1. Introduction
Pancreatic cancer is a lethal disease with very poor outcomes [
1]. Pancreatic ductal adenocarcinoma (PDAC) is the most common pancreatic cancer type accounting for more than 90% of the cases, followed by pancreatic neuroendocrine tumours (PNETs) representing <2% [
2]. PDAC five-year survival rate is <9% which falls to 3% for patients with stage IV cancer. Unfortunately, the vast majority of patients are diagnosed at a late stage and pancreatic cancer is predicted to become the second cause of cancer-related death by 2030, only surpassed by lung cancer [
3]. There are several factors associated with this poor prognosis, such as the absence of specific symptoms leading usually to late diagnosis, the high resistance of pancreatic cancer cells to available therapeutics, the highly desmoplastic and immunosuppressive tumour microenvironment (TME), the low immunogenicity and importantly, the lack of effective targets for treating early stage disease.
Currently, surgery followed by adjuvant chemotherapy remains the only curative therapeutic option for pancreatic cancer patients, although <20% of patients are resectable at the time of diagnosis (ECOG 0/1) [
4]. The main adjuvant chemotherapy used for these patients is a combination of four modified cytotoxic agents: 5-fluorouracil (5-FU), leucovorin, irinotecan and oxaliplatin, also known as mFOLFIRINOX. For patients with advance stage (ECOG 1/2) or metastatic disease (ECOG > 2), first line options consist of a combination of nab-paclitaxel (Abraxane
®, Celgene, Summit, NJ, USA) plus gemcitabine or mFOLFIRINOX for the most fit patients.
There are three main precursor lesions increasing the risk of developing pancreatic cancer [
5]. The most common are pancreatic intraepithelial neoplasms (PanINs), which are microscopic lesions arising from the small intralobular pancreatic ducts. The other two possible drivers are mucinous cystic neoplasms (MCNs) and intraductal papillary mucinous neoplasms (IPMNs). Despite their different origin, whole-genome sequencing studies have shown that their transition from precursor lesions to malignant neoplasms is caused by a first generation of point mutations in the KRAS gene, followed by mutations in tumour suppressor genes such as CDKN2A, TP53 or SMAD4 [
6]. Recent studies combining genetic and epigenetic sequencing have identified four distinct pancreatic cancer subtypes based on their molecular signatures (squamous, pancreatic progenitor, aberrantly differentiated endocrine exocrine or ADEX, and immunogenic) [
7]. Despite their differences in prognosis, these molecular subtypes have not yet been matched with specific molecular targets that could facilitate therapy selection.
In this regard, pancreatic cancer is known by its wide heterogeneous genetic mutational landscape [
8], making the development of personalised and targeted treatment strategies particularly challenging. The detailed molecular characterisation of pancreatic cancer using next-generation sequencing approaches in recent years has allowed the identification of potential molecular targets and the development of novel therapeutic strategies for precision medicine that are currently being tested in clinical trials [
9]. These targeted drugs have been designed to interfere with genes and proteins differentially expressed in cancer cells compared to healthy tissue or different components of the TME, inhibiting key factors regulating cancer cell growth, survival, and metastasis [
10]. These approaches could minimise side effects associated with conventional therapies by increasing their efficacy and selectivity for the tumour and its stroma.
The high aggressiveness and chemoresistance of pancreatic cancer urge the search for novel and more effective treatment approaches. Targeted therapies will need to overcome some key challenges before becoming first line therapy for this disease. However, several combination strategies with well-established chemotherapeutic agents are currently under development or ongoing. In this review, we have summarised the most promising targeted agents currently in phase II and III clinical trials for pancreatic cancer. The results have been classified taking into account pancreatic cancer landmarks such as surface receptors, molecular signalling pathways, mechanisms for DNA damage repair or cell cycle arrest, and TME components such as the immune system, the tumour stroma, or angiogenic factors. Thus, we will discuss the most relevant and emerging studies already in the clinical setting.
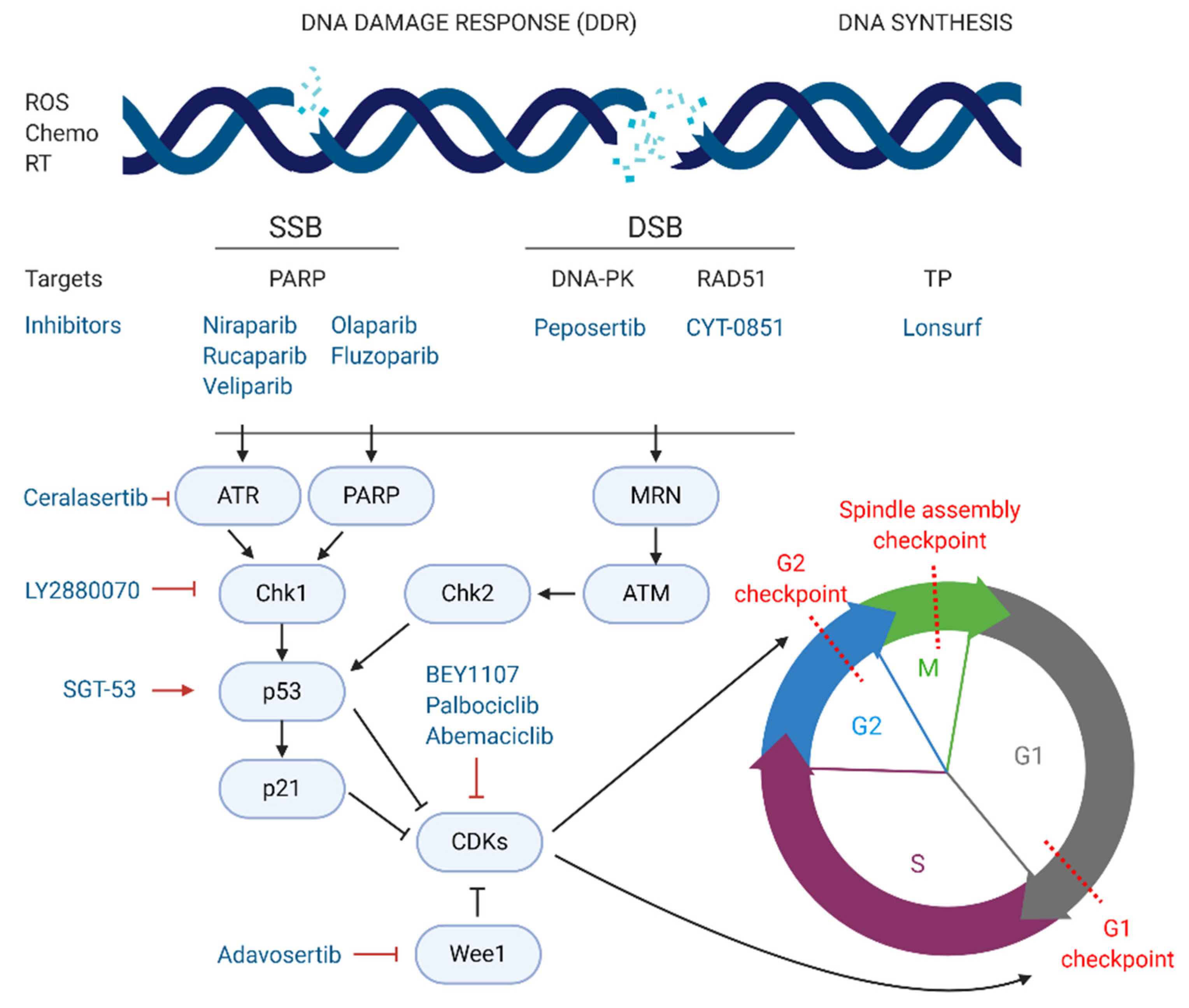
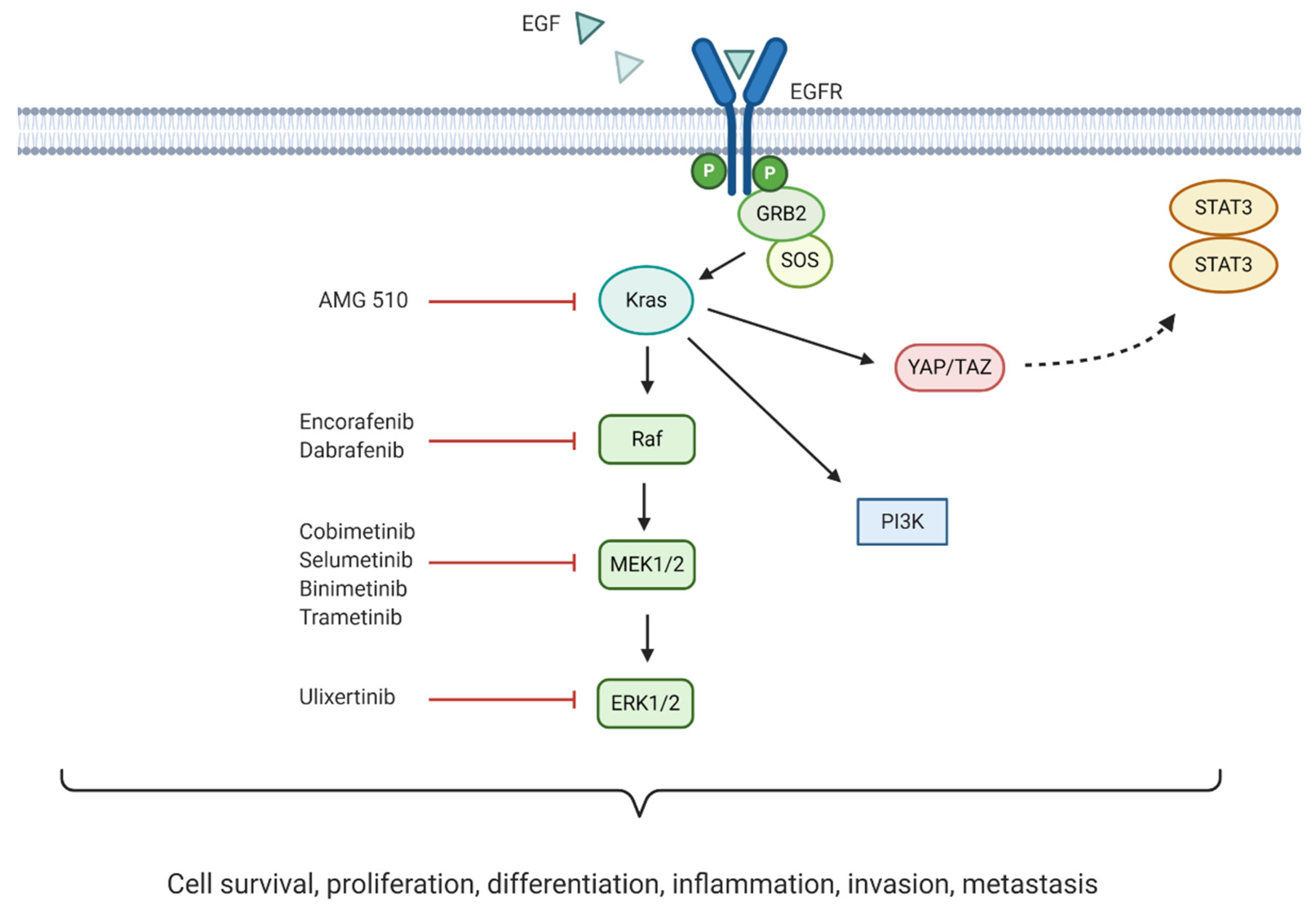
4. Targeting DNA Damage Response
DNA synthesis and replication during cell division are essential for cell proliferation. Defects in these processes may occur due to extrinsic and intrinsic factors leading to DNA damage [
71]. Cells have constitutive mechanisms to detect DNA damage. For that reason, the induction of DNA damage has been widely exploited as a promising strategy for cancer therapy, using extrinsic or intrinsic factors that impair DNA replication and activate the DNA damage response (DDR), leading to cell death. There are several types of DNA lesions that can activate the DDR. Radiation therapies for cancer treatment induce breaks in the double helix of the DNA, which can be single strand breaks (SSBs) or double strand breaks (DSBs). In SSBs the remaining undamaged strand acts as a template and guide the repair. However, for DSBs the DDR mechanisms activated are more complex often leading to more effective treatments. In normal conditions, DSBs are repaired via homologous recombination (HR) or non-homologous end joining (NHEJ) [
