
Identification of a TPP1 Q278X Mutation in an Iranian Patient with Neuronal Ceroid Lipofuscinosis 2: Literature Review and Mutations Update
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction
2.2. Whole Exome Sequencing
2.3. Polymerase Chain Reaction (PCR) and Sanger sequencing
2.4. Literature Search
3. Results
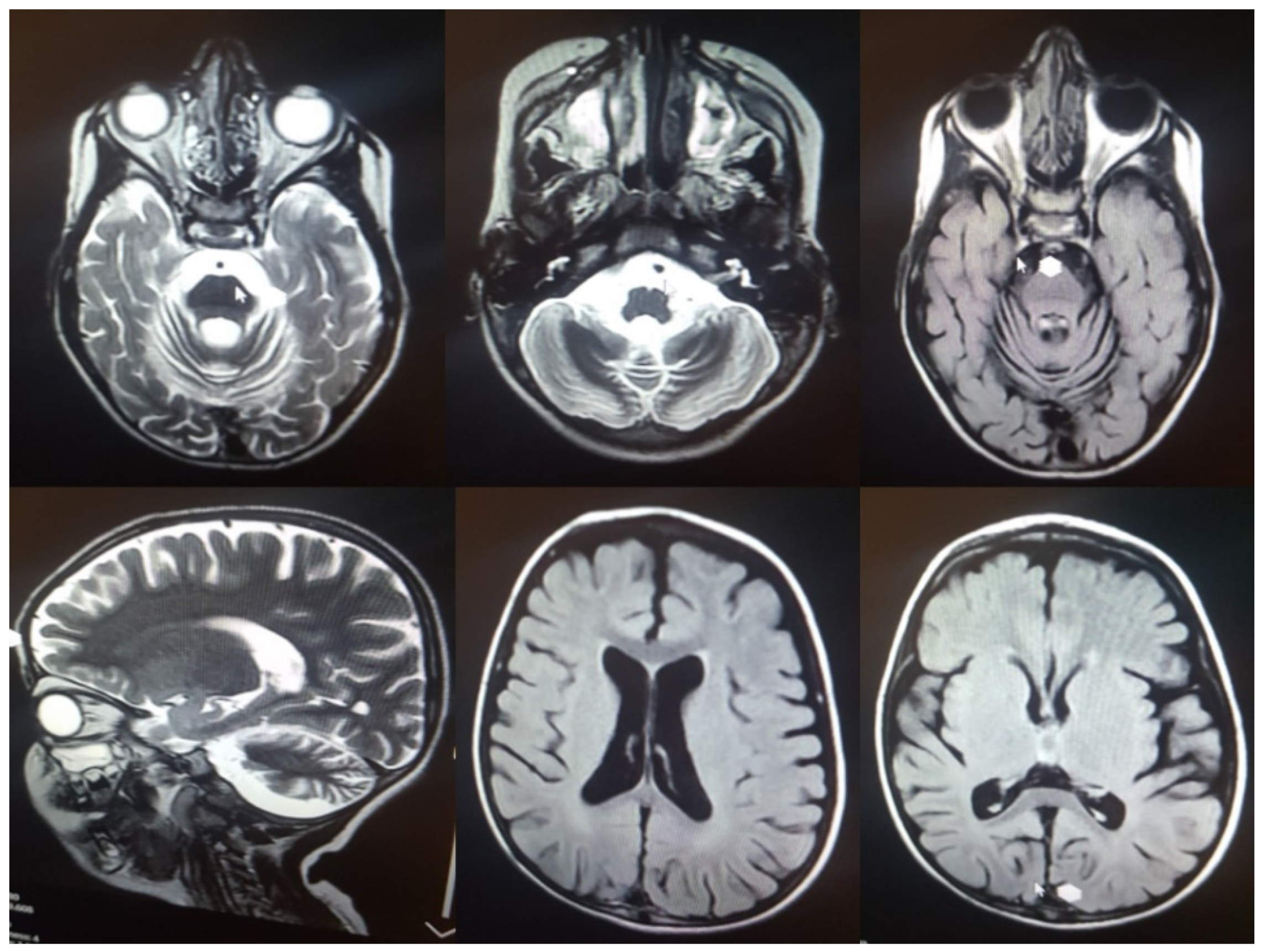
3.1. Clinical Assessment
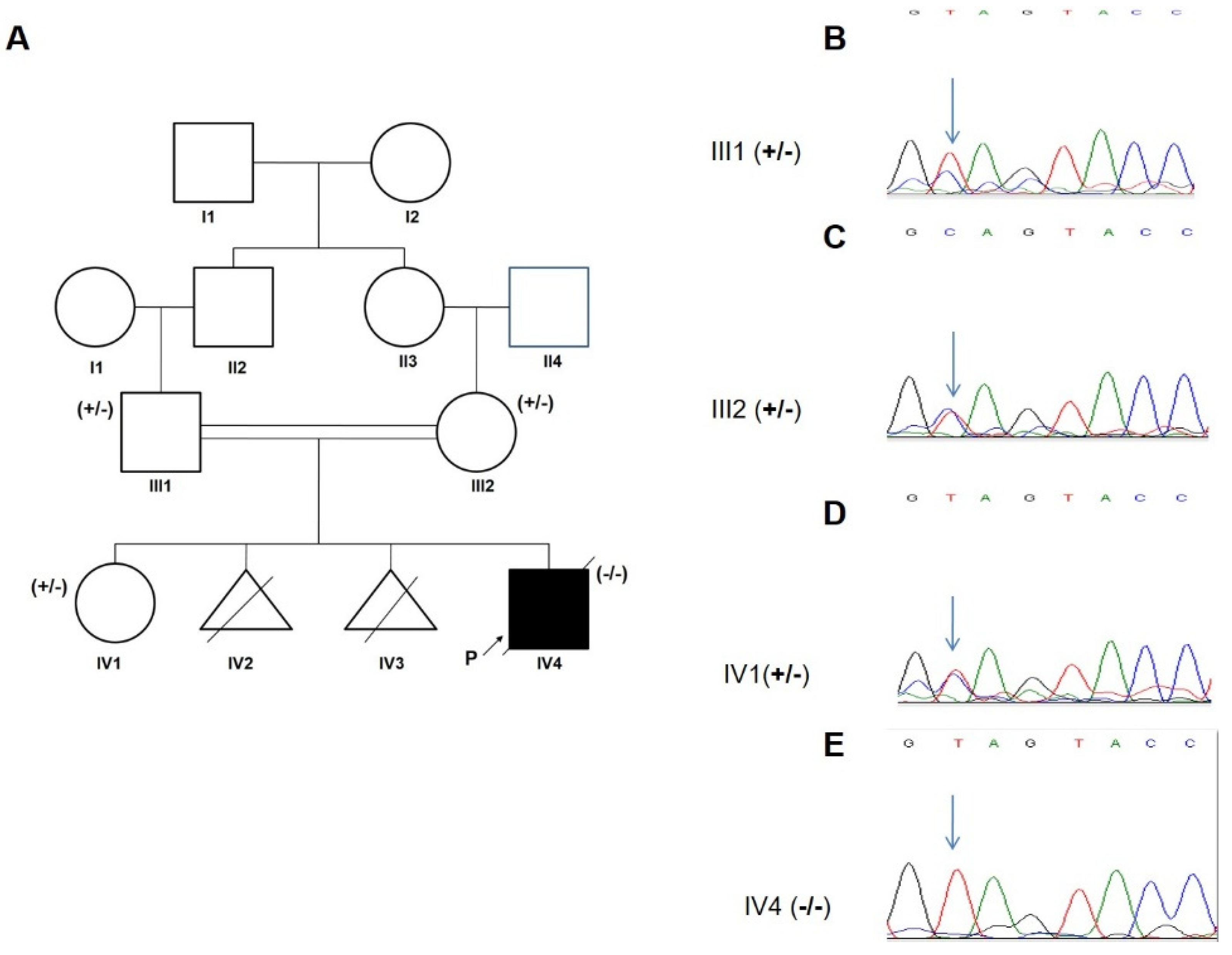
3.2. Genetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CLN2 | Neuronal Ceroid Lipofuscinoses 2 |
| NMD | Nonsense-Mediated Decay |
| ExAC | Exome Aggregation Consortium |
| ACMG | American College of Medical Genetics and Genomics |
| GCS | Glasgow Coma Scale |
| LOF | Loss of Function |
| PTC | Premature Stop Codon |
References
- Angural, A.; Ponnusamy, K.; Langeh, D.; Kumari, M.; Spolia, A.; Rai, E.; Sharma, S. Missense Variation in TPP1 Gene causes Neuronal Ceroid Lipofuscinosis Type 2 in a Family from Jammu and Kashmir-India. Preprints 2021, 2021070661. [Google Scholar] [CrossRef]
- Chen, Z.-R.; Liu, D.-T.; Meng, H.; Liu, L.; Bian, W.-J.; Liu, X.-R.; Zhu, W.-W.; He, Y.; Wang, J.; Tang, B.; et al. Homozygous missense TPP1 mutation associated with mild late infantile neuronal ceroid lipofuscinosis and the genotype-phenotype correlation. Seizure 2018, 69, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Uygur, O.; Oncel, M.Y.; Gencpinar, P.; Guvenc, M.S.; Dundar, N.O. A Case with Neonatal-onset Type 2 Neuronal Ceroid Lipofuscinosis: A Novel Mutation. J. Coll. Physicians Surg. Pak. 2020, 30, 543–544. [Google Scholar] [CrossRef] [PubMed]
- Haltia, M.; Goebel, H.H. The neuronal ceroid-lipofuscinoses: A historical introduction. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1795–1800. [Google Scholar] [CrossRef]
- Gardner, E.; Mole, S.E. The Genetic Basis of Phenotypic Heterogeneity in the Neuronal Ceroid Lipofuscinoses. Front. Neurol. 2021, 12, 754045. [Google Scholar] [CrossRef]
- Gardner, E.; Bailey, M.; Schulz, A.; Aristorena, M.; Miller, N.; Mole, S.E. Mutation update: Review of TPP1 gene variants associated with neuronal ceroid lipofuscinosis CLN2 disease. Hum. Mutat. 2019, 40, 1924–1938. [Google Scholar] [CrossRef]
- Kose, M.; Kose, E.; Ünalp, A.; Yılmaz, Ü.; Edizer, S.; Tekin, H.G.; Karaoğlu, P.; Özdemir, T.R.; Er, E.; Onay, H.; et al. Neuronal ceroid lipofuscinosis: Genetic and phenotypic spectrum of 14 patients from Turkey. Neurol. Sci. 2021, 42, 1103–1111. [Google Scholar] [CrossRef]
