
Anticipating New Treatments for Cystic Fibrosis: A Global Survey of Researchers




Abstract
:1. Introduction
2. Materials and Methods
2.1. Literature Review and Questionnaire Design
2.2. Collecting Respondents in Scientific Publications
2.3. Survey Management, Questionnaire Application, and Ethical Aspects
2.4. Statistical Analysis
2.5. Limitations of the Study
3. Results
3.1. Statistical Analysis
3.2. Descriptive Statistics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- European Cystic Fibrosis Society. ECFS Patient Registry. 2020. Available online: https://www.ecfs.eu/ecfspr (accessed on 7 October 2021).
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic Fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation. 2019 Patient Registry Annual Data Report. 2020. Available online: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2019-Patient-Registry-Annual-Data-Report.pdf (accessed on 7 October 2021).
- Cystic Fibrosis Foundation. Gene Therapy for Cystic Fibrosis. Available online: https://www.cff.org/Research/Research-Into-the-Disease/Restore-CFTR-Function/Gene-Therapy-for-Cystic-Fibrosis/ (accessed on 13 August 2021).
- Bell, S.C.; De Boeck, K.; Amaral, M. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacol. Ther. 2015, 145, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrini, G. Innovative Therapies for Cystic Fibrosis: The Road from Treatment to Cure. Mol. Diagn. Ther. 2019, 23, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Carlon, M.S.; Vidović, D.; Birket, S. Roadmap for an early gene therapy for cystic fibrosis airway disease. Prenat. Diagn. 2017, 37, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; McCray, J.P.B.; Sinn, P.L. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 2018, 9, 538. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Guerra, L.; Favia, M.; Di Gioia, S.; Laselva, O.; Bisogno, A.; Casavola, V.; Colombo, C.; Conese, M. The preclinical discovery and development of the combination of ivacaftor + tezacaftor used to treat cystic fibrosis. Expert Opin. Drug Discov. 2020, 15, 873–891. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.D.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Carnovale, V.; Iacotucci, P.; Terlizzi, V.; Colangelo, C.; Medio, P.; Ferrillo, L.; De Gregorio, F.; Francalanci, M.; Taccetti, G.; Buonaurio, S.; et al. Effectiveness and safety of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease with the Phe508del/minimal function genotype. Respir. Med. 2021, 189, 106646. [Google Scholar] [CrossRef]