72,
73].
Different targeting approaches have been directed against PDAC genetic aberrations with great importance for DDR mechanisms such as the BRCA1, PALB2, BRC2, and RAD51 genes [
74]. In a normal cell undergoing DSBs, the MRN protein complex (formed by Mre11, Rad50, and Nbs1) detects strand breaks and interacts with the breast cancer type 1 susceptibility protein (BRCA1) which initiates repair by HR. BRCA1 recruits BRCA2 and the partner and localiser of BRCA2 (PALB2) to the damage site. The complex formed by BRCA1, BRCA2, and PALB2 activates the DNA repair protein RAD51 homolog 1 (RAD51) which starts the break repair [
75]. Around 24% of PDAC tumours display mutational patterns such as the BRCAness phenotype (loss of BRCA1/2), PALB2, RAD51, and other genes involved in DSB repair. This PDAC hallmark is currently being tested in several phase II and III clinical trials, evaluating the combination of crosslinking or ionising radiation treatments, which cause DNA breaks, with targeted drugs impairing the DDR.
The most exploited target involved in DNA repair is the family of poly (ADP-ribose) polymerases or PARPs, particularly PARP-1/2 in pancreatic cancer. These nuclear proteins are responsible for the detection of SSB in the DNA and recruitment of the DNA-repair enzymatic machinery. There are several PARP inhibitors currently in phase II and III studies for the treatment of pancreatic cancer (see
Figure 3). Their mechanism of action is based on the so called PARP-trapping model, in which PARP is trapped to the DNA helix by the inhibitor, impairing the correct replication of the DNA [
76]. Pancreatic tumours harbouring mutations in genes involved in DDR are especially sensitive to PARP inhibition by targeted therapies. Some examples of successful PARP inhibitors are the small molecules niraparib, rucaparib, veliparib, olaparib, and fluzoparib (see
Table 5) which differ in their potency to trap PARP to the DNA [
77].
Niraparib is been investigated in different phase I/II and II clinical trials for pancreatic cancer treatment. The NIRA-PANC phase II study is testing niraparib in patients with metastatic disease that have previously received first line chemotherapy (NCT03553004) [
78]. Its efficacy is being tested specifically for patients bearing germline deleterious or somatic mutations in DDR genes as previously described (NCT03601923). The combination of niraparib with immunotherapeutic agents targeting PD-1, such as dostarlimab (NCT04493060), dostarlimab plus radiotherapy (NCT04409002), nivolumab, or ipilimumab (anti-CTLA-4) (NCT03404960), are currently under investigation, suggesting that the future of pancreatic cancer treatment could take advantage of novel synergistic approaches of immunotherapy and targeted PARP inhibitors.
Rucaparib is another PARP inhibitor in phase II trials for advanced and metastatic pancreatic cancer (NCT03140670, NCT04171700). This agent has been already approved for other cancers such as prostate cancer carrying the BRCA mutation and ovarian cancer. Interestingly, rucaparib is currently being tested in combination with chemotherapy (liposomal irinotecan, fluorouracil and leucovorin) in a phase I/II study, for patients with metastatic pancreatic cancer (NCT03337087).
Veliparib is also being trialled for stage III and IV pancreatic cancer, in combination with standard of care chemotherapy. A phase II multicentre randomised trial compared 50 gBRCA/PALB2+ PDAC patients undertaking veliparib alone or in combination with gemcitabine plus cisplatin (NCT01585805). In this case, even though the group receiving the triple combination showed a median PFS slightly higher than the chemotherapy alone (10.1 months), the study did not meet the proposed response rate (RR) endpoint (p = 0.55) (17). A different study tested the combination of veliparib with mFOLFIRI compared with FOLFIRI alone in 123 patients with metastatic pancreatic cancer (NCT02890355). However, the results did not show any increase in OS (5.4 vs. 6.5 months) or PFS (2.1 vs. 2.9 months). One last study has reached phase I/II stage testing veliparib with modified 5-fluorouracil and oxaliplatin (mFOLFOX-6) in metastatic pancreatic cancer with BRCA mutations, but no results are available yet (NCT01489865).
Olaparib alone (NCT02677038, NCT02184195) or in combination with pembrolizumab (anti PD-1 agent, NCT04548752), cediranib (VEGF inhibitor, NCT02498613), or ceralasertib (a potent inhibitor of the Ataxia Telangiectasia and Rad3 related (ATR) protein kinase responsible of SSB recognition, NCT03682289), are currently in phase II and III clinical trials. The POLO study (phase III), a multicentre study carried out in 12 different countries, investigated the efficacy of olaparib monotherapy compared to placebo. The study enrolled 154 metastatic patients with genomic mutations in the BRCA genes whose disease did not progress after first line platinum-based chemotherapy. The results showed a median PFS of 7.4 months in the treatment group compared to 3.8 months in the placebo arm (
p = 0.004), but no significant changes in OS (18.9 months vs. 18.1 months). The olaparib arm also showed some major adverse events including cholangitis (2.20% of patients), abdominal pain (3.30%) and anaemia (6.59%) [
79].
