Abstract
Genetic variants that result in truncation in desmoplakin (DSP) are a known cause of arrhythmogenic cardiomyopathy (AC). In homozygous carriers, the combined involvement of skin and heart muscle is well defined, however, this is not the case in heterozygous carriers. The aim of this work is to describe cutaneous findings and analyze the molecular and ultrastructural cutaneous changes in this group of patients. Four women and eight men with a mean age of 48 ± 14 years were included. Eight met definitive criteria for AC, one was borderline and three were silent carriers. No relevant macroscopic changes in skin and hair were detected. However, significantly lower skin temperature (29.56 vs. 30.97 °C, p = 0.036) and higher transepidermal water loss (TEWL) (37.62 vs. 23.95 g m 2 h 1, p = 0.028) were observed compared to sex- and age-matched controls. Histopathology of the skin biopsy showed widening of intercellular spaces and acantholysis of keratinocytes in the spinous layer. Immunohistochemistry showed a strongly reduced expression of DSP in all samples. Trichogram showed regular nodules (thickening) compatible with pseudomonilethrix. Therefore, regardless of cardiac involvement, heterozygous patients with truncation-type variants in DSP have lower skin temperature and higher TEWL, constant microscopic skin involvement with specific patterns and pseudomonilethrix in the trichogram.
1. Introduction
Arrhythmogenic cardiomyopathy (AC) is an inherited myocardial disease that predisposes to fibrofatty replacement, ventricular aneurysms and sudden cardiac death due to ventricular arrhythmias [1]. This disease is one of the leading causes of sudden cardiac death due to ventricular tachyarrhythmias, especially in people under 35 years of age and in athletes. The first genetic variants associated with this condition were described in genes encoding desmosomes, critical structures involved in adherence regulation and mechanical integrity of tissues, especially in the myocardium and the skin. Familial characterization of this disease as a genetically conditioned disorder of desmosomes was first made by studying patients with Naxos disease, an autosomal recessive cardiocutaneous syndrome caused by mutations in plakoglobin and featured by the presence of AC, palmoplantar hyperkeratosis and „woolly” hair [2]. Carvajal disease was later described as another recessive cardiocutaneous syndrome, in this case by mutations in desmoplakin (DSP). However, mutations in desmosomal genes with autosomal dominant inheritance have classically been described to cause an exclusive cardiac phenotype without skin involvement [3]. The published series have shown that heterozygous carriers can develop as severe heart disease as homozygotes, although in heterozygosis the penetrance of the disease is incomplete. The causes of this incomplete penetrance are not yet fully understood. The age at onset of AC is variable with an estimated mean age of 31 years, being rare after the age of 60 years [4]. Gender disparity in the phenotypic presentation of AC has been demonstrated in different studies, highlighting male sex as a risk factor for the development of life-threatening arrhythmias [5].
Furthermore, cutaneous keratinocytes have been shown to express all cardiac isoforms of desmosomal proteins [6,7,8]. These findings support the hypothesis that they may reflect the molecular and histological changes that occur in the myocardium of patients affected by AC. Moreover, cutaneous involvement has recently been observed in a cohort of heterozygous patients with AC and truncating DSP variants [9].
The aim of this study is to describe cutaneous findings and analyze the molecular and ultrastructural cutaneous changes in heterozygous DSP truncating variant carriers with AC. The finding of specific patterns of cutaneous and hair involvement could potentially identify new patients, even in the absence of macroscopic cardiac involvement.
2. Materials and Methods
2.1. Patients
Patients with AC and a truncating DSP variant were selected from the Inherited Cardiovascular Diseases Unit of our center. Eight of the patients fulfilled current AC Task Force criteria (TFC) for left-sided or biventricular cardiomyopathy [10], one was identified as borderline and three cases were considered silent carriers. A complete cardiological study, including an electrocardiogram (ECG), 24 h-ambulatory ECG, exercise test, echocardiogram and cardiac magnetic resonance (CMR) was performed to assess the cardiac phenotype. Genetic test ruled out any other genetic variants and complex inheritance patterns. Pathogenicity of genetic variants was classified according to the American Academy [11], taking into account frequency in public databases (including the Human Gene Mutation Database, Single Nucleotide Polymorphism Database, NHLBI GO Exome Sequencing Project and ClinVar or in the Exome Aggregation Consortium (ExAC) database), its previous literature description, and several bioinformatic predictions according to its localization and conservation. After identification of DSP pathogenic variants in the unrelated index patients, we performed a genetic cascade screening among all available relatives using Sanger DNA sequencing method. Phenocopies such as myocarditis, sarcoidosis or ischemic heart disease were reasonably ruled out.