- Salvatore, D.; Terlizzi, V.; Francalanci, M.; Taccetti, G.; Messore, B.; Biglia, C.; Pisi, G.; Calderazzo, M.A.; Caloiero, M.; Pizzamiglio, G.; et al. Ivacaftor improves lung disease in patients with advanced CF carrying CFTR mutations that confer residual function. Respir. Med. 2020, 171, 106073. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, K.A.; Wachi, S.; Drew, L.; Dukovski, D.; Green, O.; Bastos, C.; Cullen, M.D.; Hauck, S.; Tait, B.D.; Munoz, B.; et al. Use of a High-Throughput Phenotypic Screening Strategy to Identify Amplifiers, a Novel Pharmacological Class of Small Molecules That Exhibit Functional Synergy with Potentiators and Correctors. SLAS Discov. Adv. Life Sci. R&D 2018, 23, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Molinski, S.V.; Ahmadi, S.; Ip, W.; Ouyang, H.; Villella, A.; Miller, J.P.; Lee, P.; Kulleperuma, K.; Du, K.; Di Paola, M.; et al. O rkambi® and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol. Med. 2017, 9, 1224–1243. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.H.; Solomon, G.M.; Zeitlin, P.; Flume, P.A.; Casey, A.; McCoy, K.; Zemanick, E.T.; Mandagere, A.; Troha, J.M.; Shoemaker, S.A.; et al. Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. J. Cyst. Fibros. 2017, 16, 371–379. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2019, 10, 1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Alton, E.W.; Boyd, A.C.; Davies, J.; Gill, D.; Griesenbach, U.; Harrison, P.; Henig, N.; Higgins, T.; Hyde, S.C.; Innes, J.A.; et al. Genetic medicines for CF: Hype versus reality. Pediatr. Pulmonol. 2016, 51, S5–S17. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; McCray, P.B., Jr.; Engelhardt, J.F. Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet. 2019, 28, R88–R94. [Google Scholar] [CrossRef] [Green Version]
- Miah, K.M.; Hyde, S.C.; Gill, D.R. Emerging gene therapies for cystic fibrosis. Expert Rev. Respir. Med. 2019, 13, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.-S.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Bardin, P.; Sonneville, F.; Corvol, H.; Tabary, O. Emerging microRNA Therapeutic Approaches for Cystic Fibrosis. Front. Pharmacol. 2018, 9, 1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, M.A.; Lee, H.-C.; Lin, L.; Van Haasteren, J.; Schaffer, D. Novel lung tropic AAV capsids for therapeutic gene delivery. Hum. Gene Ther. 2020, 31, 996–1009. [Google Scholar] [CrossRef]
- Guggino, W.B.; Cebotaru, L. Adeno-Associated Virus (AAV) gene therapy for cystic fibrosis: Current barriers and recent developments. Expert Opin. Biol. Ther. 2017, 17, 1265–1273. [Google Scholar] [CrossRef]
- Loza, L.I.M.; Yuen, E.C.; McCray, J.P.B. Lentiviral Vectors for the Treatment and Prevention of Cystic Fibrosis Lung Disease. Genes 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Ong, V.; Mei, V.; Cao, L.; Lee, K.; Chung, E.J. Nanomedicine for Cystic Fibrosis. SLAS Technol. Transl. Life Sci. Innov. 2019, 24, 169–180. [Google Scholar] [CrossRef]