Lastly, fluzoparib, a selective PARP1/2 inhibitor, has recently started a phase III study (NCT04300114). The trial is focused on metastatic patients with BRCA1/2 or PALB2 mutations resistant to platinum-based chemotherapy. A separate phase I/II trial is testing the efficacy of fluzoparib plus mFOLFIRINOX compared to mFOLFIRINOX alone for advanced pancreatic cancer (NCT04228601).
An alternative therapeutic strategy to target pancreatic cancer cells consists on inhibiting the correct function of the DNA-dependent protein kinase (DNA-PK), responsible for repairing DSB by NHEJ [
73]. Peposertib (also known as nedisertib or M3814) inhibits the ability of DNA-PK to repair DSBs [
80]. The combination of peposertib with hypofractionated radiotherapy for locally advanced PDAC, compared to radiotherapy alone, is in phase I/II trials (NCT04172532). As previously mentioned, RAD51 is another key protein for DNA repair of DSBs via HR, and therefore a potential target for pancreatic cancer patients. In the phase I/II multi-centre clinical trial NCT03997968, CYT-0851, a potent inhibitor of RAD51, is currently being tested as a single agent in 14 different malignancies, including locally advanced or recurrent metastatic PDAC. Another strategy for the induction of DNA damage focuses on impairing DNA synthesis. Lonsurf (previously TAS-102), a combination of the drugs trifluridine and tipiracil hydrochloride, has been approved since 2015 for metastatic colon cancer and is now in phase I/II trials, in combination with liposomal irinotecan, for advanced gastrointestinal cancers including stage III and IV pancreatic cancer (NCT03368963).
Overexpression of the serine/threonine kinase, glycogen synthase kinase 3β (GSK-3β), has been associated with reduced survival in PDAC patients, via regulation of the ATR DDR pathway. Moreover, signalling of mutant Kras has been shown to increase GSK-3β expression, which ultimately aids the growth and survival of Kras-mutant tumours [
81]. Promising preclinical findings led to the initiation of an ongoing phase I/II clinical trial exploring the small molecule inhibitor 9-ING-41 to inhibit GSK-3β (NCT03678883).
5. Targeting Cell Cycle Arrest
The cell cycle is composed of four phases—G1 (growth), S (DNA synthesis), G2 (growth and preparation for mitosis) and M (mitosis), and three strongly conserved cell cycle checkpoints to minimise the accumulation of mutations—(i) at the end of G1 phase, (ii) at the G2/M transition, and (iii) during M phase [
82]. Cyclins and cyclin-dependent kinases (CDKs), such as CDK1 and CDK2, constitute the main positive regulators of the cell cycle. Complexes formed by CDKs and cyclins activate, via phosphorylation, proteins controlling the progression of cells through the cell cycle checkpoints. On the other hand, negative regulators such as p53, p21, and the retinoblastoma protein are responsible for the detection of DNA damage and recruitment of DDR repair enzymes. If the damage cannot be repaired, nuclear accumulation of high levels of p53 activates the translation of p21, a major inhibitor of CDK/cyclin complexes, blocking the progression from G1 to S phase and triggering apoptosis [
83].
In cancer, including pancreatic cancer, positive cell cycle regulators, such as CDK1, are often overexpressed and linked to poor prognosis [
84]. Thus, targeted strategies inhibiting CDKs are being explored as novel treatment options for pancreatic cancer. On the other hand, inhibition of negative regulators of CDKs, such as checkpoint kinase 1 (Chk1) or Wee1 kinase, both responsible for the blockage of CDK/cyclin-dependent cycle progression regardless of DNA damage, also constitute promising targets for PDAC [
85,
86].
BEY1107, a novel inhibitor of CDK1, is in phase I/II as monotherapy and in combination with gemcitabine for the treatment of locally advanced or metastatic pancreatic cancer (NCT03579836). Another promising targeted agent in clinical trials is adavosertib, an inhibitor of Wee1. Two clinical trials are investigating the potential of adavosertib in pancreatic cancer patients. The NCT02194829 trial is studying adavosertib in combination with nab-paclitaxel and gemcitabine for stage III-IV disease. Similarly, the above-mentioned MATCH trial (phase II) is treating PDAC patients exhibiting the BRCAness phenotype with adavosertib monotherapy (NCT02465060). The same trial is also testing palbociclib, an inhibitor of CDK4/6. In this line, abemaciclib, another CDK4/6 inhibitor is also in phase II for a variety of unresectable and metastatic neuroendocrine tumours, including PNETs, that have not responded to first line therapy (NCT03891784). Lastly, a multicentre phase I/II study carried out in Canada and USA, is testing the inhibition of Wee1-activator Chk1 by the agent LY2880070, in different solid tumours, including advanced and metastatic pancreatic cancer (NCT02632448). The study will assess the efficacy of LY2880070 alone and in combination with gemcitabine. A previous phase Ib study already showed that combination of these two agents allowed reduced dosing due to their synergistic effect [
85].
An interesting approach is being tested at the Mary Crowley Cancer Research Center (USA) and the National Taiwan University Hospital (Taiwan) (NCT02340117). In this phase II study, metastatic pancreatic cancer patients receive a combination of nab-paclitaxel, gemcitabine and the gene-therapy agent SGT-53. SGT-53 consists of wild type cDNA of the p53 gene encapsulated in a liposomal formulation, aiming at restoring the wild-type function of p53. The combination of DNA-damaging agents, like gemcitabine or nab-paclitaxel, with the active form of p53 aims at stopping tumour progression via cell cycle arrest and induction of apoptosis.
In addition to the clear function that cyclins, CDKs and their consequent activators and inhibitors, play in cell cycle regulation, there is an upcoming trend to investigate the role of epigenetic modulators in cancer development. Histone deacetylases (HDACs), together with histone acetyltransferases (HATs), are key controllers of epigenetic gene regulation. Overexpression of HDACs has been linked to the development of different cancers including pancreatic cancer. Entinostat, a selective class I HDAC inhibitor, is being tested in combination with nivolumab (anti-PD-1) in a phase II study for patients with metastatic cholangiocarcinoma and pancreatic cancer (NCT03250273). Similarly, romidepsin (or istodax), a natural product obtained from the Gram-negative bacteria Chromobacterium violaceum, has also shown anti-HDAC properties and is currently approved for the treatment of other cancers like T-cell lymphomas. Romidepsin, in combination with nab-paclitaxel and gemcitabine, is also in clinical trials for advanced pancreatic cancer (NCT04257448).
Other epigenetic modulators like DNA methyltransferases (DNMTs) represent effective targets for pancreatic cancer treatment [
87]. Azacitidine (commercialised as Vidaza and approved for acute myeloid leukaemia treatment) is an analogue of cytidine that has been described to covalently bind DNMT1 to the DNA, blocking its epigenetic function and causing DNA damage. This compound is now in phase II for the treatment of resected pancreatic cancer patients with elevated CA19-9 levels (NCT01845805). Azacitidine is also being tested in another phase II trial in combination with the immunomodulator pembrolizumab (NCT03264404).
