Abstract
The specific binding of the ubiquitous ‘marker of self’ protein CD47 to the SIRP protein anchored in the macrophage plasma membrane results in the inhibition of the engulfment of ‘self’ cells by macrophages and thus constitutes a key checkpoint of our innate immune system. Consequently, the CD47–SIRP protein complex has been recognized as a potential therapeutic target in cancer and inflammation. Here, we introduce a lattice-based mesoscale model for the biomimetic system studied recently in fluorescence microscopy experiments where GFP-tagged CD47 proteins on giant plasma membrane vesicles bind to SIRP proteins immobilized on a surface. Computer simulations of the lattice-based mesoscale model allow us to study the biomimetic system on multiple length scales, ranging from single nanometers to several micrometers and simultaneously keep track of single CD47–SIRP binding and unbinding events. Our simulations not only reproduce data from the fluorescence microscopy experiments but also are consistent with results of several other experiments, which validates our numerical approach. In addition, our simulations yield quantitative predictions on the magnitude and range of effective, membrane-mediated attraction between CD47–SIRP complexes. Such detailed information on CD47–SIRP interactions cannot be obtained currently from experiments alone. Our simulation results thus extend the present understanding of cooperative effects in CD47–SIRP interactions and may have an influence on the advancement of new cancer treatments.
1. Introduction
The adhesion of cell membranes arises from the specific binding of membrane-anchored receptor proteins to their ligands anchored in the apposing membrane. This receptor–ligand binding at the cell interface is essential for various biological processes, including tissue formation, immune responses, and signaling. An immunologically relevant example is the binding of the ubiquitous ‘marker of self’ protein CD47 to the SIRP protein anchored in the plasma membranes of macrophages. The CD47–SIRP binding results in the inhibition of the engulfment of ‘self’ cells by macrophages and thus constitutes a checkpoint of our innate immune system [1]. The binding of CD47 to SIRP has been found to play important roles in phagocytosis, auto-immunity, and host defense [1,2]. As such, the CD47–SIRP protein complex has been recognized as a potential therapeutic target in cancer [3,4] and inflammation [5].
The strength of the binding of molecules in a solution is typically quantified by the equilibrium constant , where is the volume concentration of molecular complexes, whereas and are the volume concentrations of free molecules in the solution. It is often assumed by analogy that the strength of the binding of membrane-anchored molecules is captured via the two-dimensional binding constant , where is the area concentration of receptor–ligand complexes, whereas and are the area concentrations of the free receptors and free ligands, respectively. A major difference between and is that the receptor–ligand binding that mediates membrane adhesion is determined not only with direct interactions between the receptor molecule and its ligand but also with the elastic properties of the adhering membranes [6,7]. In particular, for relatively weak adhesion and flexible membranes, has been shown to be inversely proportional to the relative roughness of the membrane surfaces brought about by thermal fluctuations [6].
The relationship derived by Hu et al. [6] is associated with a positive cooperativity in the receptor–ligand binding process, which can be explained as follows: Fluid membranes are rather soft and undergo thermally excited fluctuations. The receptor–ligand complex formation suppresses membrane fluctuations and causes the average distance between the membranes to be closer to the optimal distance for the receptor–ligand binding, which in turn facilitates the formation of additional receptor–ligand complexes between the two membranes. The feedback between the suppression of membrane fluctuations and the formation of receptor–ligand complexes leads to an effect of membrane-mediated binding cooperativity. This cooperativity effect has been predicted theoretically [8], examined in dissipative particle dynamics (DPD) simulations of a generic coarse-grained model [6], and confirmed quantitatively in fluorescence microscopy experiments with GFP-tagged CD47 on giant plasma membrane vesicles (GPMVs) binding to SIRP immobilized on a surface [9] (Figure 1A).
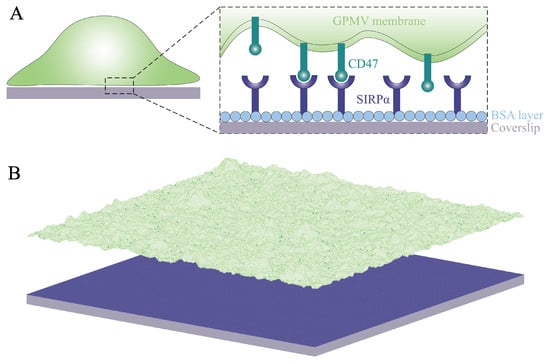
Figure 1.
(A) Cartoon of the system under study: SIRP receptors (dark blue) immobilized on a planar surface (gray) can bind CD47 ligands (dark green) on a GPMV (light green). The surface is coated with BSA (light blue) to prevent non-specific adhesion of the membrane to the surface. (B) Lattice-based mesoscale model that takes into account (i) diffusion of the membrane-anchored ligands, (ii) binding and unbinding of the receptors and their ligands, and (iii) elastic deformations and thermal undulations of the membrane. The color code is as in panel A. The lateral size of the system is 6 m.