- Nishida, K.; Smith, Z.; Rana, D.; Palmer, J.; Gallicano, G.I. Cystic fibrosis: A look into the future of prenatal screening and therapy. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 73–80. [Google Scholar] [CrossRef]
- Bañuls, L.; Pellicer, D.; Castillo, S.; Navarro-García, M.M.; Magallón, M.; González, C.; Dasí, F. Gene Therapy in Rare Respiratory Diseases: What Have We Learned So Far? J. Clin. Med. 2020, 9, 2577. [Google Scholar] [CrossRef]
- De Boeck, K.; Amaral, M. Progress in therapies for cystic fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Boyd, A.C.; Guo, S.; Huang, L.; Kerem, B.; Oren, Y.S.; Walker, A.J.; Hart, S.L. New approaches to genetic therapies for cystic fibrosis. J. Cyst. Fibros. 2020, 19, S54–S59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, S.C.; McKone, E.F. Pharmacogenetics of cystic fibrosis treatment. Pharmacogenomics 2016, 17, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Cholon, D.M.; Gentzsch, M. Recent progress in translational cystic fibrosis research using precision medicine strategies. J. Cyst. Fibros. 2018, 17, S52–S60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, R.; Paul, L.; Manickam, R.; Aronow, W.S.; Maguire, G.P. Efficacy and adverse effects of drugs used to treat adult cystic fibrosis. Expert Opin. Drug Saf. 2015, 14, 401–411. [Google Scholar] [CrossRef]
- Conese, M.; Beccia, E.; Carbone, A.; Castellani, S.; Di Gioia, S.; Corti, F.; Angiolillo, A.; Colombo, C. The role of stem cells in cystic fibrosis disease modeling and drug discovery. Expert Opin. Orphan Drugs 2018, 6, 707–717. [Google Scholar] [CrossRef]
- Cuyx, S.; De Boeck, K. Treating the Underlying Cystic Fibrosis Transmembrane Conductance Regulator Defect in Patients with Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 762–774. [Google Scholar] [CrossRef]
- Dhooghe, B.; Haaf, J.B.; Noël, S.; Leal, T. Strategies in early clinical development for the treatment of basic defects of cystic fibrosis. Expert Opin. Investig. Drugs 2016, 25, 423–436. [Google Scholar] [CrossRef]
- Fajac, I.; De Boeck, K. New horizons for cystic fibrosis treatment. Pharmacol. Ther. 2017, 170, 205–211. [Google Scholar] [CrossRef]
- Fajac, I.; Wainwright, C. New treatments targeting the basic defects in cystic fibrosis. Presse Méd. 2017, 46, e165–e175. [Google Scholar] [CrossRef]