2.2. Dermatological Evaluation and Skin Barrier Function Assessment
Two independent dermatologists evaluated skin alterations and scalp disorders in the patients. Plucked hair shafts were collected to perform a trichogram. Homeostasis parameters related to epidermal barrier function were also measured. Transepidermal water loss (in g·m−2·h−1, using Tewameter® TM 300), stratum corneum hydration (in arbitrary units (AU), using Corneometer® CM 825), pH (using Skin-pH-Meter® PH 905), erythema and melanin index (in AU, using Mexameter® MX 18) and skin temperature (in °C, using Skin-Thermometer ST 500) were measured by a Multi Probe Adapter (MPA, Courage + Khazaka electronic GmbH, Bilbao, Spain). All variables were measured ten times, using their average for analysis. All these measurements were taken following the same order. All measurements were taken in the same room at a mean room temperature of 23 ± 1 °C and ambient air humidity of 45% (range, 40–50%). All participants underwent an adaptation period of at least 20 min before the measurements were taken. No systemic or topical treatments were allowed 3 hours before the measurements were taken. These variables were measured at three body sites: the right volar forearm, an involved area on the right palm and a non-involved area on the right palm. If the patient did not have skin manifestation, measurements were only taken on the right volar forearm and on the right palm.
Healthy controls gender- and age-matched (±3 years) with patients were also measured on the right volar forearm and on the right palm. These volunteers were people who attended the Dermatology Department for trivial conditions such as melanocytic nevi or seborrheic keratoses and did not suffer from an inflammatory skin disease, such as psoriasis or atopic dermatitis.
2.3. Samples Processing
A cutaneous biopsy was taken from non lesional skin from the palm of the hand in all cases. Samples were fixed in formaldehyde 10%, cut, processed and included in paraffin. Then, 2 µm sections were obtained on high adhesion slides. The deparaffination plus antigen retrieval was automatically performed using the PTLink from Agilent Dako. The immunostaining was performed in the AutostainerLink 48 from Dako. The slides were revealed with diaminobenzidine and counterstained with hematoxylin. Sample 12 was not valid for immunohistochemical or hematoxylin-eosin study.
2.4. Immunohistochemistry
Skin biopsies were studied with hematoxylin-eosin and subsequently an immunohistochemical study was performed to assess the cutaneous expression of different proteins. Information of the antibodies used, the manufacturers as well as the conditions used during the immunohistochemical procedure is included in the Table A1 of Appendix A. Biopsies from our archives from patients without disease (normal skin) were used as controls.
2.5. Transmission Electron Microscopy
Skin samples were fixed and embedded according to standard protocols. The tissue sample was fixed by immersion in 2% glutaraldehyde in 0.1 M cacodylate buffer (PB), pH 7.4, for 4 h at 4 °C. After fixation, the samples were rinsed three times with 0.1 M cacodylate buffer (PB), pH 7.4, for 15 min at 4 °C and stored overnight until further processing. Subsequently, the samples were postfixed with 0.1% osmium tetroxide in 0.1 M PB, pH 7.2, containing 1% potassium ferrocyanide for 1 h at 4 °C, dehydrated in a graded series of alcohols and embedded in epoxy resin. Semithin sections (500 nm) were stained with toluidine blue for light microscopic examination. Ultrathin sections were stained with uranyl acetate and lead citrate and analyzed under a Zeiss LEO 906E transmission electron microscope (Zeiss, Oberkochem, Germany).