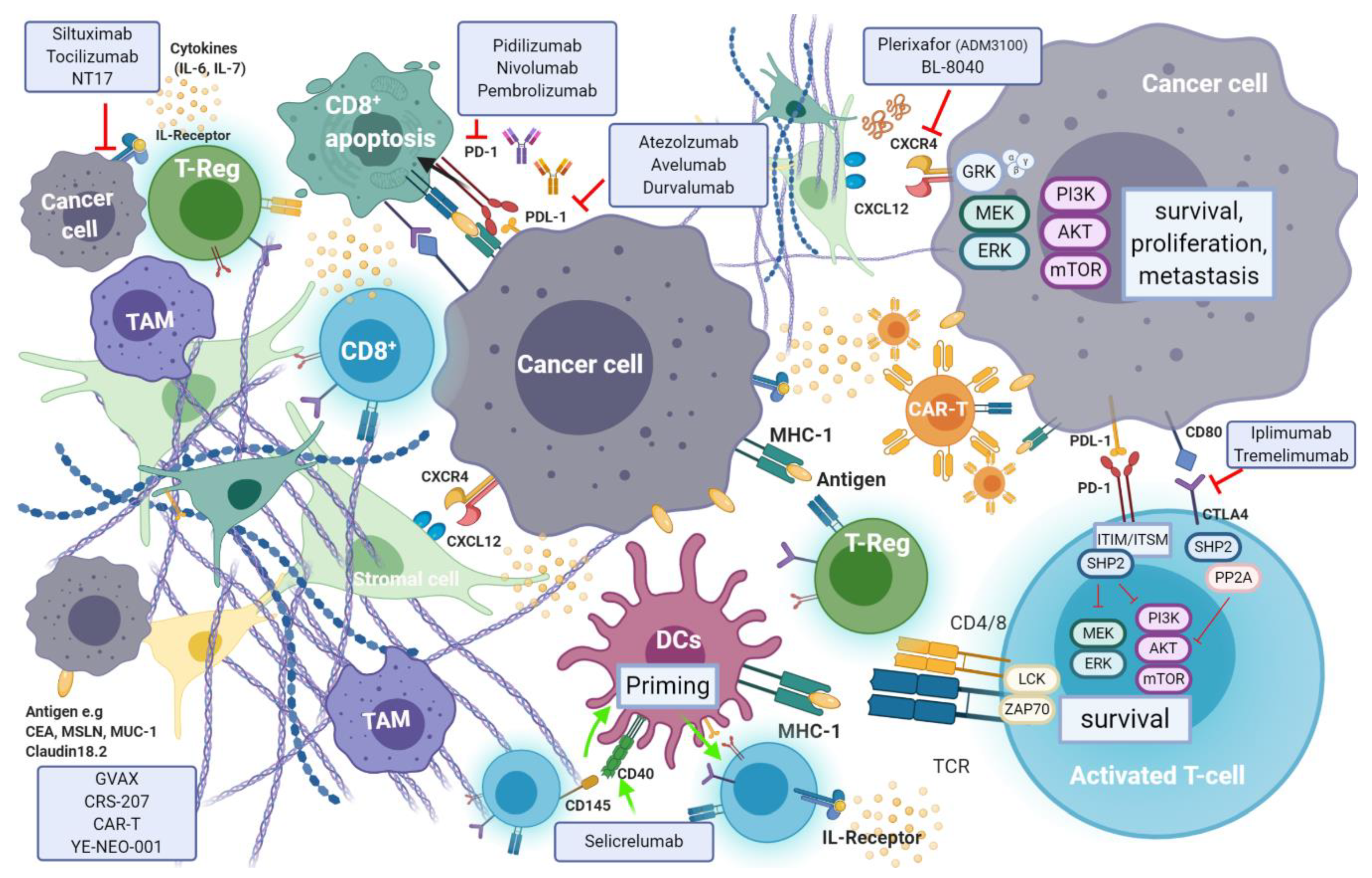
7. Immunotherapy
Recent evidence suggests that efficient immune response directed against tumour-specific neoantigens is often dampened by an immunosuppressant TME, which is a hallmark of pancreatic cancer [
9]. Cancer cell mediated activation of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs), tumour associated macrophages (TAMs) and CD4
+ T-regulatory cells (T-reg), is promoted through secretion and expression of immunosuppressive cytokines and membrane bound ligands (e.g., Programmed Death Ligand-1(PDL-1) or B7-1/2), that further hinder natural killer (NK) and T-cell anti-tumour response [
142]. Regulation of T-cell response occurs through T-cell receptor (TCRs) interaction with their respective ligands, following which signaling cascade action antagonizes T-cell activation [
143]. The induction of endogenous CD8
+ T-cell reaction is blunted by pancreatic stellate cells (PSCs) which also promote T-reg and MDSC differentiation (
Figure 7).
Moreover, a population of immature and poorly functioning antigen-presenting cells (APC) dendritic cells (DCs), that would normally induce anti-tumour immune response, are characteristic elements in pancreatic cancer. The relatively low mutational burden in pancreatic cancer, as opposed to other tumours, results in lower expression of tumour neo-antigens and therefore a reduced susceptibility to immune-surveillance [
144]. In pancreatic cancer, longer survival is observed in patients with tumours that are efficiently infiltrated by CD8
+ T-cells, FoxP3
+ and NK cells, and carry higher volumes of neo-antigens [
145]. A higher density of CD4
+ T-regulatory and T
H2-helper cells, paucity of CD8
+ T-cells and lower mutational burden are associated with impaired anti-tumour immunity and shorter survival rates. Targeted therapies seek to enhance immune effector cell response in cancer. T-cell immune responses are regulated by mechanisms known as “immune checkpoints”. Tumours exploit checkpoint mediated inhibition of cytotoxic T-cells to evade endogenous anti-tumour immune response. Targeted inhibition of specific immune checkpoints aims to disrupt this evasion mechanism and enhance tumour infiltration by cytotoxic immune effector cells. The two most frequently studied checkpoints are PD-1 and its ligand—programmed death-ligand 1 (PDL-1), as well as the cytotoxic T lymphocyte protein-4 (CTLA-4). Several antibodies targeting these checkpoints are being investigated, while others have already been approved for use.
7.1. Immune Checkpoint Inhibitors in Pancreatic Cancer
7.1.1. Targeting PD-1 and PD-L1
Programmed cell death protein 1 (PD-1) is an inhibitory receptor expressed on the surface of a variety of immune cells including T-cells, monocytes and B-cells. PD-1 is a crucial immune checkpoint molecule that inhibits the function of CD4
+ and CD8
+ T-cells in the TME and regulates its function during different physiological responses such as cancer, autoimmunity, and infection. The main PD-1 ligands are PD-L1 and PD-L2, which can lead to inhibitory cell singling resulting in the suppression of T-cell proliferation and cytokine production leading to the inhibition of mediated T-cell immune response [
146] (see
Figure 6). PD-L1 is expressed on the surface of many cell types, including APC, T and B cells, monocytes, and epithelial cells. Moreover, it is upregulated in response to proinflammatory cytokines such as IFNγ, IL-4, STAT1, and IFN regulatory factor-1 (IRF1). In addition, PD-L1 upregulation has been described in different types of cancers [
147]. In contrast to PD-L1 expression, PD-L2 expression is limited mainly to APC cells.
The overexpression of PD-L1 in cancer cells allows them to escape T-cell mediated immune response [
148]. Blocking the interaction between PD-1 and its ligand PD-L1 improves T-cell function leading to cancer cell immune recognition [
149]. Therefore, this particular interaction has gained much popularity as a therapeutic target for the treatment of several cancers, including pancreatic cancer, as it has been linked to poor prognosis. The KEYNOTE-028 trial (NCT02054806) evaluated the effect of the PD-1 antagonist pembrolizumab (Keytruda
®) in a cohort of 20 patients with various advanced solid tumours [
150]. In the subset population with pancreatic cancer, the use of pembrolizumab in monotherapy resulted in lack of efficient ORR (0–14%) and PFS of 1.7 months (95% CI, 1.5–2.9 months).
Resistance mechanisms are attributed to the often low mutational burden, the high population of suppressant immune cell population and CD8
+ T-cell sequestration. This has inspired a shift toward a multi-faceted approach, utilising combinations of immunomodulatory agents in an attempt to simultaneously target several aspects of drug resistance in PDAC. However, boosted PD-L1 expression does not necessarily sensitise PDAC to anti PD-1/PD-L1 therapies. In contrast to vaccine therapy, where it induced infiltration of PD-1 effector T-cells, chemo- or radio-therapy did not have the same effect. As a result, T-cell inducing agents may be crucial for the combination of chemo- or radio-therapy and immune checkpoint inhibitors, to improve the outcome of PDAC patients. A phase I/II trial completed in 2018 (NCT02331251), reported the use of gemcitabine, nab-paclitaxel and pembrolizumab (arm 3) as safe for first line treatment in PDAC. However, treatment efficacy reported only faintly improved outcomes compared to outcomes with a 28-day course, three treatment cycle, with gemcitabine and nab-paclitaxel [
151].