- Griesenbach, U.; Davies, J.C.; Alton, E. Cystic fibrosis gene therapy: A mutation-independent treatment. Curr. Opin. Pulm. Med. 2016, 22, 602–609. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Siegwart, D.J.; Strug, L.J.; Cebotaru, L.; Torres, M.J.; Sofoluwe, A.; Beekman, J.M. Translational research to enable personalized treatment of cystic fibrosis. J. Cyst. Fibros. 2018, 17, S46–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, C.A.; Conlon, R.A. Delivering on the promise of gene editing for cystic fibrosis. Genes Dis. 2019, 6, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Hubert, D.; Bui, S.; Marguet, C.; Colomb-Jung, V.; Murris-Espin, M.; Corvol, H.; Munck, A. Nouvelles thérapeutiques de la mucoviscidose ciblant le gène ou la protéine CFTR. Rev. Mal. Respir. 2016, 33, 658–665. [Google Scholar] [CrossRef]
- Kim, N.; Duncan, G.; Hanes, J.; Suk, J.S. Barriers to inhaled gene therapy of obstructive lung diseases: A review. J. Control. Release 2016, 240, 465–488. [Google Scholar] [CrossRef] [Green Version]
- Loring, H.S.; ElMallah, M.K.; Flotte, T.R. Development of rAAV2-CFTR: History of the First rAAV Vector Product to be Used in Humans. Hum. Gene Ther. Methods 2016, 27, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marangi, M.; Pistritto, G. Innovative Therapeutic Strategies for Cystic Fibrosis: Moving Forward to CRISPR Technique. Front. Pharmacol. 2018, 9, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martiniano, S.L.; Sagel, S.D.; Zemanick, E. Cystic fibrosis: A model system for precision medicine. Curr. Opin. Pediatr. 2016, 28, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Maule, G.; Arosio, D.; Cereseto, A. Gene Therapy for Cystic Fibrosis: Progress and Challenges of Genome Editing. Int. J. Mol. Sci. 2020, 21, 3903. [Google Scholar] [CrossRef]
- Mondejar-Lopez, P.; Pastor-Vivero, M.D.; Sanchez-Solis, M.; Escribano, A. Cystic fibrosis treatment: Targeting the basic defect. Expert Opin. Orphan Drugs 2016, 5, 181–192. [Google Scholar] [CrossRef]
- Parsons, D.; Donnelley, M. Will Airway Gene Therapy for Cystic Fibrosis Improve Lung Function? New Imaging Technologies Can Help Us Find Out. Hum. Gene Ther. 2020, 31, 973–984. [Google Scholar] [CrossRef]
- Paul-Smith, M.C.; Bell, R.V.; Alton, W.E.; Alton, E.W.; Griesenbach, U. Gene therapy for cystic fibrosis: Recent progress and current aims. Expert Opin. Orphan Drugs 2016, 4, 649–658. [Google Scholar] [CrossRef]
- Prakash, V.; Moore, M.; Yáñez-Muñoz, R.J. Current Progress in Therapeutic Gene Editing for Monogenic Diseases. Mol. Ther. 2016, 24, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafeeq, M.M.; Murad, H.A.S. Cystic fibrosis: Current therapeutic targets and future approaches. J. Transl. Med. 2017, 15, 84. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.; Chanson, M.; Gawenis, L.R.; Liu, J.; Sofoluwe, A.; Zoso, A.; Engelhardt, J.F. Animal and model systems for studying cystic fibrosis. J. Cyst. Fibros. 2018, 17, S28–S34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider-Futschik, E.K. Beyond cystic fibrosis transmembrane conductance regulator therapy: A perspective on gene therapy and small molecule treatment for cystic fibrosis. Gene Ther. 2019, 26, 354–362. [Google Scholar] [CrossRef]
