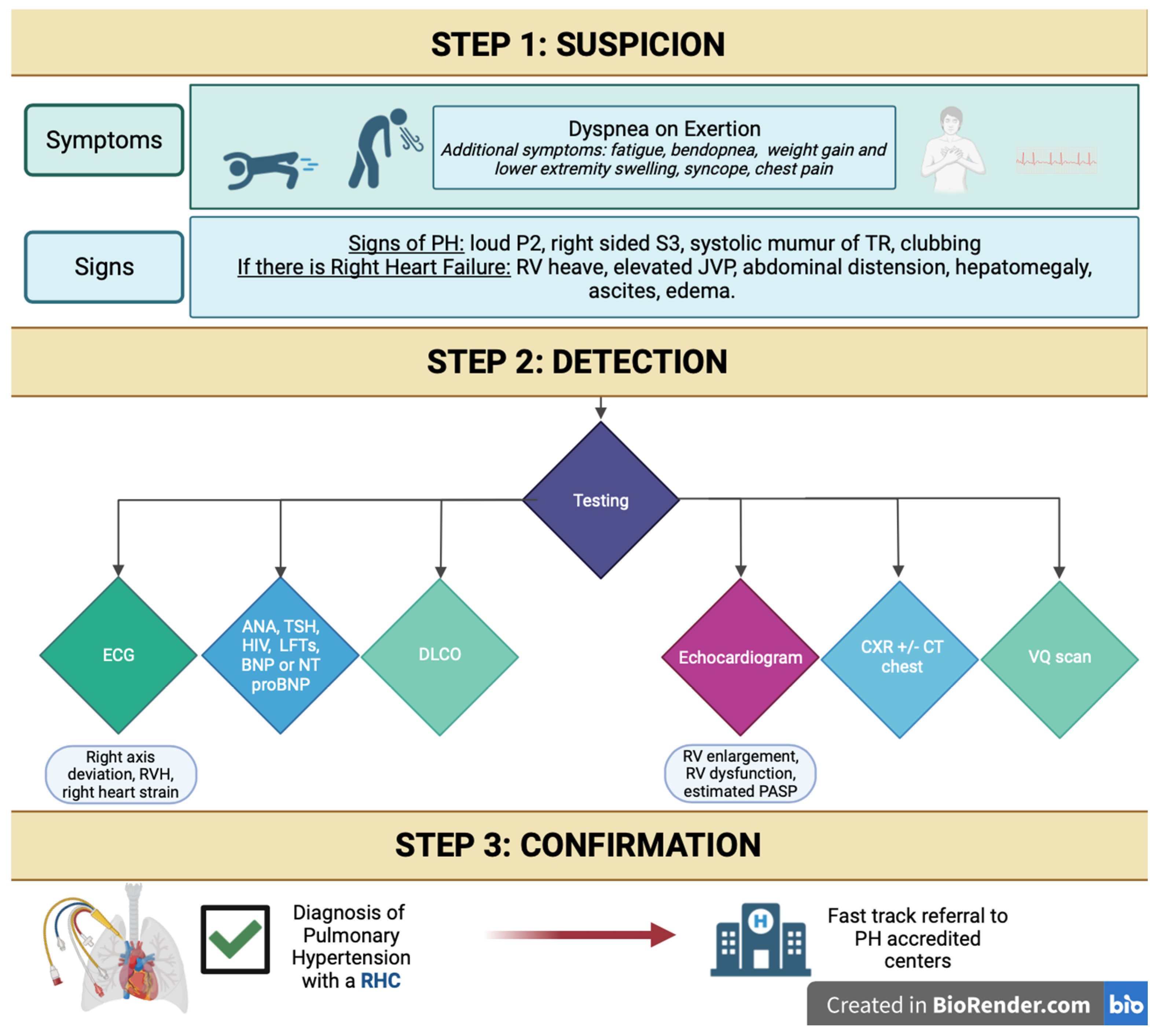
6. Suggested Initial Evaluation by the Primary Care Provider for Suspected PH and Indications for Referral to Expert PH Center
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.; Brida, M.; Carlsen, J.; Coats, A.J.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
- Brown, L.M.; Chen, H.; Halpern, S.; Taichman, D.; McGoon, M.D.; Farber, H.W.; Frost, A.E.; Liou, T.G.; Turner, M.; Feldkircher, K.; et al. Delay in recognition of pulmonary arterial hypertension: Factors identified from the REVEAL Registry. Chest 2011, 140, 19–26. [Google Scholar] [CrossRef]
- Deano, R.C.; Glassner-Kolmin, C.; Rubenfire, M.; Frost, A.; Visovatti, S.; McLaughlin, V.V.; Gomberg-Maitland, M. Referral of patients with pulmonary hypertension diagnoses to tertiary pulmonary hypertension centers: The multicenter RePHerral study. JAMA Intern. Med. 2013, 173, 887–893. [Google Scholar] [CrossRef]
- Vizza, C.D.; Badagliacca, R.; Messick, C.R.; Rao, Y.; Nelsen, A.C.; Benza, R.L. The impact of delayed treatment on 6-minute walk distance test in patients with pulmonary arterial hypertension: A meta-analysis. Int. J. Cardiol. 2018, 254, 299–301. [Google Scholar] [CrossRef]
- Gaine, S.; Sitbon, O.; Channick, R.N.; Chin, K.M.; Sauter, R.; Galiè, N.; Hoeper, M.M.; McLaughlin, V.V.; Preiss, R.; Rubin, L.J.; et al. Relationship Between Time From Diagnosis and Morbidity/Mortality in Pulmonary Arterial Hypertension: Results From the Phase III GRIPHON Study. Chest 2021, 160, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Mandras, S.A.; Ventura, H.O.; Corris, P.A. Breaking Down the Barriers: Why the Delay in Referral for Pulmonary Arterial Hypertension? Ochsner J. 2016, 16, 257–262. [Google Scholar] [PubMed]
- Gomberg-Maitland, M.; Dufton, C.; Oudiz, R.J.; Benza, R.L. Compelling evidence of long-term outcomes in pulmonary arterial hypertension? A clinical perspective. J. Am. Coll. Cardiol. 2011, 57, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Archer, S.L. The right ventricle in pulmonary arterial hypertension: Disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef]
- van Wolferen, S.A.; Marcus, J.T.; Westerhof, N.; Spreeuwenberg, M.D.; Marques, K.M.; Bronzwaer, J.G.; Henkens, I.R.; Gan, C.T.; Boonstra, A.; Postmus, P.E.; et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur. Heart J. 2008, 29, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Oldroyd, S.H.; Manek, G.; Sankari, A.; Bhardwaj, A. Pulmonary Hypertension; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Braganza, M.; Shaw, J.; Solverson, K.; Vis, D.; Janovcik, J.; Varughese, R.A.; Thakrar, M.V.; Hirani, N.; Helmersen, D.; Weatherald, J. A Prospective Evaluation of the Diagnostic Accuracy of the Physical Examination for Pulmonary Hypertension. Chest 2019, 155, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Reddy, Y.N.; Kaye, D.M.; Handoko, M.L.; Van De Bovenkamp, A.A.; Tedford, R.J.; Keck, C.; Andersen, M.J.; Sharma, S.; Trivedi, R.K.; Carter, R.E.; et al. Diagnosis of Heart Failure with Preserved Ejection Fraction Among Patients with Unexplained Dyspnea. JAMA Cardiol. 