2.6. Statistical Analysis
Descriptive statistics were used to present the sample characteristics. Continuous data was expressed as the mean ± standard deviation. The absolute and relative frequency distributions were estimated for qualitative variables. The Shapiro–Wilk test was used to check the normality of data distribution and Levene’s test to check the homogeneity of variance. The Student’s t-test for independent samples or Welch’s test was used to compare homeostasis parameters between patients and controls. The Student’s t-test for independent samples was used to compare homeostasis parameters between affected and non-affected areas in the same subject. A p-value of ≤ 0.05 was considered statistically significant. Statistical Analyses were performed using the SPSS package (SPSS for Windows, Version 24.0 Chicago: SPSS Inc.).
3. Results
Twelve heterozygous carriers of genetic variants leading to truncation in DSP from seven different families were included—four females (33%) and eight males (66%)—with a mean age of 48 ± 14 years. Eight patients met definitive criteria for the diagnosis of AC according to the 2010 TFC: two with exclusive left ventricular involvement, two with right ventricular involvement and four with biventricular involvement. Most of these cases showed segregation of the DSP mutation with a phenotype consisting of left ventricular fibrofatty replacement, low QRS complex voltages and an increased arrhythmic risk (Figure A1 of Appendix B). Patients 4, 6 and 9 had no pathological findings on ECG, echocardiogram or CMR, did not present events during follow-up, and were classified as silent carriers. Finally, one case showed a borderline AC status according to the TFC. A summary of the baseline cardiac phenotype of the enrolled patients is included in Table 1.

Table 1.
Baseline cardiac features.
3.1. Dermatological Evaluation and Skin Barrier Function Assessment
Clinically, only 33.33% (4/12) of patients had dermatological manifestations: 16.67% (2/12) had androgenic alopecia, Ludwig grade I, and 16.67% hyperkeratosis in pressure areas of the hands. Regarding skin barrier function (Table 2), we observed that transepidermal water loss was significantly higher on the patients’ palm than in controls’ (37.62 vs. 23.94 g·m−2·h−1, p = 0.028) while temperature was lower (29.56 vs. 30.97 °C, p = 0.036). No differences in stratum corneum hydration, erythema, melanin or pH were found on the palm. Moreover, no differences in skin barrier function on the volar forearm were found between patients and controls. Differences between the involved and the non-involved area on the right palm were not observed in any skin barrier function parameter.
Table 2.
Main histopathological findings.
3.2. Histopathology
Table 2 shows the main histopathological findings in each patient. The most remarkable feature was a significant widening of the intercellular spaces of the keratinocytes, which was evident in all the cases. This was most noticeable in the spinous layer and, in consequence, the spines of the keratinocytes were more evident than usual. Focally, acantholysis with separation of the keratinocytes from each other and open intercellular clefts (Figure 1A), with evidence of round acantholytic cells (Figure 1B) were observed.
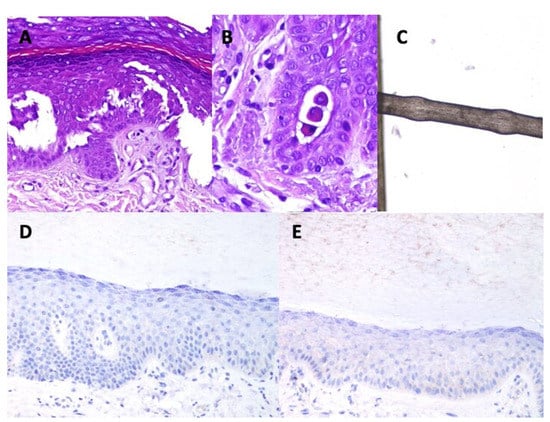
Figure 1.
Main findings in the immunohistochemistry study. (A): acantholytic areas evidenced in case 9 (Hematoxylin-Eosin ×200). (B): Acantholytic round cells. Some desmosomes (spines) are still observed between the round cells and adjacent keratinocytes (hematocillin-eosin ×400). (C): monilethrix. (D): negative expression of desmoplakin in case 4 (×200). (E): weak expression of desmoplakin in case 6 (×200).
In all the analyzed cases, the keratinocytes showed a very unusual appearance, with a striated cytoplasm which reminded the lines that shape fingerprints. For this reason, the term ‘fingerprint sign’ has been proposed (Figure 2). In addition, papillomatosis, dyskeratosis, transition cells, pale cytoplasm and cytoplasmic eosinophilic globules were absent.