- Skov, M.; Hansen, C.R.; Pressler, T. Cystic fibrosis—An example of personalized and precision medicine. APMIS 2019, 127, 352–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondhi, D.; Stiles, K.M.; De, B.P.; Crystal, R.G. Genetic Modification of the Lung Directed Toward Treatment of Human Disease. Hum. Gene Ther. 2017, 28, 3–84. [Google Scholar] [CrossRef]
- Turnbull, A.R.; Davies, J. New drug developments in the management of cystic fibrosis lung disease. Expert Opin. Pharmacother. 2016, 17, 1103–1112. [Google Scholar] [CrossRef]
- Van Haasteren, J.; Hyde, S.C.; Gill, D.R. Lessons learned from lung and liver in-vivo gene therapy: Implications for the future. Expert Opin. Biol. Ther. 2018, 18, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Velino, C.; Carella, F.; Adamiano, A.; Sanguinetti, M.; Vitali, A.; Catalucci, D.; Bugli, F.; Iafisco, M. Nanomedicine Approaches for the Pulmonary Treatment of Cystic Fibrosis. Front. Bioeng. Biotechnol. 2019, 7, 406. [Google Scholar] [CrossRef] [Green Version]
- Villate-Beitia, I.; Zarate, J.; Puras, G.; Pedraz, J.L. Gene delivery to the lungs: Pulmonary gene therapy for cystic fibrosis. Drug Dev. Ind. Pharm. 2017, 43, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Cleophas, T.J.; Zwinderman, A.H. Non-Parametric Tests. In Statistical Analysis of Clinical Data on a Pocket Calculator; Cleophas, T.J., Zwinderman, A.H., Eds.; Springer: Dordrecht, The Netherlands, 2011; pp. 9–13. ISBN 978-94-007-1210-2. [Google Scholar]
- Rosner, B.; Glynn, R.J.; Lee, M.-L.T. The Wilcoxon Signed Rank Test for Paired Comparisons of Clustered Data. Biometrics 2006, 62, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Cabral, B.P.; Braga, L.; Mota, F. Expert Opinions on the Most Promising Treatments and Vaccine Candidates for COVID-19: Global Cross-sectional Survey of Virus Researchers in the Early Months of the Pandemic. JMIR Public Health Surveill. 2021, 7, e22483. [Google Scholar] [CrossRef] [PubMed]
- Mota, F.; Braga, L.; Rocha, L.; Cabral, B. 3D and 4D bioprinted human model patenting and the future of drug development. Nat. Biotechnol. 2020, 38, 689–694. [Google Scholar] [CrossRef]
- Mota, F.; Braga, L.A.M.; Cabral, B.P.P.; Filho, C.G.C. What is the future of lab-on-a-chip diagnostic devices? Assessing changes in experts’ expectations over time. Foresight 2021, 23, 640–654. [Google Scholar] [CrossRef]