- Segura, O.E.; Hernández, Z.; Mancilla, N.; Naranjo, R.; Tavera, L. Real world effectiveness of cerliponase alfa in classical and atypical patients. A case series. Mol. Genet. Metab. Rep. 2021, 27, 100718. [Google Scholar] [CrossRef]
- Kurachi, Y.; Oka, A.; Itoh, M.; Mizuguchi, M.; Hayashi, M.; Takashima, S. Distribution and development of CLN2 protein, the late-infantile neuronal ceroid lipofuscinosis gene product. Acta Neuropathol. 2001, 102, 20–26. [Google Scholar] [CrossRef]
- Radwan, A.; Fateen, E.; Gouda, A.; Zaki, M.; Ali, O. Enzyme based diagnosis of type 1 and 2 neuronal ceroid lipofuscinoses. Azhar Int. J. Pharm. Med. Sci. 2021, 1, 40–48. [Google Scholar] [CrossRef]
- Santorelli, F.M.; Garavaglia, B.; Cardona, F.; Nardocci, N.; Bernardina, B.D.; Sartori, S.; Suppiej, A.; Bertini, E.; Claps, D.; Battini, R.; et al. Molecular epidemiology of childhood neuronal ceroid-lipofuscinosis in Italy. Orphanet J. Rare Dis. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Kohan, R.; Carabelos, M.N.; Xin, W.; Sims, K.; Guelbert, N.; Cismondi, I.A.; Pons, P.; Alonso, G.I.; Troncoso, M.; Witting, S.; et al. Neuronal ceroid lipofuscinosis type CLN2: A new rationale for the construction of phenotypic subgroups based on a survey of 25 cases in South America. Gene 2013, 516, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.-T.; Wang, X.-H.; Ding, C.-H.; Shen, X.; Zhang, H.; Zhang, W.-H.; Li, J.-W.; Ren, C.-H.; Fang, F. Next-Generation Sequencing Analysis Reveals Novel Pathogenic Variants in Four Chinese Siblings with Late-Infantile Neuronal Ceroid Lipofuscinosis. Front. Genet. 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Kozina, A.A.; Okuneva, E.G.; Baryshnikova, N.V.; Kondakova, O.B.; Nikolaeva, E.A.; Fedoniuk, I.D.; Mikhailova, S.V.; Krasnenko, A.Y.; Stetsenko, I.F.; Plotnikov, N.A.; et al. Neuronal ceroid lipofuscinosis in the Russian population: Two novel mutations and the prevalence of heterozygous carriers. Mol. Genet. Genom. Med. 2020, 8, e1228. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Zhong, R.; Moore, S.; Moroziewicz, D.; Currie, J.R.; Parfrey, P.; Brown, W.T.; Zhong, N. Identification of novel CLN2 mutations shows Canadian specific NCL2 alleles. J. Med. Genet. 2002, 39, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Barisić, N.; Logan, P.; Pikija, S.; Skarpa, D.; Blau, N. R208X mutation in CLN2 gene associated with reduced cerebrospinal fluid pterins in a girl with classic late infantile neuronal ceroid lipofuscinosis. Croat. Med. J. 2003, 44, 489–493. [Google Scholar]