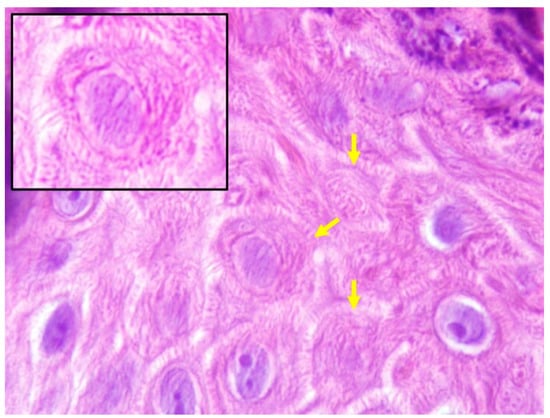
Figure 2.
The fingerprint sign. Keratinocytes with striated cytoplasm (Hematoxylin-Eosin ×1000).
3.3. Trichogram
All trichogram findings are summarized in the Table A2 of Appendix B. Some patients showed a prominent shift towards catagen/telogen. There was no hair shaft dysplasia or cuticular defects. However, seven showed non-uniform thickness of hair shafts and pseudomonilethrix (Figure 1C).
3.4. Immunohistochemistry
3.5. Transmission Electron Microscopy
In transmission electron microscopy analysis of skin biopsies, 50% of patients showed that keratinocytes of the spinous layer had a normal structure, characterized by a polygonal/elongated morphology with abundant bundles of tonofilaments (keratin filaments) in the cytoplasm, sometimes associated with masses of keratohyalin (Figure 3A). In these samples, desmosomes showed normal ultrastructural features with the presence of undisturbed electron-dense midline, inner plaques and keratin filament junctions (Figure 3B). For the other 50% of patients, ultrastructural analysis of the spinous layer showed an enlargement of the intercellular space between keratinocytes and an increase in the length of the cytoplasmic bridges (Figure 3C,D), as was found in the histological analysis of the samples. In these cases, a decrease in the number of desmosomes was observed, although they showed an unaffected structure (Figure 3E).
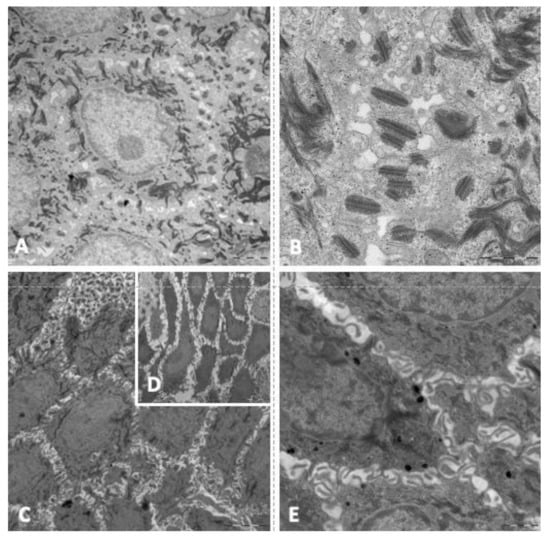
Figure 3.
Main findings in the electron microscope study. (A): keratinocytes of the spinous layer with abundant keratin filaments in cytoplasm. (B): keratinocytes with normal ultrastructural features (electron-dense midline, inner plaques and keratin filament junctions). (C): enlargement of the intercellular space between keratinocytes. (D): increase in the length of the cytoplasmic bridges. (E): Keratinocytes with unaffected structure.
4. Discussion
Truncating variants in DSP have repeatedly been related with a high penetrance cardiac syndrome with left ventricular fibrofatty replacement, predisposition to malignant arrhythmias and sudden cardiac death [12,13,14,15]. The Carvajal Syndrome reflects a recessive inheritance pattern with clinical affection of DSP not only at the cardiac level, but also affecting the skin. A major hallmark of the disease pathophysiology is the presence of cell death leading to fibrosis, predominantly at the epicardium. This location has classically made it difficult to obtain pathological samples to observe typical AC changes. The presence of cutaneous involvement when these variants are present in heterozygosis has previously been reported is small studies, but the real prevalence of skin and ultrastructural underlying cellular changes is yet to be described. Our study confirms the existence of a non-clinically relevant cutaneous phenotype, but in the presence of a highly depletion (or even complete absence) of DSP at the cutaneous level. We have been able to describe novel and potentially very specific cutaneous DSP changes that might be useful for the diagnosis and follow up, as they may mirror ultrastructural changes of the heart. These changes may help, as well, in the interpretation of variants of uncertain significance in DSP in patients with AC.