2022, 7, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, M.; Torbicki, A.; Gopalan, D.; Sitbon, O.; Klok, F.A.; Lang, I.; Jenkins, D.; Kim, N.H.; Humbert, M.; Jais, X.; et al. ERS statement on chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2021, 57, 2002828. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Müller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Doelberg, M.; et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 2014, 73, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Gering, L.E.; Knilans, T.K.; Surawicz, B.; Tavel, M.E. Chou’s Electrocardiography in Clinical Practice, 6th ed.; Surawicz, B., Knilans, T.K., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2008; p. ix. [Google Scholar]
- Balieva, I.; Dzudie, A.; Thienemann, F.; Mocumbi, A.O.; Karaye, K.; Sani, M.U.; Ogah, O.S.; Voors, A.A.; Kengne, A.P.; Sliwa, K. Prevalence and predictive value of electrocardiographic abnormalities in pulmonary hypertension: Evidence from the Pan-African Pulmonary Hypertension Cohort (PAPUCO) study. Cardiovasc. J. Afr. 2017, 28, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Janda, S.; Shahidi, N.; Gin, K.; Swiston, J. Diagnostic accuracy of echocardiography for pulmonary hypertension: A systematic review and meta-analysis. Heart 2011, 97, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Ratanawatkul, P.; Oh, A.; Richards, J.C.; Swigris, J.J. Performance of pulmonary artery dimensions measured on high-resolution computed tomography scan for identifying pulmonary hypertension. ERJ Open Res. 2020, 6, 00232–2019. [Google Scholar] [CrossRef]
- Tunariu, N.; Gibbs, S.J.; Win, Z.; Gin-Sing, W.; Graham, A.; Gishen, P.; Adil, A.N. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J. Nucl. Med. 2007, 48, 680–684. [Google Scholar] [CrossRef]
- He, J.; Fang, W.; Lv, B.; He, J.G.; Xiong, C.M.; Liu, Z.H.; He, Z. Diagnosis of chronic thromboembolic pulmonary hypertension: Comparison of ventilation/perfusion scanning and multidetector computed tomography pulmonary angiography with pulmonary angiography. Nucl. Med. Commun. 2012, 33, 459–463. [Google Scholar] [CrossRef]
- McLure, L.E.; Peacock, A.J. Cardiac magnetic resonance imaging for the assessment of the heart and pulmonary circulation in pulmonary hypertension. Eur. Respir. J. 2009, 33, 1454–1466. [Google Scholar] [CrossRef]
- Kovacs, G.; Avian, A.; Foris, V.; Tscherner, M.; Kqiku, X.; Douschan, P.; Bachmaier, G.; Olschewski, A.; Matucci-Cerinic, M.; Olschewski, H. Use of ECG and Other Simple Non-Invasive Tools to Assess Pulmonary Hypertension. PLoS ONE 2016, 11, e0168706. [Google Scholar] [CrossRef]