- Goetz, M.; Istrail, S. Alternative Splicing Analysis of Tripeptidyl Peptidase 1 Mutations in CLN2 Batten Disease Models; Presented at the Data Science Initiative; Brown University: Providence, RI, USA, 14 April 2021. [Google Scholar]
- Ma, L.; Prada, A.; Schmidt, M.; Morrow, E.M. Generation of Pathogenic TPP1 Mutations in Human Stem Cells as a Model for CLN2 Disease. Stem Cell Res. 2021, 53, 102323. [Google Scholar] [CrossRef]
- Wujek, P.; Kida, E.; Walus, M.; Wisniewski, K.E.; Golabek, A.A. N-Glycosylation Is Crucial for Folding, Trafficking, and Stability of Human Tripeptidyl-peptidase I. J. Biol. Chem. 2004, 279, 12827–12839. [Google Scholar] [CrossRef]
- Golabek, A.A.; Kida, E. Tripeptidyl-peptidase I in health and disease. Biol. Chem. 2006, 387, 1091–1099. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Chandra, S.; Roy, A.; Dasarathi, S.; Kundu, M.; Pahan, K. Upregulation of tripeptidyl-peptidase 1 by 3-hydroxy-(2, 2)-dimethyl butyrate, a brain endogenous ligand of PPARα: Implications for late-infantile Batten disease therapy. Neurobiol. Dis. 2019, 127, 362–373. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, C.M.; Pessoa, A.; Mendes, C.C.; Rivera-Nieto, C.; Vergara, D.; Troncoso, M.; Mole, S.E. Revealing the clinical phenotype of atypical neuronal ceroid lipofuscinosis type 2 disease: Insights from the largest cohort in the world. J. Paediatr. Child Health 2021, 57, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Bellusci, M.; Pietrafusa, N.; Trivisano, M.; De Palma, L.; Vigevano, F. Photosensitivity is an early marker of neuronal ceroid lipofuscinosis type 2 disease. Epilepsia 2017, 58, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Liu, X.-M.; Chen, Y.-H.; Zhang, S.-Q.; Wang, K. A novel CLN2/TPP1 mutation in a patient with late infantile neuronal ceroid lipofuscinosis. Neurol. Sci. 2015, 36, 1917–1919. [Google Scholar] [CrossRef] [PubMed]
- Helman, G.; Taylor, L.E.; Walkiewicz, M.; Le Moing, M.; Eggers, S.; Yaplito-Lee, J.; Fuller, M.; Dabscheck, G.; Rodriguez-Casero, V.; White, S.M.; et al. Aberrant splicing and transcriptional activity of TPP1 result in CLN2-like disorder. Eur. J. Med. Genet. 2021, 64, 104259. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.C.; Hsu, C.-J.; Lee, W.-T. Perampanel attenuates myoclonus in a patient with neuronal ceroid lipofuscinoses type 2 disease. Brain Dev. 2019, 41, 817–819. [Google Scholar] [CrossRef]