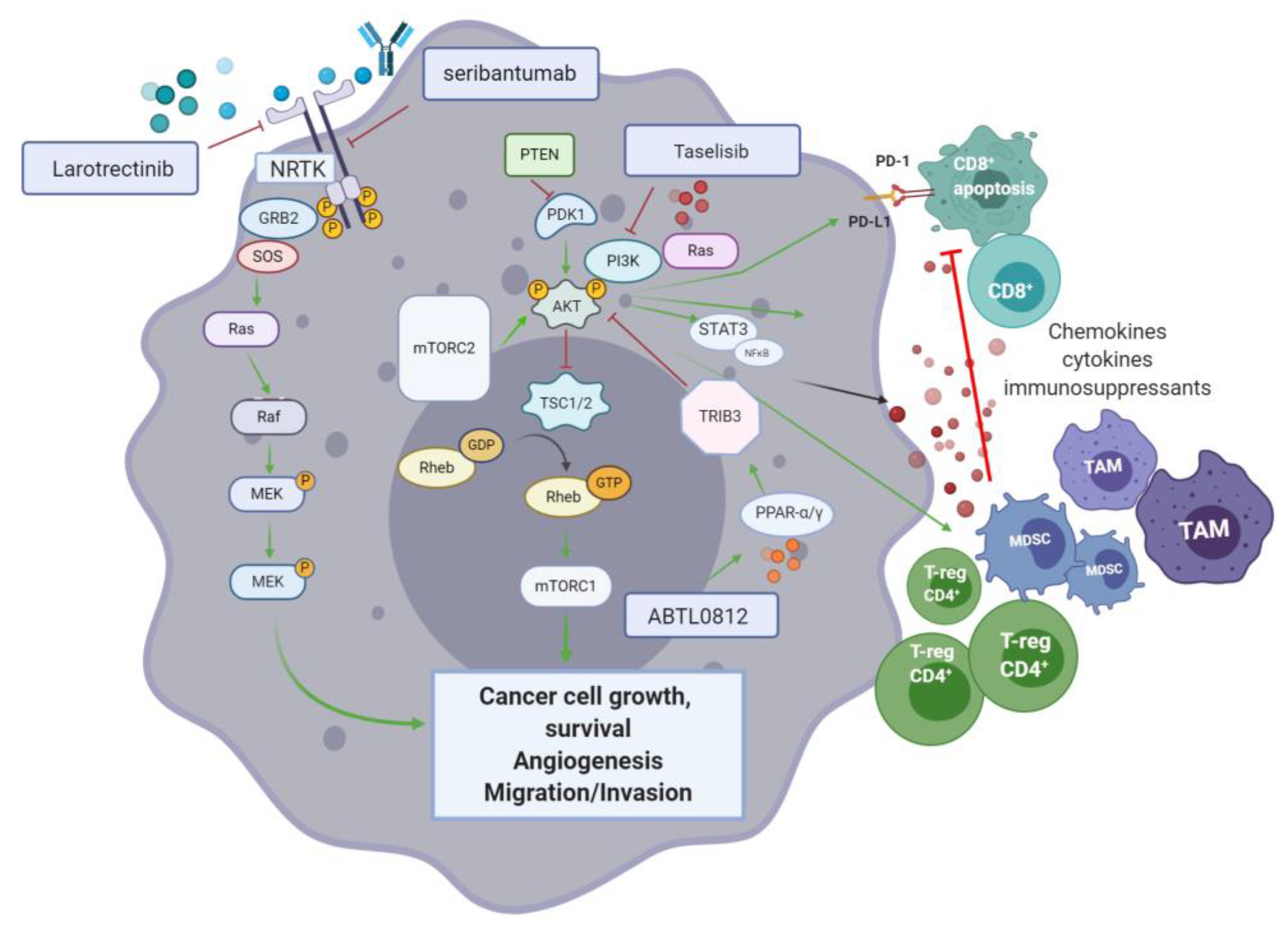
An ongoing randomised phase II study (NCT03727880) is looking into the effectiveness and safety of combining standard pancreatic cancer chemotherapy, prior and post-surgery, and pembrolizumab with or without the FAK inhibitor defactinib, in patients with high-risk resectable pancreatic cancer. The aim is to investigate whether reprograming the tumour microenvironment (FAK targeting) followed by chemotherapy can improve the effect of pembrolizumab. FAK inhibition by defactinib, could block the activation of downstream signalling pathways such as RAS/MEK/ERK and PI3K, inhibiting tumour proliferation and migration. Defactinib is also included in the phase II MATCH trial.
A 2-armed randomised phase III study (NCT03983057) is currently comparing the effect of modified-FOLFIRINOX alone in combination with the anti-PD-1 monoclonal antibody nivolumab, in patients with borderline resectable and locally advanced pancreatic cancer. The safety and efficacy of the co-administration of nivolumab and ipilimumab (anti-CTLA-4) together with high dose radiation therapy, in patients with metastatic pancreatic cancer, biliary tract cancer or in patients with previous gemcitabine intolerance, will be assessed in another phase II randomised open label trial (NCT02866383). Results are expected towards the end of 2021. A phase I/II trial (NCT03767582) is investigating the safety of nivolumab in combination with BMS-813160 (a CCR2/CCR5 dual antagonist) and GVAX (pancreatic cancer vaccine) in locally advanced pancreatic cancer patients. Those patients would have already received chemotherapy and radiotherapy, but the study aims to evaluate whether this combination therapy enhances CD8+ CD137+ cells infiltration in PDAC.
7.1.2. Targeting the Cytotoxic T-Lymphocyte Associated Antigen-4 (CTLA-4)
Activated T-cells express the cell surface receptor CTLA-4 (cluster of differentiation (CD)152), which upon binding to its activating ligands, B7-1 (CD80) and B7-2 (CD86), triggers a signal cascade that results in T-cell inactivation and apoptosis. Impairment of T-cell mediated anti-tumoural immune response therefore follows. An inverse correlation between expression levels of B7 by cancer cells (and tumour infiltrating monocytes, APCs and T-cells) and survival exists, which varies between different tumour types [
152]. In PDAC, expression of B7 ligand by tumour cells is upregulated as the disease progresses.
The only anti CTLA-4 blocking monoclonal antibody approved for clinical use is a direct antagonist of CTLA-4—iplimumab (YERVOY
®, Bristol-Mayers Squibb, New York, NY, USA). Promising preclinical studies formed basis for its regulatory approval for the treatment of advanced stage malignant melanoma in 2011 [
153], and is currently being investigated in other cancers such as NSCLC, advanced prostate and bladder cancers. Three phase Ib dose escalation studies (NCT01473940, NCT01473940 and NCT01473940) of iplimumab and gemcitabine in patients with late stage (III and IV) pancreatic cancer, showed minimal benefit over gemcitabine alone (RECIST study; ORR 14, 15% and stable disease in seven, five, and five patients, respectively). In a phase II study of iplimumab as a single agent in locally advanced or metastatic PDAC, no response was observed apart from one delayed response after initial disease progression (
Table 9). The one patient with a delayed response was reported to have a reduction in both primary and metastatic lesion tumour burden [
154].
Tremelimumab (CP-675 206) is a CTLA-4 specific fully humanised monoclonal antibody. Tremelimumab has been investigated in various cancers including gastric, pancreatic, mesothelioma, renal, melanoma and breast [
143]. In a dose escalation study (NCT00556023) in 34 patients with metastatic PDAC, the combined administration of gemcitabine and tremelimumab led to a partial response in only two patients (OR of 7.5 months (95% CI, 5.6–9.4 months)). This observation was reported in the cohort of patients subjected to the highest tremelimumab dose studied (15 mg/Kg). A phase II study of tremelimumab (NCT02527434) as monotherapy however, reported poor efficacy in a metastatic disease cohort of patients with previous treatment with 5-FU or gemcitabine-based chemotherapy. Ninety percent of patients (18/20) had disease progression with a poor median OS of 4 months (95% CI, 5.8–9.4 months) [
155].
7.2. The CXCL12-CXCR4 Axis
CXCR4 is a pan-cancer G-coupled receptor expressed in both solid and haematological malignancies. Cancer stromal cells in PDAC abundantly express C-X-C Motif Ligand 12 (CXCL12)—A ligand of CXCR4. CXCL12-CXR4 axis activation results in tumour progression, survival, enhanced invasion and metastasis, as well as TME modulation [
156]. Through increased expression of this ligand in metastatic sites of pancreatic cancer, CXCL12 is also thought to have a cancer cell-homing function at the target organs. In more aggressive subtypes, invasive CXCR4
+ cancer stem cells have also been identified at distant sites, suggestive of the CXCL12-CXCR4 axis role in invasion and metastasis [
157]. The role of CXCL12-mediated activation of FAK, ERK and AKT, has also been associated with chemoresistance in pancreatic cancer, among other gastrointestinal cancers [
157]. PDAC is described as a “cold” tumour in which an imbalance between immunosuppressive populations of CD4
+ T-cells and cytotoxic CD8
+ exists. Chemotaxis of immunosuppressive cells (MDSCs, T-Regs, PDL-1+ mast cells) and CD8
+ T-cell sequestration is enabled through the activation of the CXCL12-CXCR4 axis, and its interruption results in higher CD8
+ T-cell infiltration and intra-tumoural density, especially when used in combination with immune checkpoint inhibitors.