Compared to previous publications [9], in our cohort the macroscopic skin findings are not apparent, but the differences observed in skin homeostasis, with lower temperature and greater transepidermal water loss (TEWL), which had not been described until now, are noteworthy. High TEWL values are related with some skin diseases, such as atopic dermatitis or psoriasis [16]. So, our results might be reflecting subclinical skin impairment in AC patients. On the contrary, Maruthappu et al., found a fully penetrant cardio-cutaneous syndrome in heterozygous DSP loss of function mutation carriers [9]. These differences may reflect a different spectrum of DSP mutations, as their families carried variants affecting both DSP isoforms, while ours affected predominantly the cardiac isoform (DSP I). It is likely that our population represent in a better way the real-world AC-DSP-related patients, and we have provided evidence of ultrastructural changes in cell-cell adhesion that might have clinical implications in the future.
The histopathological changes evidenced in Carvajal syndrome and Naxos disease are usually described in the literature as epidermolysis with dyskeratosis and epidermal pseudospongiosis, respectively [17,18]. However, it is difficult to find a more accurate description with detailed histopathologic imaging. In fact, several of the most relevant studies on these conditions do not show the cutaneous histopathological findings [19,20,21,22]. Our patients showed negative (or almost negative) results in the immunohistochemical expression of desmoplakin. Since desmoplakin is located in the central part of the desmosomal plaque, it is not surprising that disruption in the protein lead to the observed findings: widening of the interepidermal space and eventual acantholytic foci [23]. A similar decrease in DSP expression has been involved in other acantholytic conditions, such as acantholytic squamous cell carcinoma [24], Hailey-Hailey disease [25], Darier’s disease [25], or epidermolysis bullosa [26]. In contrast, desmoplakin is not decreased (even in acantholytic cells) in the case of autoimmune acantholysis [25]. Winik et al. published very interesting results in two children with acantholytic ectodermal dysplasia. Their histopathological findings shown by electron microscopy are very similar to ours, with widening of the intercellular spaces between adjacent keratinocytes [18]. However, in our study those observations are accompanied by an atypical appearance of the keratinocytes with a striated cytoplasm, which we have termed the fingerprint sign. All these observations can be explained by the altered function of cellular desmosomes that may be caused by the truncation of DSP. It is particularly interesting that other series of heterozygous patients with pathogenic variants in DSP do not show a reduction in DSP expression in the skin, but only alterations in the distribution of plakoglobin and connexin 43 [9].
On the other hand, a very high prevalence of pseudomonilethrix was observed, which did not correlate with a curly hair phenotype that was highly prevalent in the previously mentioned study [9].
We found no differences in cutaneous and hair involvement between borderline and silent carriers compared to those who met definitive diagnostic criteria for AC. According to Table 2, the three silent carriers and the patient with a borderline diagnosis showed the same features as those with a definitive diagnosis, including a widening of the intercellular spaces of the keratinocytes, a striated appearance of the keratinocyte cytoplasm (‘fingerprint sign’) and a significant reduction of desmoplakin in the immunohistochemical study. Concerning the study of hair samples, one of the silent carriers showed pseudomonilethrix, while the other two did not. The sample from the patient with borderline diagnosis could not be taken.
These findings suggest that the cutaneous–pilose involvement in these patients is independent of the degree of cardiac involvement, which could be particularly helpful in the diagnosis in early stages of the disease or when cardiac tests are inconclusive.
5. Conclusions
Regardless of cardiac involvement, heterozygous patients with AC and truncation-type variants in DSP present lower skin temperature and higher transepidermal water loss, constant microscopic cutaneous findings and hair shaft involvement (pseudomonilethrix) with specifical patterns such as the “fingerprint sign”, in the absence of relevant clinically cutaneous findings. Although studies with larger cohorts are needed, as there are notable differences from the findings published before, these results open the possibility to include the skin and hair study as a marker of the disease even in the absence of cardiocutaneous affection.