- Bossone, E.; Paciocco, G.; Iarussi, D.; Agretto, A.; Iacono, A.; Gillespie, B.W.; Rubenfire, M. The prognostic role of the ECG in primary pulmonary hypertension. Chest 2002, 121, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Lafitte, S.; Pillois, X.; Reant, P.; Picard, F.; Arsac, F.; Dijos, M.; Coste, P.; Dos Santos, P.; Roudaut, R. Estimation of pulmonary pressures and diagnosis of pulmonary hypertension by Doppler echocardiography: A retrospective comparison of routine echocardiography and invasive hemodynamics. J. Am. Soc. Echocardiogr. 2013, 26, 457–463. [Google Scholar] [CrossRef]
- Fisher, M.R.; Forfia, P.R.; Chamera, E.; Housten-Harris, T.; Champion, H.C.; Girgis, R.E.; Corretti, M.C.; Hassoun, P.M. Accuracy of Doppler echocardiography in the hemodynamic assessment of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2009, 179, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Kowal-Bielecka, O.; Avouac, J.; Pittrow, D.; Huscher, D.; Behrens, F.; Denton, C.P.; Foeldvari, I.; Humbert, M.; Matucci-Cerinic, M.; Nash, P.; et al. Echocardiography as an outcome measure in scleroderma-related pulmonary arterial hypertension: A systematic literature analysis by the EPOSS group. J. Rheumatol. 2010, 37, 105–115. [Google Scholar] [CrossRef]
- Opotowsky, A.R.; Ojeda, J.; Rogers, F.; Prasanna, V.; Clair, M.; Moko, L.; Vaidya, A.; Afilalo, J.; Forfia, P.R. A simple echocardiographic prediction rule for hemodynamics in pulmonary hypertension. Circ. Cardiovasc. Imaging 2012, 5, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Forfia, P.R. Diagnosis and assessment of pulmonary vascular disease by Doppler echocardiography. Pulm. Circ. 2011, 1, 160–181. [Google Scholar] [CrossRef]
- Forfia, P.R.; Fisher, M.R.; Mathai, S.C.; Housten-Harris, T.; Hemnes, A.R.; Borlaug, B.A.; Chamera, E.; Corretti, M.C.; Champion, H.C.; Abraham, T.P.; et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 1034–1041. [Google Scholar] [CrossRef]
- Vaidya, A.; Golbus, J.R.; Vedage, N.A.; Mazurek, J.; Raza, F.; Forfia, P.R. Virtual echocardiography screening tool to differentiate hemodynamic profiles in pulmonary hypertension. Pulm. Circ. 2020, 10, 2045894020950225. [Google Scholar] [CrossRef]
- Vedage, N.A.; Forfia, P.R.; Grafstrom, A.; Vaidya, A. Virtual Echocardiography Screening Tool Identifies Pulmonary Arterial Hypertension Significantly Earlier Than High-Risk Clinical Diagnosis. Am. J. Cardiol. 2023, 201, 328–334. [Google Scholar] [CrossRef]
- Remy-Jardin, M.; Ryerson, C.J.; Schiebler, M.L.; Leung, A.N.; Wild, J.M.; Hoeper, M.M.; Alderson, P.O.; Goodman, L.R.; Mayo, J.; Haramati, L.B.; et al. Imaging of pulmonary hypertension in adults: A position paper from the Fleischner Society. Eur. Respir. J. 2021, 57. [Google Scholar] [CrossRef]
- Ascha, M.; Renapurkar, R.D.; Tonelli, A.R. A review of imaging modalities in pulmonary hypertension. Ann. Thorac. Med. 2017, 12, 61–73. [Google Scholar] [PubMed]