Plerixafor (AMD3100; Mozobil; Sanofi Genzyme, Cambridge, MA, USA) is the only FDA approved CXRC4 inhibitor. It has been widely studied in the context of treatment of acute myeloid leukemia, and in preclinical mouse models of PDAC [
158,
159]. Encouraging preclinical findings formed basis for clinical studies of plerixafor in patients with advanced PDAC, with a phase I (NCT02179970) and an open label phase II (NCT04177810) studies currently underway. The latter will assess the response of plerixafor in combination with the anti PD-1 antibody cemiplimab (REG-2810; Regeneron) in metastatic PDAC patients and is expected to report findings in late 2024.
BL-8040 (Motixafortide; BiolineRX, Modiin, Israel) is a high affinity, synthetic inhibitory peptide of CXCR4 which promotes bone marrow and lymph-node lymphocyte and NK cell mobilisation alongside inhibition of T-reg cell function in various solid tumours. Moreover, a synergistic effect was observed when BL-8040 was used in combination with PD-1 blockade, in preclinical models of PDAC. In a single arm phase IIa study (the COMBAT study; NCT02826486) the efficacy of BL-8040 together with pembrolizumab (keytruda®, Merk, Kenilworth, NJ, USA) as second or third line therapy in pancreatic cancer (cohort 1, n = 37), was compared with a second cohort receiving both agents together with chemotherapy (the NAPOLI-1 regimen: liposomal irinotecan, fluorouracil, and leucovorin) (cohort 2, n = 16). The highest benefit was observed when used in a second line setting, with the triple-therapy combination showing an ORR of 32% and a disease control rate of 77%. A median duration of response of 7.8 months was recorded in this cohort. The COMBAT/KEYNOTE-202 trial (NCT02826486) is assessing BL-8040 in combination with pembrolizumab in patients with metastatic pancreatic cancer. BL-8040 is also being evaluated in a phase I/II trial investigating multiple combinations of immunotherapy agents in treatment naïve, and in those with one-line prior systemic therapy (NCT03193190).
7.3. The Role of CD40, Immunocytokine and the Adenosine Pathway in PDAC Immunotherapy
CD40—A member of the tumour necrosis factor receptor superfamily, is a surface marker expressed by B-cells, antigen presenting DCs, monocytes as well as various cancer cells [
160]. The activation of CD40 occurs through T-cell presentation of its ligand CD154, by triggering downstream signalling cascades that result in the priming of CD40 expressing DCs. These in turn further induce the activation of cytotoxic T-cells and the release of pro-inflammatory cytokines, as part of cell mediated immune response. Cytokines (e.g., G-CSF, IL-2, IL-21, and IL-15) can be harnessed to stimulate an anti-tumour immune response by activation of DCs, NK and effector T-cells in combination with immune checkpoint inhibition, to transform the immunosuppressant TME of PDAC into an inflammatory one in vivo [
161]. Clinical studies of CD40 and cytokine-mediated therapy have been less successful. Selective activation of CD40 using the fully human monoclonal antibody selicrelumab, as single agent, showed limited clinical efficacy in 27 patients with advanced solid tumours [
162]. A more recent phase Ib study (NCT02760797) similarly reported lack of objective clinical response to CD40-targeted activation with selicrelumab, not even when used in combination with CSF-1R blockade (emactuzumab) [
163].
Several pro-inflammatory cytokines have been associated with disease progression, chemoresistance and poor outcomes in pancreatic cancer. In particular, IL-6 is currently being studied as a potential target for anti-inflammatory therapy. Siltuximab and tocilizumab are IL-6 targeting mAbs, which are FDA approved for treatment of several rheumatic, lymphoproliferative, and cytokine release syndromes. They have also been tested in various solid tumours (renal, ovarian, and multiple-myeloma) [
164]. The effects of IL-6 blockade in combination with other checkpoint inhibitors (e.g., anti PD-1 and anti CTLA-4) is currently being evaluated in phase I/II trials in metastatic pancreatic cancer (NCT04191421 and NCT04258150). The benefit of using tocilizumab in combination with gemcitabine and nab-paclitaxel is also being assessed in a randomised multi-national phase II study (PACTO; NCT02767557) in 140 patients, as first line treatment in locally advanced or metastatic PDAC. IL-7 plays a part in lymphocyte (T-cell and B-cell) development and homeostasis and therefore, its dysregulation supports autoimmunity and carcinogenesis. Overexpression of IL-7R in pancreatic cancer strongly correlated with disease progression following tumour resection, as well as with low survival rates [
165]. The effects of NT17 (efineptakin-α)—a long acting IgG fusion (recombinant IL-7) protein, is currently being tested in advanced, relapsed and refractory to previous treatment solid tumours (breast, lung, colorectal and pancreatic—or any other advanced solid tumours). This phase Ib/IIa study (KEYNOTE A60, NCT04332653) will also assess the effect of combining immunotherapy with pembrolizumab (anti PD-1). Results from the KEYNOTE A60 study are expected in 2023.
The adenosine pathway has recently been studied as a novel strategy to enhance anti-tumour immune response in cancers, due to its key role in immunosuppression, angiogenesis and ischaemic preconditioning. Activation of adenosine-mediated immunomodulation interestingly, has shown to be both of immunosuppressant and stimulant nature [
166]. Extracellular levels of ATP peak with rising cellular stress associated with inflammation, hypoxia and necrosis in the TME. Adenosine is the end product of ATP degradation by specific ecto-nucleosidases in both canonical (CD73, CD39) and non-canonical mediated pathways (CD38 and CD203a). Through the former, ATP is degraded into AMP by CD39 and later dephosphorylated to form adenosine by CD73. CD39 expression on T-regulatory cells, DCs, and macrophages is induced in a hypoxic environment. CD73 is expressed on tumour and immunosuppressant cells such as MDSCs and T-reg cells [
166]. By binding specific purinergic receptors (A2sR and A2bR), adenosine induces an immunosuppressive downstream signalling cascade as a consequence of intracellular cAMP accumulation. CD39 independent (non-canonical) adenosine production is enabled through NAD
+ conversion into AMP by CD38 (NAD
+ nucleosidase) and CD203a (ecto-nucleotide pyrophosphatase/phosphodiesterase 1), which is complemented by CD73 mediated AMP to adenosine conversion [
166]. With respect to pancreatic cancer, adenosine signalling plays a key role in pancreatic exocrine physiology, and together with CD73 and CD39, has been extensively reported to be upregulated in PDAC and its TME, in preclinical work [
167]. Interestingly however, in a murine model (BxPC-3-GFP cells in BALB/c mice) of pancreatic cancer, adenosine was shown to also have anti-tumourigenic effects by inducing cancer cell apoptosis. This effect, which was linked with structural similarities between adenosine and gemcitabine, was further augmented by inhibition of the AKT/p21 axis using the Akt inhibitor GSK690693 (GlaxoSmithKline, London, UK) [
168]. The evaluation of adenosine pathway targeting in clinical trials is however in its early stages and active clinical trials are limited to phase I or phase I/II studies—the majority of which have not yet started subject recruitment.
Oleclumab is a fully human IgG antibody, non-competitive inhibitor of CD73, currently being tested in 20 NCT registered clinical trials for treatment of various solid malignancies. It is currently being evaluated in a phase Ib/II in combination with durvalumab (anti PDL-1) and chemotherapy, for the treatment of 339 patients with metastatic pancreatic cancer. The study is expected to complete in late 2022 (NCT03611556). CD38 inhibition using the anti-CD38 antibody daratumumab (Darzalex®, Genmab, Copenhagen, Denmark) together with nivolumab (anti PD-1 IgG4) is being analysed in a phase I/II study of 120 patients with advanced pancreatic, lung and breast cancer (NCT3098550).
The Adenosine A2A receptor (A2aR) is the second most commonly used target. Initially investigated as an anti-Parkinson’s agent, A2aR inhibition is thought to play a role in tumour growth and metastasis inhibition. Effector T-cells increased expression of A2aR in inflammation, is inhibitory to T-cell cytotoxicity and inhibits cytokine release. In NK cells, A2aR activation results in their suppression while in regulatory T-cells, expansion and increased immunosuppressant activity follows [
167]. The role of A2aR in pancreatic cancer and its treatment is not yet well established. An open label phase II study (NCT03207867) is evaluating the A2aR antagonist NIR178 in combination with spartalizumab (anti PD-1) in multiple solid tumours (including pancreatic).