- Cabral, B.P.; Bonventre, J.V.; Wieringa, F.; Mota, F.B. Probing expert opinions on the future of kidney replacement therapies. Artif. Organs 2021, 45, 79–87. [Google Scholar] [CrossRef]
- Cabral, B.P.; da Graça Derengowski Fonseca, M.; Mota, F.B. What is the future of cancer care? A technology foresight assessment of experts’ expectations. Econ. Innov. New Technol. 2019, 28, 635–652. [Google Scholar] [CrossRef]
- Cabral, B.P.; da Graça Derengowski Fonseca, M.; Mota, F.B. Long term prevention and vector control of arboviral diseases: What does the future hold? Int. J. Infect. Dis. 2019, 89, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; Van Der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Kerem, E.; Corey, M.; Kerem, B.-S.; Rommens, J.; Markiewicz, D.; Levison, H.; Tsui, L.-C.; Durie, P. The Relation between Genotype and Phenotype in Cystic Fibrosis—Analysis of the Most Common Mutation (ΔF508). N. Engl. J. Med. 1990, 323, 1517–1522. [Google Scholar] [CrossRef] [Green Version]
- Robinson, E.; MacDonald, K.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steines, B.; Dickey, D.D.; Bergen, J.; Excoffon, K.J.; Weinstein, J.R.; Li, X.; Yan, Z.; Alaiwa, M.A.; Shah, V.S.; Bouzek, D.C.; et al. CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight 2016, 1, e88728. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, F.; Uchibori, R.; Mato, N.; Sehara, Y.; Saga, Y.; Urabe, M.; Mizukami, H.; Sugiyama, Y.; Kume, A. Optimization of adeno-associated virus vector-mediated gene transfer to the respiratory tract. Gene Ther. 2017, 24, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Vidović, D.; Carlon, M.; Da Cunha, M.F.; Dekkers, J.F.; Hollenhorst, M.I.; Bijvelds, M.J.C.; Ramalho, A.S.; van den Haute, C.; Ferrante, M.; Baekelandt, V.; et al. rAAV-CFTRΔR Rescues the Cystic Fibrosis Phenotype in Human Intestinal Organoids and Cystic Fibrosis Mice. Am. J. Respir. Crit. Care Med. 2016, 193, 288–298. [Google Scholar] [CrossRef]
- Yan, Z.; Feng, Z.; Sun, X.; Zhang, Y.; Zou, W.; Wang, Z.; Jensen-Cody, C.; Liang, B.; Park, S.-Y.; Qiu, J.; et al. Human Bocavirus Type-1 Capsid Facilitates the Transduction of Ferret Airways by Adeno-Associated Virus Genomes. Hum. Gene Ther. 2017, 28, 612–625. [Google Scholar] [CrossRef]
- Cooney, A.L.; Abou Alaiwa, M.H.; Shah, V.S.; Bouzek, D.C.; Stroik, M.R.; Powers, L.S.; Gansemer, N.D.; Meyerholz, D.K.; Welsh, M.J.; Stoltz, D.A.; et al. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight 2016, 1, e88730. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Ouyang, H.; Laselva, O.; Bartlett, C.; Zhou, Z.P.; Duan, C.; Gunawardena, T.; Avolio, J.; Bear, C.E.; Gonska, T.; et al. A helper-dependent adenoviral vector rescues CFTR to wild-type functional levels in cystic fibrosis epithelial cells harbouring class I mutations. Eur. Respir. J. 2020, 56, 2000205. [Google Scholar] [CrossRef]