- Saini, A.G.; Sankhyan, N.; Singhi, P. Chorea in Late-Infantile Neuronal Ceroid Lipofuscinosis: An Atypical Presentation. Pediatr. Neurol. 2016, 60, 75–78. [Google Scholar] [CrossRef]
- Chang, X.; Huang, Y.; Meng, H.; Jiang, Y.; Wu, Y.; Xiong, H.; Wang, S.; Qin, J. Clinical study in Chinese patients with late-infantile form neuronal ceroid lipofuscinoses. Brain Dev. 2012, 34, 739–745. [Google Scholar] [CrossRef]
- Bessa, C.; Teixeira, C.A.; Dias, A.; Alves, M.; Rocha, S.; Lacerda, L.; Ribeiro, M.G. CLN2/TPP1 deficiency: The novel mutation IVS7-10A> G causes intron retention and is associated with a mild disease phenotype. Mol. Genet. Metab. 2008, 93, 66–73. [Google Scholar] [CrossRef]
- Ługowska, A.; Purzycka-Olewiecka, J.K.; Płoski, R.; Truszkowska, G.; Pronicki, M.; Felczak, P.; Bednarska-Makaruk, M. Tripeptidyl Peptidase 1 (TPP1) Deficiency in a 36-Year-Old Patient with Cerebellar-Extrapyramidal Syndrome and Dilated Cardiomyopathy. Life 2021, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.W.; Miu-Kuen Poon, P.; Tong, S.F.; Ko, C.H. Two novel CLN2 gene mutations in a Chinese patient with classical late-infantile neuronal ceroid lipofuscinosis. Am. J. Med. Genet. 2001, 99, 161–163. [Google Scholar] [PubMed]
- Amadori, E.; Scala, M.; Cereda, G.S.; Vari, M.S.; Marchese, F.; Di Pisa, V.; Mancardi, M.M.; Giacomini, T.; Siri, L.; Vercellino, F.; et al. Targeted re-sequencing for early diagnosis of genetic causes of childhood epilepsy: The Italian experience from the ‘beyond epilepsy’ project. Ital. J. Pediatr. 2020, 46, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hartikainen, J.; Ju, W.; Wisniewski, K.E.; Moroziewicz, D.N.; Kaczmarski, A.L.; McLendon, L.; Zhong, D.; Suarez, C.T.; Brown, W.; Zhong, N. Late Infantile Neuronal Ceroid Lipofuscinosis Is Due to Splicing Mutations in the CLN2 Gene. Mol. Genet. Metab. 1999, 67, 162–168. [Google Scholar] [CrossRef]
- Gall, K.; Izzo, E.; Seppälä, E.H.; Alakurtti, K.; Koskinen, L.; Saarinen, I.; Alastalo, T.P. Next-generation sequencing in childhood-onset epilepsies: Diagnostic yield and impact on neuronal ceroid lipofuscinosis type 2 (CLN2) disease diagnosis. PLoS ONE 2021, 16, e0255933. [Google Scholar]
- Elleder, M.; Dvořáková, L.; Stolnaja, L.; Vlášková, H.; Hůlková, H.; Druga, R.; Mikuláštík, J. Atypical CLN2 with later onset and prolonged course: A neuropathologic study showing different sensitivity of neuronal subpopulations to TPP1 deficiency. Acta Neuropathol. 2008, 116, 119–124. [Google Scholar]