The use of immune checkpoint inhibitors has shown promising results in various cancers. In pancreatic cancer, the tumour favouring immune landscape challenges the use of single agent checkpoint inhibition and therefore, a multi-faceted approach is recommended. The Morpheus-Pancreatic cancer trial (NCT03193190) is a phase Ib/II multicentre open label study that will assess multiple combinations of immunotherapy agents in 290 patients with metastatic PDAC.
7.4. Cancer Vaccines
Priming of T-cells in order to stimulate an efficient anti tumour immune response can be induced using vaccines [
169]. While promising results have been observed in other cancers (cervical, melanoma, renal, and breast), the immunosuppressant TME, the low mutational burden (and therefore the paucity of immunity inducing neoantigens), highlight the need for further research of such an approach in pancreatic cancer [
170]. Vaccine based antigen delivery aims to sensitise antigen presenting DCs using the introduction of tumour cells, DNA or specific peptides that are further presented to T-cells, triggering their activation. Activated, antigen specific T-cells are then able to infiltrate the tumour and assert their cytotoxic effects as well as stimulate anti-tumour immunity.
Tissue specificity is crucial when inducing a localised immune reaction. Tissue markers that are differentially expressed in pancreatic cancer (CEA, mucin-1 (MUC-1)), mesothelin, mutated KRAS G12, CA19-9, carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) have been recently studied as potentials vaccine targets [
171]. Despite of the abundance of various reports of preclinical studies of anti-cancer vaccines in pancreatic cancer, the use of vaccines in the treatment of pancreatic cancer is an emerging field, and to date no such are approved, with only a handful being investigated in phase II/III clinical trials [
169]. In this section we will discuss those which have shown in particular promise in PDAC as well as those that reported promising findings in more advanced clinical studies.
7.4.1. BN-CV301
BN-CV301 is a poxviral based vaccine which contains transgenes that encode for the cancer associated antigens CEA and MUC-1 (an inducer of EMT and chemoresistance) that are both highly expressed in several solid tumours, including PDAC. BN-CV301 is a modified version of PANVAC which previously showed to increase antitumour T-cell (CD8
+) response against highly expressing MUC-1 and CEA tumours in vivo. These translated into clinical benefit in patients treated for breast, ovarian, colon and appendiceal cancers in terms of survival and response to further treatments. In view of lower safety profiles in patients with atopic dermatitis or who are immune-suppressed, as well as, potential cardiac risks associated with a replication capable smallpox delivery vector, PANVAC was later modified to its currently used attenuated version BN-CV301. Favourable safety profile with no dose limiting toxicities was reported in a dose escalation phase I study (NCT02840994). Antigen specific T-cell generation was observed in most patients with a prolonged stable disease most notably observed in those with KRAS mutated tumours, as well as those treated with immune checkpoint inhibitors following BN-CV301. The pancreatic cancer arm of a phase II study (NCT03376659) will evaluate the effects of combining BN-CV301 together with immune checkpoint inhibition (durvalumab) and capecitabine chemotherapy in a metastatic disease setting (
Table 10).
7.4.2. GVAX
GVAX is composed of two human pancreatic cancer cell lines genetically modified to secrete granulocyte-macrophage colony stimulating factor (G-CSF). GVAX stimulates long lasting T-cell infiltration into tumours as well as the activation of myeloid cells. Following limited toxicity and augmentation of anti-tumour immune response observed in 14 patients who underwent resection of PDAC [
172], GVAX was further tested in a phase II study in 60 patients with resectable disease (NCT00084383). In this study, GVAX was given together with 5-FU. Improved median overall and disease-free survival were observed (24.8 months (95% CI 21.2–31.6) and 17.3 months (95% CI 14.6–22.8), respectively) alongside induction of mesothelin specific CD8
+ T-cell response. The benefit of combining GVAX with cyclophosphamide (single injection or multiple doses) versus GVAX alone, is currently under study in a 72-patient cohort—phase II study in resected (or stage I-II) disease led by the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NCT01088789).
The combined use of GVAX and iplimumab (anti-CTLA-4) in a cohort of patients with metastatic PDAC previously receiving FOLFIRINOX, was evaluated in another randomised phase II study (NCT01896869) which reported findings only recently [
173]. In this cohort which was composed of two arms: iplimumab plus GVAX (arm A) versus continuation chemotherapy (FOLFIRINOX) (arm B), inferior survival of 9.38 months (95% CI, 5.0–12.2) versus 14.7 months (95% CI, 11.6–20.0) were reported. Furthermore, differentiation and augmentation of anti-tumour T-cell and macrophage (M1) responses were observed in arm A of the trial. Taken together with the aforementioned findings reported by Lutz et al., the combination of GVAX with iplimumab was superior to iplimumab monotherapy, however, inferior to FOLFIRINOX as maintenance therapy [
173]. The combination of GVAX with single (nivolumab) or several other checkpoint inhibitors (nivolumab, iplimumab, urelumab and pembrolizumab), cyclophosphamide with or without radiotherapy in surgically resected cases, is currently evaluated in four phase II trials (NCT03161379, NCT03190265, NCT02451982, NCT02648282). All four are expected to complete enrolment and report findings between 2021 and 2023.
The C-C motif chemokine ligand-2 (CCL2) also enables pancreatic cancer immune evasion. The CC receptor 2 (CCR2)/CCL2 axis facilitates inflammatory cell mobilisation and suppressant macrophage (CCR2
+) recruitment, further promoting a non-immunogenic TME [
174]. The CCR5/CCL5 axis, apart from having a similar immunomodulatory role to CCR2/CCL2, has also been implicated in cancer progression and metastasis as well as a marker of poor prognosis in several cancers. Phase I/II trials evaluating clinical outcomes following nivolumab and a dual CCR2/CCR5 antagonist (BMS-813160) together with GVAX with (NCT03184870) and without (NCT03767582) chemotherapy, in locally advanced and metastatic PDAC, respectively, are currently recruiting.
7.4.3. CRS-207
CRS-207 is a bacterial based vaccine of attenuated
Listeria monocytogenes which was modified to express mesothelin (MSLN)—A tumour associated antigen (cell-surface glycoprotein) expressed in PDAC. MSLN facilitates cancer cell attachment on mesothelial surfaces, MMP upregulation and autocrine signalling—playing multiple roles in PDAC metastasis [
175]. Early favourable results reported in a phase II study (NCT01417000) of 90 patients with metastatic PDAC [
176], who previously had a combination of cyclophosphamide and GVAX, were randomly assigned to further receive Cyclo/GVAX or four doses of CRS-207, were not translatable in a larger phase IIb cohort study. The authors reported an OS of 9.7 months with CRS-207 versus 4.6 months in the Cyclophosphamide/GVAX arm [
176]. The ECLIPSE study (NCT02004262) however, evaluated disease response in 303 patients with similar disease stage, to single agent chemotherapy, CRS-207 alone or CRS-207 plus Cyclo/GVAX. This larger, three-armed randomised phase IIb clinical trial failed to reach its primary efficacy point with OS of 3.7 (2.9–5.3), 5.4 (4.2–6.4), and 4.6 (4.2–5.7) months in these arms, respectively. No significant difference between chemotherapy and the combined treatments (HR = 1.17; 95% CI, 0.84–1.64) were observed [
177].
7.4.4. GI-4000
GI-4000 (GlobeImmune Inc., Louisville, CO, USA) is composed of recombinant yeasts which are engineered to express mutant KRAS epitopes in order to stimulate DCs, that have previously demonstrated to elicit an immune response in vivo [
178]. Mutated proteins are seen as antigens by CD8
+ T-cells which illicit a specific cytotoxic response against cells expressing such targets [
178]. A phase II study (NCT00300950) of 176 RAS mutant (mRAS) pancreatic cancer positive subjects, randomised (1:1) patients to receive gemcitabine or gemcitabine in combination with GI-4000. A priming injection (or placebo) was given prior to gemcitabine cycles until the primary endpoints were reached. An anti mRAS (G12 codon mutation)-specific immune response (INFγ) was measured in patients with microscopically positive tumour resection margins (R1), as opposed to those with an R0 (with negative margins). When compared to those receiving the placebo (8.3%; 1/12), 46% of patients with R1 resection (7/15) demonstrated a significant response (
p = 0.043) to gemcitabine plus GI-4000. Interestingly, longer median survival rates were observed in RAS mutated patients randomised to receive GI-4000 (by 773 days) than those which did not bear the mutation. With respect to RAS G12 mutated subjects receiving GI-4000 vs. placebo, those that were treated with GI-4000 showed a median survival which was longer by 568 days. In a subset of tumours with a specific proteomic signature (BDX-001) in this study, GI-4000 was shown to be of particular benefit. There are currently no actively recruiting trials in which GI-4000 is being further evaluated; however, a phase I (NCT03552718) trial will assess a novel yeast based vaccine (YE-NEO-001), in a subset of patients with pancreatic, colon and lung cancers who are in surveillance following a potentially curative treatment.