Author Contributions
Conceptualization: E.C.-B., J.J.-J., S.A.-S., F.J.B.-J., J.T.-S., T.M.-V., M.S.-D., M.M.-J., F.J.C.-G., E.F.-S., R.M.-R., A.F.-F. and L.T.-S.; Data curation: E.C.-B., T.M.-V., M.S.-D., F.J.C.-G. and A.F.-F.; Formal analysis: T.M.-V., M.S.-D., S.A.-S. and L.T.-S.; Funding acquisition: E.C.-B., M.M.-J., J.J.-J., F.J.B.-J. and R.M.-R.; Investigation: E.C.-B., T.M.-V., M.S.-D., S.A.-S., J.J.-J., F.J.C.-G., E.F.-S. and A.F.-F.; Methodology: E.C.-B., J.J.-J., S.A.-S., J.T.-S. and F.J.B.-J.; Project administration: E.C.-B., J.J.-J., S.A.-S. and M.M.-J.; Resources: E.C.-B., M.M.-J., T.M.-V., M.S.-D., F.J.C.-G., E.F.-S. and A.F.-F.; Supervision: E.C.-B.; J.J.-J. and S.A.-S.; Validation: T.M.-V., M.S.-D., S.A.-S., A.F.-F., F.J.C.-G. and E.F.-S.; Visualization: E.C.-B., J.J.-J., F.J.B.-J. and S.A.-S.; Writing: E.C.-B., J.J.-J., F.J.B.-J., S.A.-S., T.M.-V., A.F.-F. and F.J.C.-G.; Writing: E.C.-B., J.J.-J., F.J.B.-J., S.A.-S. and T.M.-V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Andalusian Society of Cardiology with a “beca de Investigación general” which was granted on 2 October 2020.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of Hospital Virgen de las Nieves (protocol code Desmoplaquina y piel and date of approval 23 December 2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
Datasets related to this article can be found at https://data.mendeley.com/datasets/3mw76vz26s/1, an open-source online data repository hosted at Mendeley Data (30 July 2021). This article would be part of the PhD thesis research line of Eva Cabrera-Borrego.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Appendix A
Table A1.
Information on the antibodies used in the study as well as on the immunohistochemical procedure.
Table A1.
Information on the antibodies used in the study as well as on the immunohistochemical procedure.
| Antibody | Dilution | Incubation Time (minutes) | Antigen Retrieval pH | Visualization System | Manufacturer | Isotype | Type | Identification Number | Clone |
|---|---|---|---|---|---|---|---|---|---|
| Plakoglobin | 1/1000 | 30 | Low | Flex | BioNova | IgG1 | Monoclonal | A303-717A | 3728 |
| Desmoplakin | 1/25 | 30 | Low | Flex | PROGEN | IgG1 | Monoclonal | 61003 | DP1 + 2-2.15 |
| Conexin 43 | 1/400 | 30 | Low | Flex | Sigma Aldrich | IgG1 | Monoclonal | 045M4882V | C6219 |
| Filamin C | 1/50 | 45 | Low | Flex + Mouse | LSBio | IgG1 | Monoclonal | LS-B6314/47000 | N-terminus |
Appendix B
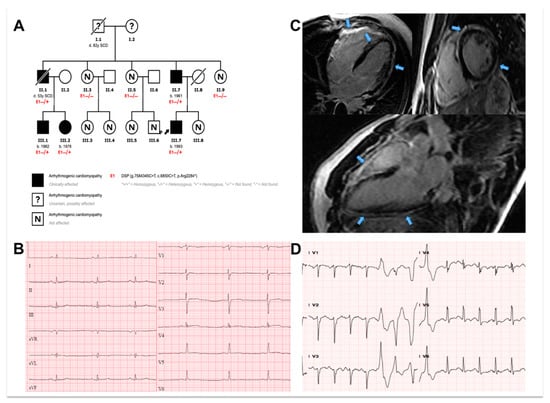
Figure A1.