- Hoeper, M.M.; Dwivedi, K.; Pausch, C.; A Lewis, R.; Olsson, K.M.; Huscher, D.; Pittrow, D.; Grünig, E.; Staehler, G.; Vizza, C.D.; et al. Phenotyping of idiopathic pulmonary arterial hypertension: A registry analysis. Lancet Respir. Med. 2022, 10, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Meyer, K.; Rademacher, J.; Fuge, J.; Welte, T.; Olsson, K.M. Diffusion Capacity and Mortality in Patients With Pulmonary Hypertension Due to Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.P.; Johnson, B.D.; Borlaug, B.A. Impaired Pulmonary Diffusion in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Fuge, J.; Meyer, K.; Welte, T.; Hoeper, M.M. More on idiopathic pulmonary arterial hypertension with a low diffusing capacity. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Trip, P.; Nossent, E.J.; de Man, F.S.; van den Berk, I.A.; Boonstra, A.; Groepenhoff, H.; Leter, E.M.; Westerhof, N.; Grünberg, K.; Bogaard, H.-J.; et al. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: Patient characteristics and treatment responses. Eur. Respir. J. 2013, 42, 1575–1585. [Google Scholar] [CrossRef]
- Jilwan, F.N.; Escourrou, P.; Garcia, G.; Jais, X.; Humbert, M.; Roisman, G. High occurrence of hypoxemic sleep respiratory disorders in precapillary pulmonary hypertension and mechanisms. Chest 2013, 143, 47–55. [Google Scholar] [CrossRef]
- Swift, A.J.; Dwivedi, K.; Johns, C.; Garg, P.; Chin, M.; Currie, B.J.; Rothman, A.M.; Capener, D.; Shahin, Y.; A Elliot, C.; et al. Diagnostic accuracy of CT pulmonary angiography in suspected pulmonary hypertension. Eur. Radiol. 2020, 30, 4918–4929. [Google Scholar] [CrossRef]
- Grünig, E.; Peacock, A.J. Imaging the heart in pulmonary hypertension: An update. Eur. Respir. Rev. 2015, 24, 653–664. [Google Scholar] [CrossRef]
- Kasai, H.; Tanabe, N.; Fujimoto, K.; Hoshi, H.; Naito, J.; Suzuki, R.; Matsumura, A.; Sugiura, T.; Sakao, S.; Tatsumi, K.; et al. Mosaic attenuation pattern in non-contrast computed tomography for the assessment of pulmonary perfusion in chronic thromboembolic pulmonary hypertension. Respir. Investig. 2017, 55, 300–307. [Google Scholar] [CrossRef]
- Kligerman, S.; Horowitz, M.; Hahn, L.; Hsiao, A.; Weihe, E. Multimodality Imaging of Pulmonary Hypertension. Adv. Pulm. Hypertens. 2019, 18, 115–125. [Google Scholar] [CrossRef]
- Giordano, J.; Khung, S.; Duhamel, A.; Hossein-Foucher, C.; Bellevre, D.; Lamblin, N.; Remy, J.; Remy-Jardin, M. Lung perfusion characteristics in pulmonary arterial hypertension (PAH) and peripheral forms of chronic thromboembolic pulmonary hypertension (pCTEPH): Dual-energy CT experience in 31 patients. Eur. Radiol. 2017, 27, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ma, R.; Wu, D.; Xiong, C.; He, J.; Wang, L.; Sun, X.; Fang, W. Value of lung perfusion scintigraphy in patients with idiopathic pulmonary arterial hypertension: A patchy pattern to consider. Pulm. Circ. 2019, 9, 2045894018816968. [Google Scholar] [CrossRef] [PubMed]