7.5. Adoptive Cell Therapies
Tumour infiltrating lymphocytes (TIL) are often found in surgically resected specimens of PDAC. As part of an anti-tumour immune response, infiltration by CD8
+ and CD4
+ T-cells results in recognition of tumour associated antigens presented on cancer cell surface using MHC-1 molecules, and a subsequent antitumour cytotoxic response. Density of T-cell infiltration has been directly correlated with improved survival in pancreatic cancer [
179]. In TIL therapy, tumour reactive T-cells are isolated, expanded ex-vivo and genetically programmed to target specific antigens. T-cells can be “reprogrammed” by means of modifications applied to either T-cells with antigen specific receptors (TCRs) or a genetic modification that induces expression of chimeric antigen receptors (CAR) of interest. While TCRs are natively expressed by T-cells and are MHC presentation dependent, CARs are artificially induced and can both recognise specific antigens as well as further activate T-cells—without the need for antigen presentation using MHC molecules [
180]. MHC-1 are able to present intracellularly degraded peptides, while CARs are only designed to recognise specific cell surface antigens.
MHC-1 expression, however, is often lost in cancer cells which challenges immune mediated tumour rejection using the TCR approach [
181]. Despite numerous reports of successful use of TILs in treatment of mostly haematologic malignancies, their use in solid tumours however is underdeveloped [
180]. Encouraging clinical response observed (complete remission) in ~22% of patients with metastatic melanoma that were treated using TIL infusions, have motivated their application in other cancers [
182]. The expansion of TILs ex-vivo has been successful in several solid tumours (e.g., pancreatic, melanoma, ovarian, breast, and lung) and showed significant anti-tumour reactivity in preclinical and early clinical studies of TILs [
182]. Specifically, in pancreatic cancer, isolation yield and anti-PDAC reactivity of TILs (CD4
+, CD8
+) can be improved by adjuvant use of immune checkpoint blockade (PD-1). TILs have been modified to recognise several neoantigens (including CEA, MUC-1, MSLN, prostate stem cell antigen (PSCA), claudin 18.2 and CD47, among others) in PDAC [
183]. Despite promising results in pre-clinical study reports, the translation of such findings in clinical settings is still challenging. A phase I (NCT01212887) trial that evaluated anti-CEA CAR-T (CEACAM5
+) treatment for CEA positive solid tumours, including PDAC, was halted due to observed acute respiratory toxicity, resulting from T-cell response directed against CEA co-expressing pulmonary epithelium.
In another study described by Beatty et al., the use of mRNA-based CAR-T generated T-cells directed against MSLN, in six patients with metastatic PDAC, showed stable disease in 30% of patients. In another case, the compassionate use of this approach in one patient with advanced PDAC, resulted in treatment tolerance as well as an objective evidence of antitumour humoral immune response. Multiple phase I/II studies of CAR-T treatment in pancreatic cancer are currently active (NCT04348643, NCT04581473, NCT04348643). A phase II trial (NCT04037241) is currently evaluating the benefit of combining anti-CEA CAR-T with different regimens of gemcitabine, nab-paclitaxel and capecitabine in 167 patients with liver metastatic lesions of CEA positive PDAC.
8. Targeting the Tumour Microenvironment
A hallmark of pancreatic cancer is the presence of a dense desmoplastic stroma, also known as the tumour microenvironment (TME), which can comprise up to 90% of the tumour volume. This stroma is composed of heterogeneous populations of cells, including cancer cells but also stromal components (e.g., fibroblasts, stellate cells, endothelial, neuronal, and immune cells) which are tightly embedded in a fibrotic extracellular matrix (ECM). This abundant ECM mediates elevations in interstitial fluid pressure and solid stress, thereby resulting in blood vessel constriction and vasculature collapse, reducing micro-vessel density and increasing intra-tumour hypoxia [
10]. Several studies have confirmed the key role of the TME in PDAC disease progression, metastasis niche formation and therapeutic resistance by favouring immune evasion or acting as a mechanical barrier for drug delivery [
184]. In the last years, it has also been shown the presence of cancer-associated fibroblast (CAF) phenotypic and functional heterogeneity (e.g., tumour-restraining and tumour-promoting CAFs), making the development of therapies targeting the crosstalk between cancer cells and stromal components challenging [
185]. Thus, stromal-targeting therapy in PDAC is an emerging research field aiming at achieving the optimal balance between stromal depletion and TME reprogramming. In this section, we have summarised the main clinical trials currently evaluating this strategy.
A phase Ib/II open-label, single arm clinical trial (NCT04203641) is evaluating the TME modifier L-DOS47 in combination with doxorubicin, in advanced pancreatic cancer patients previously treated. L-DOS47 is a urease conjugated to an anti-CEACAM6 (carcinoembryonic antigen-related cell adhesion molecule 6) monoclonal antibody, which exploits the acidic TME to locally increase pH and produce ammonia toxicity. CEACAM6 is overexpressed in PDAC cases and is linked to poor prognosis. A phase I trial has already shown promise using L-DOS47 to treat NSCLC (NCT02309892) [
186] and the phase I/II study has just been completed (NCT02340208).
The PDAC TME is characterised by high levels of hyaluronic acid (HA) that elevates interstitial pressure and impairs perfusion. A phase II study (HALO-109-202) evaluated the HA modulator PEGPH20 (pegylated recombinant human hyaluronidase), in combination with gemcitabine and nab-paclitaxel, compared with chemotherapy alone, in patients with metastatic untreated PDAC (NCT01839487) [
187]. Promising results led to a subsequent phase III study (NCT02715804) testing the same combination. However, in this study, PEGPH20+gemcitabine+nab-paclitaxel did not significantly improve OS. PEGPH20 was also tested in a phase Ib/II trial in combination with mFOLFIRINOX versus mFOLFIRINOX alone, in patients with metastatic PDAC (NCT01959139). The addition of PEGPH20 to mFOLFIRINOX caused increased toxicity and treatment duration needed to be decreased compared to mFOLFIRINOX alone [
188].
Different phase II trials have also evaluated inhibiting the Hedgehog signalling pathway as it regulates stroma deposition. However, compared to the promising results observed in preclinical studies, only a few targeted drugs improved patient survival, and their use was often accompanied by drug-related toxicity. For example, IPI-926 (NCT01130142) failed to show significant therapeutic benefits in PDAC patients. Similarly, vismodegib (Erivedge
®, Genentech, South San Francisco, CA, USA), the first FDA-approved small molecule inhibiting the hedgehog pathway, was used in combination with gemcitabine in a randomised phase I/II trial (NCT01064622). The combination did not improve OS or PFS in patients with recurrent or metastatic pancreatic cancer compared to gemcitabine alone [
189].
In addition to the above-mentioned drugs, other drugs targeting the stroma in phase II-III clinical trials include connective tissue growth factor (CTGF) antagonists, such as pamrevlumab (FG-3019; an anti-fibrotic agent). The randomised, double-blind phase III trial NCT03941093, is evaluating a neoadjuvant treatment with pamrevlumab or placebo in combination with gemcitabine plus nab-paclitaxel, for the treatment of locally advanced, non-resectable pancreatic cancer patients. CTGF antagonists have shown better results than anti-fibrotic agents (e.g., pirfenidone, still in preclinical stage) or metalloproteinase inhibitors (e.g., marimastat). A promising approach, still in phase I/II trials, is galectin targeted therapy. Galectins are members of the lectin family, and play a fundamental role in proliferation, metastasis, immunomodulation and angiogenesis. One galectin inhibitor specifically targeting galectin-9 is the monoclonal antibody LYT200. LYT200 alone or in combination with anti-PD1 or chemotherapy (gemcitabine+nab-paclitaxel), will be evaluated in a recently opened phase I/II open-label, multi-centre study (NCT04666688) in patients with relapsed/refractory metastatic solid tumours, including pancreatic cancer.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}