- Goldberg-Stern, H.; Halevi, A.; Marom, D.; Straussberg, R.; Bloch, A.M. Late Infantile Neuronal Ceroid Lipofuscinosis: A New Mutation in Arabs. Pediatr. Neurol. 2009, 41, 297–300. [Google Scholar] [CrossRef]
- Johnson, A.M.; Mandelstam, S.; Andrews, I.; Boysen, K.; Yaplito-Lee, J.; Fietz, M.; Ellaway, C. Neuronal ceroid lipofuscinosis type 2: An Australian case series. J. Paediatr. Child Health 2020, 56, 1210–1218. [Google Scholar]
- Sun, Y.; Almomani, R.; Breedveld, G.J.; Santen, G.W.; Aten, E.; Lefeber, D.J.; Maat-Kievit, A.J. Autosomal recessive spinocerebellar ataxia 7 (SCAR 7) is caused by variants in TPP1, the gene involved in classic late-infantile neuronal ceroid lipofuscinosis 2 disease (CLN 2 disease). Hum. Mutat. 2013, 34, 706–713. [Google Scholar] [CrossRef]
- Sheth, J.; Mistri, M.; Bhavsar, R.; Pancholi, D.; Kamate, M.; Gupta, N.; Kabra, M.; Mehta, S.; Nampoothiri, S.; Thakker, A.; et al. Batten disease: Biochemical and molecular characterization revealing novel PPT1 and TPP1 gene mutations in Indian patients. BMC Neurol. 2018, 18, 1. [Google Scholar] [CrossRef]
- Karimi, S.; Rezaei-moghadam, M.; Mosavi, S.A. Juvenile Neuronal Ceroid Lipofuscinosis in a Patient of Iranian Origin. Razavi Int. J. Med. 2018, 6, 42–45. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
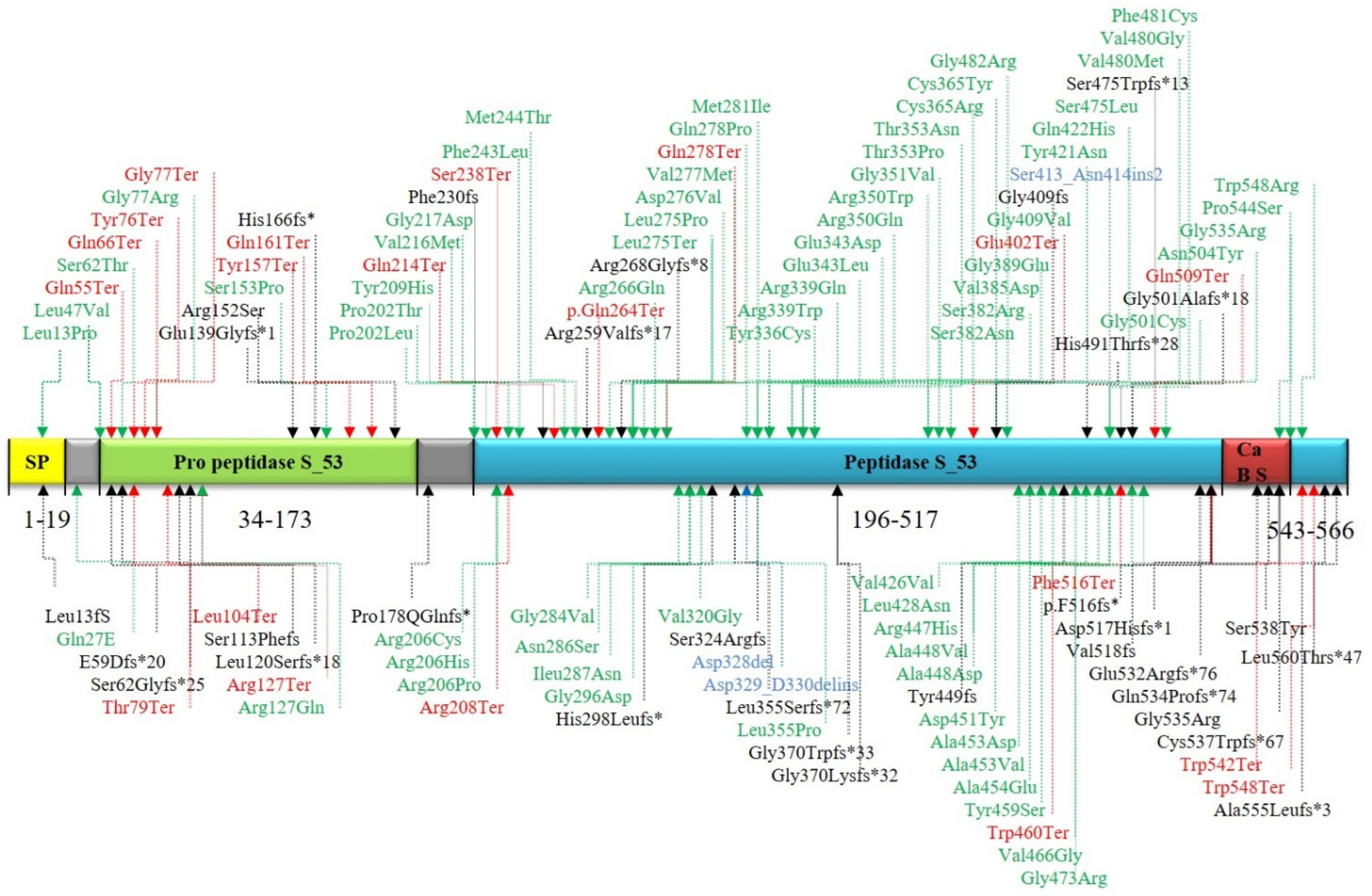{kind=link}
| Age/ Sex | First Symptoms/Age at Onset | Additional Symptoms | Consan-Guineous | HGVS cDNA | HGVS Protein | Exon/ Intron | ACMG Scoring | ACMG Prediction | Zygosity | Ethnicity | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5y/M | Seizures/4y | Cerebellar atrophy, ataxia, epilepsy, vision loss, spasticity, tremor, motor difficulties, swallowing problems, sleep disorder, cognitive decline, language regression | yes | c.C832T | p.Q278X | 7 | PVS1, PM2 | LP | Hom | Iran | This study |
| 36y/M | Dysarthria, cardiac arrhythmia/4y | Severe cerebellar atrophy, dysarthria, ataxia, dystonia, right-hand tremor | No | c.38T > C c.1523A > G | p.L13P p.T508C | 2 12 | PM2 PM2 | VUS VUS | Comp het | Poland | [32] |
| 4y/F | Seizure/4y | Cognitive decline, motor deterioration, epilepsy, language regression, generalized hypotonia, cerebral and cerebellar atrophy | No | c.89 + 1G > A | Splice effect | 2 | PVS1, PM2, PP5 | LP | Hom | India | [29] |