- Alabed, S.; Shahin, Y.; Garg, P.; Alandejani, F.; Johns, C.S.; Lewis, R.A.; Condliffe, R.; Wild, J.M.; Kiely, D.G.; Switft, A.J. Cardiac-MRI Predicts Clinical Worsening and Mortality in Pulmonary Arterial Hypertension: A Systematic Review and Meta-Analysis. JACC Cardiovasc. Imaging 2021, 14, 931–942. [Google Scholar] [CrossRef]
- Cerne, J.W.; Pathrose, A.; Gordon, D.Z.; Sarnari, R.; Veer, M.; Blaisdell, J.; Allen, B.D.; Avery, R.; Markl, M.; Ragin, A.; et al. Evaluation of Pulmonary Hypertension Using 4D Flow MRI. J. Magn. Reason. Imaging 2022, 56, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Lee, S.H.; Voswinckel, R.; Palazzini, M.; Jais, X.; Marinelli, A.; Barst, R.J.; Ghofrani, H.A.; Jing, Z.-H.; Opitz, C.; et al. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J. Am. Coll. Cardiol. 2006, 48, 2546–2552. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Kovacs, G.; Vaidya, A.; Bhatt, D.L.; Nishimura, R.A.; Mak, S.; Guazzi, M.; Tedford, R.J. Cardiopulmonary Hemodynamics in Pulmonary Hypertension and Heart Failure: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 76, 2671–2681. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Preston, I.R. Right heart catheterisation: Best practice and pitfalls in pulmonary hypertension. Eur. Respir. Rev. 2015, 24, 642–652. [Google Scholar] [CrossRef]
- Kovacs, G.; Douschan, P.; Maron, B.A.; Condliffe, R.; Olschewski, H. Mildly increased pulmonary arterial pressure: A new disease entity or just a marker of poor prognosis? Eur. J. Heart Fail 2019, 21, 1057–1061. [Google Scholar] [CrossRef]
- Maron, B.A.; Brittain, E.L.; Hess, E.; Waldo, S.W.; Barón, A.E.; Huang, S.; Goldstein, R.H.; Assad, T.; Wertheim, B.W.; Alba, A.G.; et al. Pulmonary vascular resistance and clinical outcomes in patients with pulmonary hypertension: A retrospective cohort study. Lancet Respir. Med. 2020, 8, 873–884. [Google Scholar] [CrossRef]
- Xanthouli, P.; Jordan, S.; Milde, N.; Marra, A.; Blank, N.; Egenlauf, B.; Gorenflo, M.; Harutyunova, S.; Lorenz, H.-M.; Nagel, C.; et al. Haemodynamic phenotypes and survival in patients with systemic sclerosis: The impact of the new definition of pulmonary arterial hypertension. Ann. Rheum. Dis. 2020, 79, 370–378. [Google Scholar] [CrossRef]
- Vachiéry, J.L.; Galiè, N.; Barberá, J.A.; Frost, A.E.; Ghofrani, H.A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Blair, C.; et al. Initial combination therapy with ambrisentan + tadalafil on pulmonary arterial hypertension-related hospitalization in the AMBITION trial. J. Heart Lung Transpl. 2019, 38, 194–202. [Google Scholar] [CrossRef]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.