- Cooney, A.L.; Thornell, I.; Singh, B.; Shah, V.S.; Stoltz, D.A.; McCray, P.B., Jr.; Zabner, J.; Sinn, P.L. A Novel AAV-mediated Gene Delivery System Corrects CFTR Function in Pigs. Am. J. Respir. Cell Mol. Biol. 2019, 61, 747–754. [Google Scholar] [CrossRef]
- Guggino, W.B.; Cebotaru, L. Gene Therapy for Cystic Fibrosis Paved the Way for the Use of Adeno-Associated Virus in Gene Therapy. Hum. Gene Ther. 2020, 31, 538–541. [Google Scholar] [CrossRef]
- Choi, S.H.; Reeves, R.E.; Romano Ibarra, G.S.; Lynch, T.J.; Shahin, W.S.; Feng, Z.; Gasser, G.N.; Winter, M.C.; Evans, T.I.A.; Liu, X.; et al. Detargeting Lentiviral-Mediated CFTR Expression in Airway Basal Cells Using miR-106b. Genes 2020, 11, 1169. [Google Scholar] [CrossRef]
- Osman, G.; Rodriguez, J.; Chan, S.Y.; Chisholm, J.; Duncan, G.; Kim, N.; Tatler, A.L.; Shakesheff, K.M.; Hanes, J.; Suk, J.S.; et al. PEGylated enhanced cell penetrating peptide nanoparticles for lung gene therapy. J. Control. Release 2018, 285, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, D.J.; Turner, D.L.; Ng, P. A Single “All-in-One” Helper-Dependent Adenovirus to Deliver Donor DNA and CRISPR/Cas9 for Efficient Homology-Directed Repair. Mol. Ther. Methods Clin. Dev. 2020, 17, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Erwood, S.; Laselva, O.; Bily, T.M.; Brewer, R.A.; Rutherford, A.H.; Bear, C.E.; Ivakine, E.A. Allele-Specific Prevention of Nonsense-Mediated Decay in Cystic Fibrosis Using Homology-Independent Genome Editing. Mol. Ther. Methods Clin. Dev. 2020, 17, 1118–1128. [Google Scholar] [CrossRef]
- Shaikh, S.B.; Bhandary, Y.P. CRISPR/Cas9 Genome Editing Tool: A Promising Tool for Therapeutic Applications on Respiratory Diseases. Curr. Gene Ther. 2020, 20, 333–346. [Google Scholar] [CrossRef]
- Graham, C.; Hart, S. CRISPR/Cas9 gene editing therapies for cystic fibrosis. Expert Opin. Biol. Ther. 2021, 21, 767–780. [Google Scholar] [CrossRef]
- Santos, L.; Mention, K.; Cavusoglu-Doran, K.; Sanz, D.J.; Bacalhau, M.; Lopes-Pacheco, M.; Harrison, P.T.; Farinha, C.M. Comparison of Cas9 and Cas12a CRISPR editing methods to correct the W1282X-CFTR mutation. J. Cyst. Fibros. 2021, 21, 181–187. [Google Scholar] [CrossRef]