| 9y/M | Anxiety, hypervigilance, sleep disorder/5y | Language regression, motor difficulties, microcephaly, spasticity, mild cortical and cerebellar atrophy | No | c.225A > G c.1012C > G | p.Q75Q p.Q338E | 3 8 | PM2, PP5 PM1, PP2 | LP VUS | Comp het | Australia | [27] |
| 6y/F | Seizures/3.5y | Mental and motor deterioration, ataxia, cerebellar atrophy, vision loss, Retinitis pigmentosa, myoclonic epilepsy | NA | c.229G > T IVS4-17~-4delTGTTCTCTGACCTC | p.G77X; Splice effect | 3 | PVS1, PM2 | LP | Comp het | China | [30] |
| 4.6y/M | Speech delay, mild mental retardation, autistic features/2y | Seizure, myoclonic jerks, ataxia, motor difficulties, bilateral optic atrophy | NA | c.183_184 delCT c.417G > A | p.S61fsX25 p.G473R | 3 11 | PVS1,PM2 PM2, PP2 | LP LP | Com het | China | [33] |
| 8y/M | Movement disorder, neuroregression/2y | Cerebellar atrophy, microcephaly | No | c.341C > T | p.A114V | 4 | PP2, PM2 | VUS | Hom | Turkey | [7] |
| 6y/M | Seizure/3y | Mental and motor deterioration, vision loss, ataxia, cerebellar atrophy, myoclonic epilepsy | NA | c.409-410insGCTG c.1546-1547insTTCA | p.E139G p.D517H | 5 12 | PM2. PP2 | VUS VUS | Comp het | China | [30] |
| 50M/F | Seizures/45M | Tremor, ataxia, cerebellar atrophy | No | c.509-1G > C | Splice effect | 6 | PVS1, PM2 | P | Hom | Italy | [34] |
| -/F | Seizures/3.5y | Ventricle and axial fluid spaces, ataxia, dysarthria, dementia, language regression, vision loss, tremor | No | c.T523-1G > A | Splice effect | NA NA | PVS1, PS4 | P | Hom | Scottish Irish English | [35] |
| 20y/F | Vision failure/7y | Seizures | No | c.T523-1G > C | Splice effect | NA | PVS1, PS4 | P | Hom | Lebanese | [35] |
| NA/F | Seizures/44M | Speech delay, regression, ataxia, myoclonus | NA | c.533del | p.P178Q | 6 | PVS1, PM2, PP5 | P | Hom | NA | [36] |
| 15y/M | Motor dysfunction/3y | Dementia, mental deterioration, speech difficulties, blindness, spasticity | NA | c.622C > T c.1439T > G | p.R208 X p.V480G | 6 12 | PVS1, PM2 | P | Comp het | Czech Republic | [37] |
| 5y/F | Seizures/ 4y | Focal abnormality, hypsarrhythmia, tremor, generalized ataxia, motor and mental regression, cerebellar atrophy, optic nerves bilateral atrophy | No | c.622C > T | p.R208X | 6 | PVS1, PM2 | P | Hom | Swiss | [16] |
| 5y/F | Seizures/2y | Mental and motor deterioration, ataxia, cerebellar atrophy, myoclonic epilepsy | NA | c.622C > T: c.640C > T | p.R208X; p.Q214X | 6 6 | PVS1, PM2; PVS1, PM2, PP5 | P P | Comp het | China | [30] |