-A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Rubin, L.J.; Long, W.A.; McGoon, M.D.; Rich, S.; Badesch, D.B.; Groves, B.M.; Tapson, V.F.; Bourge, R.C.; Brundage, B.H.; et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N. Engl. J. Med. 1996, 334, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, A.R.; Mubarak, K.K.; Li, N.; Carrie, R.; Alnuaimat, H. Effect of balloon inflation volume on pulmonary artery occlusion pressure in patients with and without pulmonary hypertension. Chest 2011, 139, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.G.; Hansen, J.E.; Oudiz, R.J.; Wasserman, K. Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation 2001, 104, 429–435. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tests | Sensitivity | Specificity | Benefit | Limitation |
|---|---|---|---|---|
| ECG | 20% [15] | 79.3–100% [16] | Easy to obtain, provides important clues to PH when symptoms are present, helps detect arrhythmia. | ECG considered inadequate for screening. |
| TTE [17] | 83% | 72% | Useful initial noninvasive modality for screening and measurement of pulmonary pressures. | Dependence on the quality of imaging, difficulty in image acquisition with increased RV volumes, steady heart rate, and experience of the laboratory staff. |
| CT chest [18] | 74–79% | 81–83% | CT chest allows for comprehensive evaluation of the pulmonary vasculature and lung parenchyma. | Radiation exposure. |
| VQ scan [19,20] | 90–100% | 94–100% | Allows us to distinguish CTEPH from other forms of PH, negative test helpful for ruling out CTEPH. | Low utility in diagnosing causes of PH other than thromboembolic disease. |
| CMR [21] | 84% | 71% | Provides a comprehensive evaluation of the heart, good for quantification of right ventricular volumes, mass and function. | CMR is expensive, not widely available, and requires significant operator expertise.Also limited lung parenchyma evaluation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anand, S.; Sadek, A.; Vaidya, A.; Oliveros, E. Diagnostic Evaluation of Pulmonary Hypertension: A Comprehensive Approach for Primary Care Physicians. J. Clin. Med. 2023, 12, 7309. https://doi.org/10.3390/jcm12237309
Anand S, Sadek A, Vaidya A, Oliveros E. Diagnostic Evaluation of Pulmonary Hypertension: A Comprehensive Approach for Primary Care Physicians. Journal of Clinical Medicine. 2023; 12(23):7309. https://doi.org/10.3390/jcm12237309
Chicago/Turabian StyleAnand, Suneesh, Ahmed Sadek, Anjali Vaidya, and Estefania Oliveros. 2023. "Diagnostic Evaluation of Pulmonary Hypertension: A Comprehensive Approach for Primary Care Physicians" Journal of Clinical Medicine 12, no. 23: 7309. https://doi.org/10.3390/jcm12237309
APA StyleAnand, S., Sadek, A., Vaidya, A., & Oliveros, E. (2023). Diagnostic Evaluation of Pulmonary Hypertension: A Comprehensive Approach for Primary Care Physicians. Journal of Clinical Medicine, 12(23), 7309. https://doi.org/10.3390/jcm12237309