- Fleischer, A.; Vallejo-Díez, S.; Martín-Fernández, J.M.; Sánchez-Gilabert, A.; Castresana, M.; del Pozo, A.; Esquisabel, A.; Ávila, S.; Castrillo, J.L.; Gaínza, E.; et al. iPSC-Derived Intestinal Organoids from Cystic Fibrosis Patients Acquire CFTR Activity upon TALEN-Mediated Repair of the p.F508del Mutation. Mol. Ther. Methods Clin. Dev. 2020, 17, 858–870. [Google Scholar] [CrossRef]
- Lee, C.; Flynn, R.; Hollywood, J.; Scallan, M.F.; Harrison, P.T. Correction of the ΔF508 Mutation in the Cystic Fibrosis Transmembrane Conductance Regulator Gene by Zinc-Finger Nuclease Homology-Directed Repair. BioRes. Open Access 2012, 1, 99–108. [Google Scholar] [CrossRef]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S.; et al. Targeted Correction and Restored Function of the CFTR Gene in Cystic Fibrosis Induced Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | Results | |
|---|---|---|
| Normality test | Shapiro–Wilk | The sample did not show normal distribution. |
| Kolmogorov–Smirnov | The sample did not show normal distribution. | |
| Binomial test | The sample was not homogeneously distributed | |
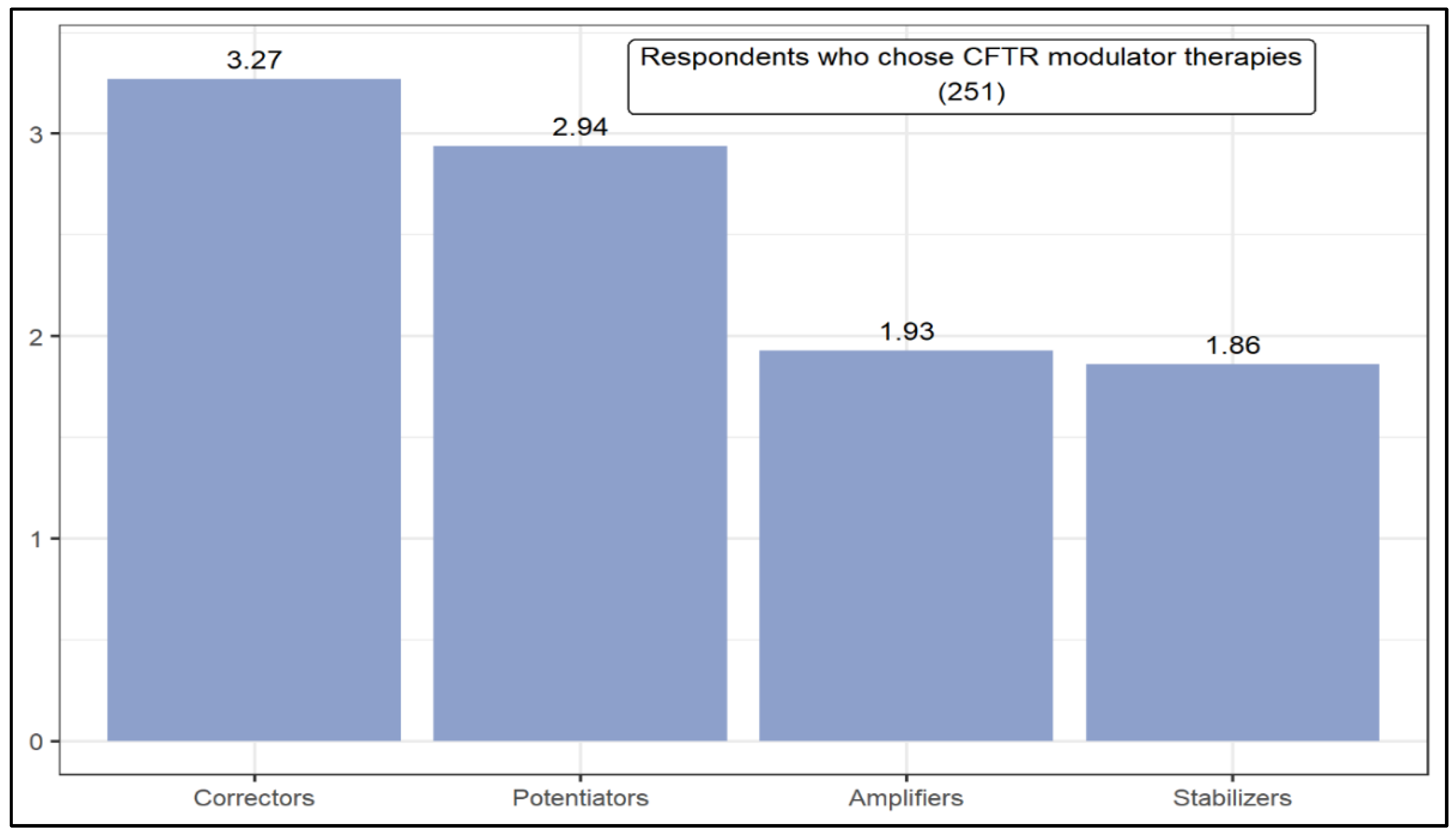
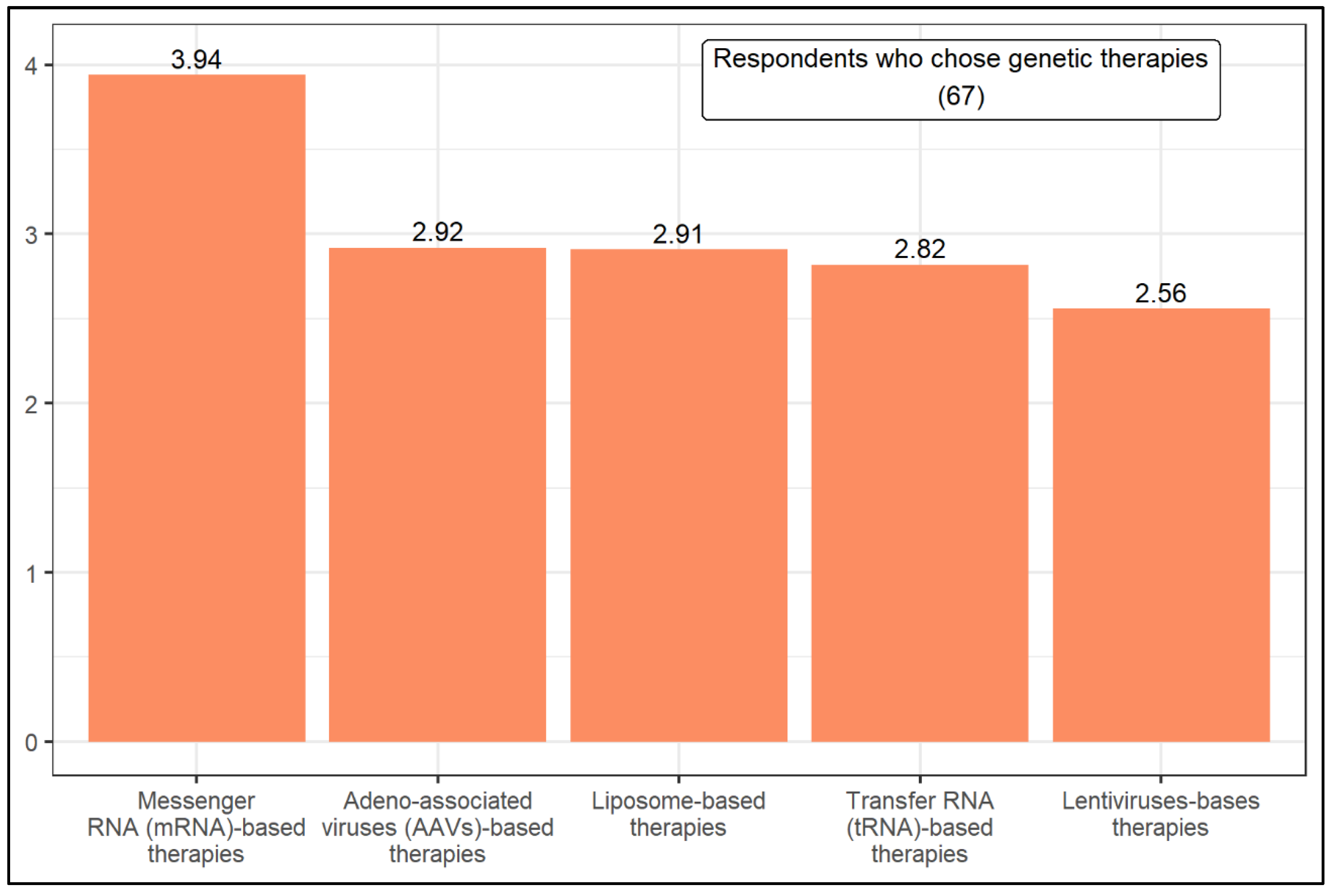
| Mann–Whitney U | The level of knowledge influences the result in the following situations: (1) Which therapeutic option is most likely to be successful in treating cystic fibrosis in the next 15 years. (2) CFTR modulator therapies: potentiators, correctors, and stabilizers. (3) Genetic therapies: Adeno-associated viruses-based therapies. | |
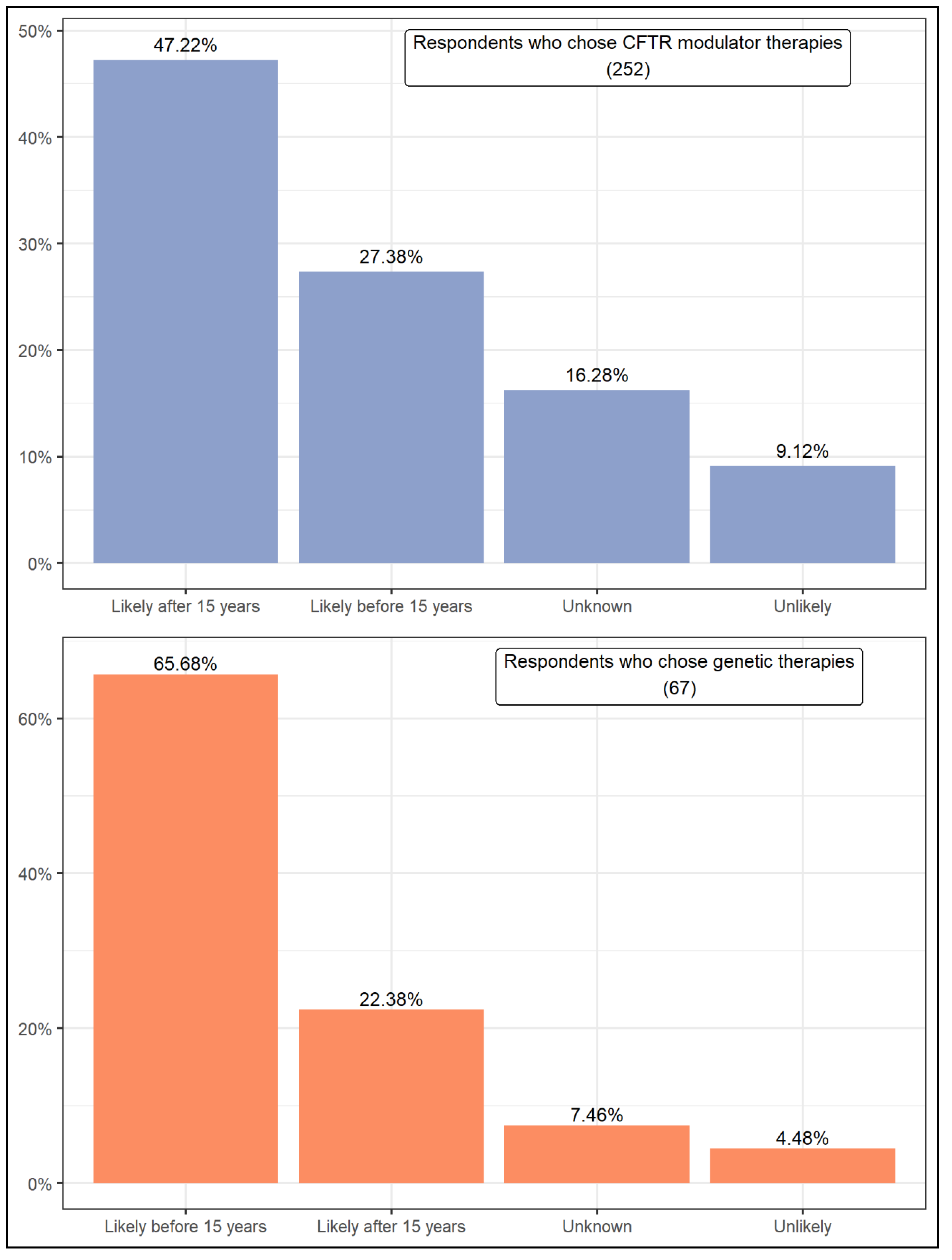
| Wilcoxon rank test | (1) CFTR modulator therapies are the predominant option. (2) Respondents who choose CFTR modulator therapies expect that a cure will take more than 15 years. (3) Respondents who choose genetic therapies expect that a cure will take less than 15 years. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabral, B.; Terlizzi, V.; Laselva, O.; Conte Filho, C.; Mota, F. Anticipating New Treatments for Cystic Fibrosis: A Global Survey of Researchers. J. Clin. Med. 2022, 11, 1283. https://doi.org/10.3390/jcm11051283
Cabral B, Terlizzi V, Laselva O, Conte Filho C, Mota F. Anticipating New Treatments for Cystic Fibrosis: A Global Survey of Researchers. Journal of Clinical Medicine. 2022; 11(5):1283. https://doi.org/10.3390/jcm11051283
Chicago/Turabian StyleCabral, Bernardo, Vito Terlizzi, Onofrio Laselva, Carlos Conte Filho, and Fabio Mota. 2022. "Anticipating New Treatments for Cystic Fibrosis: A Global Survey of Researchers" Journal of Clinical Medicine 11, no. 5: 1283. https://doi.org/10.3390/jcm11051283
APA StyleCabral, B., Terlizzi, V., Laselva, O., Conte Filho, C., & Mota, F. (2022). Anticipating New Treatments for Cystic Fibrosis: A Global Survey of Researchers. Journal of Clinical Medicine, 11(5), 1283. https://doi.org/10.3390/jcm11051283