| 12y/F | Seizures/2y | Cerebellar atrophy, ataxia, cognitive deterioration, myoclonic epilepsy, blindness, language delay | No | c.622C > T | p.R208X | 6 | PVS1, PM2 | P | Hom | Taiwan | [28] |
| NA/F | Seizures/43M | Delayed speech development, behavioral and sleep abnormalities, cerebellar atrophy | NA | c.622C > T | p.R208X | 6 | PVS1, PM2 | P | Hom | NA | [36] |
| NA/M | Seizures/36M | Cerebellar and cerebral Atrophy, language delay, motor difficulty, behavioral abnormalities | NA | c.622C > T | p.R208X | 6 | PVS1, PM2 | P | Hom | NA | [36] |
| NA /F | Seizures/35M | Motor and language regression, ataxia, choreoathetosis, enlargement of subarachnoidspace, cerebral and cerebellar atrophy | NA | c.622C > T | p.R208X | 6 | PVS1, PM2 | P | Hom | NA | [36] |
| NA/F | Seizures/36M | Language delay, motor disturbance, cerebellar atrophy | NA | c.622C > T | p.R208X | 6 | PVS1, PM2 | P | Hom | NA | [36] |
| NA/F | Seizures/38M | Hypotonia of right upperlimb, delayed speech | NA | c.622C > T | p.R208X | 6 | PVS1, PM2 | P | Hom | NA | [36] |
| 4y/M | Seizures/2y | Cognitive and motor deterioration, speech delay, tonic-clonic seizures, muscular hypotonia, cerebral atrophy, cerebellum hypoplasia, optic nerves damage, hand tremor, ataxia | NA | c.622C > T | p.R208X | 6 | PVS1, PM2 | P | Hom | Russian | [14] |
| 9y/M | Seizures/3y | Mental and motor deterioration, ataxia, cerebellar atrophy, myoclonic epilepsy | NA | c.640C > T; c.650G > T | p.Q214X; p.G217D | 6 6 | PVS1, PM2, PP5; PM2, PM5, PM1, PP2, PP5 | P P | Comp het | China | [30] |
| -/M | Seizures/3.5y | Mild cerebellar atrophy | No | c.646G > A | p.V216M | 6 | PVS1, PM2 | LP | Hom | China | [2] |
| 8y/F | Seizures/3y | Visual abnormalities, cognitive regression, mild atrophy of the cerebellum | No | c.646G > A | p.V216M | 6 | PVS1, PM2 | LP | Hom | China | [2] |
| 40y | -/10y | Cognitive and motor dysfunction, epilepsy, seizure. | NA | IVS7-10A > G | NA | 7 | NA | LP | Hom | Portugal | [31] |
| 10y/F | Seizures, vomiting/3y | Ataxia, language regression, hyperreflexia, cerebral and cerebellar atrophy | No | c.775delC | p.Arg259fs | 7 | PVS1, PM2 | LP | Hom | Arab | [38] |
| -/M | Seizures/3y | Stopped walking and talking | No | c.775delC | p.Arg259fs | 7 | PVS1, PM2 | LP | Hom | Arab | [38] |
| 4.5/F | Seizures/2.5y | Cognitive and motor impairment, loss of ambulation, developmental delay, speech deterioration, hypertonia, hypereflexia | No | c.1016G > A | p.R339Q | 8 | PM2, PM5, PM1, PP2 | P | Hom | India | [1] |