Example of cardiac phenotype of included patients. (A): Pedigree of family according to the carrier status of the p.Arg2284* desmoplakin gene variant. Males and females are marked with squares and circles, respectively. Mutation carriers are marked with (E1-/+) and non-carriers with (E1-/-). Deceased individuals are annotated by a slash. Proband is indicated with black arrow. (B) and (C): classic ECG and CMR abnormalities for DSP gene carriers are illustrated (cases III-2 and III-7, respectively). The ECG (B) is characterized by low voltages and inverted/flattened T-waves. CMR demonstrates extensive circumferential subepicardial late gadolinium enhancement (C). (D): Non-sustained ventricular tachycardia (case III.2) during cardiopulmonary exercise testing.
Table A2.
Trichogram.
Table A2.
Trichogram.
| Patient | Anagen/Telogen (%) | Dystrophic Hairs | Keratotis Material at the Proximal Areas of the Hair Shaft | Hair Dysplasia | Uniform Diameters of the Shafts | Cuticular Defects | Other Findings |
|---|---|---|---|---|---|---|---|
| 1 * | a | a | a | a | a | a | a |
| 2 | 0/100 | Pseudomonilethrix | No | No | No | Yes | None |
| 3 | 100/0 | Pseudomonilethrix | Yes | No | No | No | White hairs |
| 4 | 100/0 | Pseudomonilethrix. Fractures in trichoschisis. Periodic stem crunches. | Yes | No | Yes | No | White hairs |
| 5 | 40/60 | Pseudomonilethrix | Yes | No | No | No | None |
| 6 | 90/10 | No | No | No | No | No | None |
| 7 | 0/100 | Pseudomonilethrix | Yes | No | No | No | None |
| 8 | 80/20 | No | No | No | No | No | White hairs |
| 9 | 20/80 (small sample) | No | No | No | No | No | None |
| 10 | 95/5 | No | No | No | No | No | None |
| 11 | 0/100 | Pseudomonilethrix | No | No | No | No | White hairs |
| 12 | 98/2 | Pseudomonilethrix | Yes | No | No | No | None |
* In patient 1 it was not possible to take a hair draft.
References
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm 2019, 16, e373–e407. [Google Scholar] [CrossRef] [Green Version]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Smith, E.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Mazzanti, A.; Ng, K.; Faragli, A.; Maragna, R.; Chiodaroli, E.; Orphanou, N.; Monteforte, N.; Memmi, M.; Gambelli, P.; Novelli, V.; et al. Arrhythmogenic right ventricular cardiomyopathy: Clinical course and predictors of arrhythmic risk. J. Am. Coll. Cardiol. 2016, 68, 2540–2550. [Google Scholar] [CrossRef]
- Desai, B.V.; Harmon, R.M.; Green, K.J. Desmosomes at a glance. J. Cell Sci. 2009, 122, 4401–4407. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.W.; McGrath, J.A. Mutations in genes encoding desmosomal proteins: Spectrum of cutaneous and extracutaneous abnormalities. Br. J. Dermatol. 2021, 184, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.B.; Nissen, P.H.; Palmfeldt, J.; Gehmlich, K.; Dalagen, S.; Jensen, U.B.; Kim, W.Y.; Heickendorff, L.; Mølgaard, H.; Jensen, H.K.; et al. Truncating plakophilin-2 mutations in arrhythmogenic cardiomyopathy are associated with protein haploinsufficiency in both myocardium and epidermis. Circ. Cardiovasc. Genet. 2014, 7, 230–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruthappu, T.; Posafalvi, A.; Castelletti, S.; Delaney, P.J.; Syrris, P.; O’Toole, E.A.; Green, K.J.; Elliott, P.M.; Lambiase, P.D.; Tinker, A.; et al. Loss-of-function desmoplakin I and II mutations underlie dominant arrhythmogenic cardiomyopathy with a hair and skin phenotype. Br. J. Dermatol. 2019, 180, 1114–1122. [Google Scholar] [CrossRef] [Green Version]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.P.J.; Daubert, J.P. Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia Proposed Modification of the Task Force Criteria Special Report. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- López-Ayala, J.M.; Gomez-Milanes, I.; Sanchez Munoz, J.J.; Ruiz-Espejo, F.; Ortíz, M.; González-Carrillo, J.; López-Cuenca, D.; Oliva-Sandoval, M.J.; Monserrat, L.; Valdés, M.; et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: Characterizing a phenotype. Europace 2014, 16, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Manchón, J.; Fernández, E.; Igual, B.; Asimaki, A.; Syrris, P.; Osca, J.; Salvador, A.; Zorio, E. Left Dominant Arrhythmogenic Cardiomyopathy Caused by a Novel Nonsense Mutation in Desmoplakin. Rev. Española Cardiol. 2011, 64, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.; Simpson, M.; Mogensen, J.; Shaw, A.; Hughes, S.; Syrris, P.; Sen-Chowdhry, S.; Rowland, E.; Crosby, A.; McKenna, W.J. Novel Mutation in Desmoplakin Causes Arrhythmogenic Left Ventricular Cardiomyopathy. Circulation 2005, 112, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Montero-Vílchez, T.; Segura-Fernández-Nogueras, M.-V.; Pérez-Rodríguez, I.; Soler-Góngora, M.; Martínez-López, A.; Fernández-González, A.; Molina-Leyva, A.; Arias-Santiago, S. Skin Barrier Function in Psoriasis and Atopic Dermatitis: Transepidermal Water Loss and Temperature as Useful Tools to Assess Disease Severity. J. Clin. Med. 2021, 10, 359. [Google Scholar] [CrossRef]
- Protonotarios, N.; Tsatsopoulou, A. Naxos disease and Carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 2004, 13, 185–194. [Google Scholar] [CrossRef]
- Winik, B.C.; Asial, R.A.; McGrath, J.A.; South, A.P.; Boente, M.C. Acantholytic ectodermal dysplasia: Clinicopathological study of a new desmosomal disorder. Br. J. Dermatol. 2009, 160, 868–874. [Google Scholar] [CrossRef]
- Ahranjani, B.M.; Moradi, P.; Nazari, S.; Farshadfar, S. A nine-year-old girl with left ventricle non-compaction and skin lesions (Carvajal syndrome). Iran J. Pediatr. 2015, 25, e370. [Google Scholar] [CrossRef] [Green Version]
- Baykan, A.; Olgar, Ş.; Argun, M.; Özyurt, A.; Pamukçu, Ö.; Üzüm, K.; Narin, N. Different clinical presentations of Naxos disease and Carvajal syndrome: Case series from a single tertiary center and review of the literature. Anatol J Cardiol. 2015, 15, 404–408. [Google Scholar] [CrossRef]
- Salam, A.A.; Remadevi, K.S.; Kurup, R.P. Naxos disease and Carvajal variant. Indian Pediatr. 2013, 50, 596–598. [Google Scholar]
- Sun, Q.; Wine Lee, L.; Hall, E.K.; Choate, K.A.; Elder, R.W. Hair and skin predict cardiomyopathies: Carvajal and erythrokeratodermia cardiomyopathy syndromes. Pediatr. Dermatol. 2021, 38, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Chidgey, M.A. Desmosomes and disease: An update. Histol. Histopathol. 2002, 17, 1179–1192. [Google Scholar] [CrossRef]
- Jurčić, V.; Kukovič, J.; Zidar, N. Expression of desmosomal proteins in acantholytic squamous cell carcinoma of the skin. Histol. Histopathol. 2015, 30, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Setoyama, M.; Choi, K.C.; Hashimoto, K.; Ishihara, M.; Predeteanu, G.S.; Dinehart, S.; Predeteanu, C.; Hamzavi, L.H.; Etoh, H. Desmoplakin I and II in acantholytic dermatoses: Preservation in pemphigus vulgaris and pemphigus erythematosus and dissolution in Hailey-Hailey’s disease and Darier’s disease. J. Dermatol. Sci. 1991, 2, 9–17. [Google Scholar] [CrossRef]
- Jonkman, M.F.; Pasmooij, A.M.G.; Pasmans, S.G.M.A.; Van Den Berg, M.; Ter Horst, H.; Timmer, A.; Pas, H.H. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am. J. Hum. Genet. 2005, 77, 653–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).