| 11M/M | Hypotonic/26day | Severe cerebellar and cerebral atrophy and no newborn reflexes | No | c.1145G > A | p.S382N | 9 | PM2,PM5,PP3,PP2 | VUS | Hom | Turkey | [3] |
| 5y/F | Myoclonus, nystagmus/6M | Cerebral atrophy, movement disorder, epilepsy | Yes | c.1204G > T | p.E402X | 10 | PVS1, PM2 | LP | Hom | Turkey | [7] |
| 7y/M | Seizures/2.5y | Drop attacks, low vision, speech and cognitive degeneration | No | c.1551 + 1insTGAT | Splice effect | 12 | PVS1, PM2, PP5 | P | Hom | China | [13] |
| 7y/F | Seizures/3y | Ataxia, vision loss, cerebellar atrophy, brain stem atrophy, general epileptic discharges, drop attacks | No | c.1551 + 1insTGAT | Splice effect | 12 | PVS1, PM2, PP5 | P | Hom | China | [13] |
| 3.7/M | Seizures/8M | Frequent spike and wave, unsteady gait, myoclonic jerks, | No | c.1551 + 1insTGAT | Splice effect | 12 | PVS1, PM2, PP5 | P | Hom | China | [13] |
| 5y/F | Seizures/ 3.2y | Ataxia, mild brain atrophy, myoclonic seizures, cognitive decline, motor dysfunction. | NA | c.1551 + 1G > T; c.1613C > A | Splice effect; p.S538Y | 12 13 | PVS1, PP5, PM2 PM2, PP3, PP2 | P VUS | Comp het | China | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baranzehi, T.; Kordi-Tamandani, D.M.; Najafi, M.; Khajeh, A.; Schmidts, M. Identification of a TPP1 Q278X Mutation in an Iranian Patient with Neuronal Ceroid Lipofuscinosis 2: Literature Review and Mutations Update. J. Clin. Med. 2022, 11, 6415. https://doi.org/10.3390/jcm11216415
Baranzehi T, Kordi-Tamandani DM, Najafi M, Khajeh A, Schmidts M. Identification of a TPP1 Q278X Mutation in an Iranian Patient with Neuronal Ceroid Lipofuscinosis 2: Literature Review and Mutations Update. Journal of Clinical Medicine. 2022; 11(21):6415. https://doi.org/10.3390/jcm11216415
Chicago/Turabian StyleBaranzehi, Tayebeh, Dor Mohammad Kordi-Tamandani, Maryam Najafi, Ali Khajeh, and Miriam Schmidts. 2022. "Identification of a TPP1 Q278X Mutation in an Iranian Patient with Neuronal Ceroid Lipofuscinosis 2: Literature Review and Mutations Update" Journal of Clinical Medicine 11, no. 21: 6415. https://doi.org/10.3390/jcm11216415
APA StyleBaranzehi, T., Kordi-Tamandani, D. M., Najafi, M., Khajeh, A., & Schmidts, M. (2022). Identification of a TPP1 Q278X Mutation in an Iranian Patient with Neuronal Ceroid Lipofuscinosis 2: Literature Review and Mutations Update. Journal of Clinical Medicine, 11(21), 6415. https://doi.org/10.3390/jcm11216415