The adhesion of cell membranes involves multiple length scales ranging from Angstroms to micrometers. Namely, the specific binding of the receptor proteins to their ligands occurs on the Angstrom length scale. The thickness of the lipid membrane is about 5 nanometers. The extension of the extracellular domains of the receptor and ligand proteins is typically of the order of 10 nanometers. The typical distance between the receptor–ligand complexes involved, e.g., in immune responses or signaling is of the order of 100 nanometers. Finally, the linear extension of the interface between cell membranes is of the order of micrometers. Because all of these length scales are relevant, theoretical and computational studies on membrane adhesion are challenging and require multi-scale modeling and suitable approximations that capture the essential physics of the system under study. Here, we use a lattice-based mesoscale model that captures the relevant length scales [10,11,12,13]. In particular, it takes into account (i) the diffusion of the membrane-anchored adhesion molecules, (ii) the binding and unbinding of the receptors and their ligands, and (iii) the elastic deformations and thermal undulations of fluid lipid membranes. We adapt this model to carry out large-scale simulations of the SIRP–CD47 system studied in the fluorescence microscopy experiments [9]. The system under study and the model in use are illustrated in Figure 1. Our simulations not only reproduce several different experiments [1,9,14] but also yield quantitative predictions on the range and magnitude of fluctuation-induced, membrane-mediated attraction between CD47–SIRP complexes. Since the SIRP–CD47 protein complex has been recognized as a promising therapeutic target in cancer [3,4], our detailed studies on the cooperative binding of CD47 to SIRP may have an influence on the advancement of new cancer treatments.
2. Model and Methods
We model the system used in the fluorescence microscopy experiments, where GST-tagged SIRP molecules (receptors) immobilized on a planar surface bind GFP-labeled CD47 molecules (ligands) on a GPMV [9], as illustrated in Figure 1A. The surface is coated with BSA to prevent non-specific adhesion of the GPMV membrane to the surface. The area concentration of the receptors on the substrate is , which provides a characteristic length scale nm.
The model is based on representing the GPMV membrane as a two-dimensional elastic sheet and discretizing this sheet into ‘patches’ of linear size a larger than the membrane thickness [8,15,16], as illustrated in Figure 1B. The choice of the patch size a is somewhat ambiguous. One option is to take nm to capture the complete spectrum of the bending deformations of fluid membranes [8,17]. Another option is to take a somewhat larger patch size, nm, to match it to an average exclusion radius of membrane proteins [12,18]. For comparison, Weikl and Lipowsky used nm in their studies on pattern formation during T-cell adhesion [19]. Here, we take nm to have, on average, one surface-immobilized receptor per membrane patch.
Membrane patches are labeled with index , which is a set of two integer numbers that specify the Cartesian coordinates in a reference plane. Here, we take the reference plane to coincide with the planar surface coated with BSA. The distance between membrane patch i and the BSA-coated surface is denoted by . The configuration of the membrane is thus given by a set .
The spatial distribution of ligands on the membrane is described using a set of binary variables with or indicating, respectively, the absence or presence of a ligand at membrane patch i. It is assumed at this point that any patch can accommodate only one ligand, which is in contrast with the model for pattern formation during T-cell adhesion [19], where multiple adhesion proteins could occupy a single patch. It should be noted, however, that the patch size a used in that model is about 5 times larger than in our present model.
To ensure the specific receptor–ligand binding, one ligand on membrane patch i only binds one receptor if is within a certain binding range, namely, . We define parameter , where denotes the length of the receptor–ligand complex and denotes the thickness of the BSA layer on the surface. Parameter reflects the flexibility of the receptor–ligand complex. Here, nm, as determined in molecular simulations of the surface-immobilized GST-tagged SIRP in complex with the membrane-anchored GFP-labeled CD47 [9].
The total energy of receptor–ligand interactions reads
where the sum is performed over all membrane patches and the receptor–ligand binding potential
is a square-well potential of depth U, width , and range . Here, denotes the Heaviside step function, i.e., if and otherwise. The depth U of potential can be interpreted as the receptor–ligand binding energy.
The three-dimensional binding constant of the soluble variants of CD47 and SIRP has been determined experimentally, yielding the dissociation constant [1]. An estimate for the binding energy U can be obtained from the relation , where and T denote the Boltzmann constant and room temperature, respectively. Taking nm, nm, and , we obtain .
In addition to the receptor–ligand interaction energy, , the Hamiltonian of the system under study also comprises the energy of membrane bending. We adapt the Helfrich theory for membrane elasticity [20] and use the formula derived by Weikl and Lipowsky [21,22] to compute the energy of membrane bending
Here, is the bending rigidity modulus of the membrane and denotes a discretized Laplacian which is equal to twice the local mean curvature of the membrane surface times [23]. It is implicitly assumed here that the membrane is not under tension and has no spontaneous curvature. The bending rigidity modulus of the GPMV membrane has been determined experimentally using flicker spectroscopy, leading to [9].
It should be noted that unspecific membrane–surface interactions (i.e., interactions not related to the specific receptor–ligand binding) are not included in the model, except for the short-ranged steric repulsion between the membrane and the BSA-coated surface, which is taken into account using a constraint . This assumption can be justified because the GPMVs in the fluorescence microscopy experiments have been observed not to adhere to the BSA-coated surface in the absence of the GST-tagged SIRP molecules immobilized on the surface [9].
2.1. Monte Carlo Simulations
Monte Carlo (MC) simulations were performed with Hamiltonian in the canonical ensemble, where the temperature T, the number of membrane patches, the number of immobile receptors, and the number of mobile ligands were fixed. Periodic boundary conditions were applied in the directions parallel to the planar surface. Two types of trial moves were used: (i) vertical local displacements of single patches to take into account deformations and thermal undulations of the membrane and (ii) horizontal shifts of single ligands to capture their diffusion within the membrane. The trial moves of type (i) caused variations in the field l of local distances between the membrane and the BSA-coated surface. Here, the maximal displacement of any membrane patch was 1.5 nm. In the trial moves of type (ii), the ligands were attempted to hop between neighboring patches, which led to variations in the composition field n. All of the trial moves were accepted according to the standard Metropolis criterion. Any trial move of type (i) leading to was rejected to prevent any overlaps of the membrane with the BSA-coated surface.
The proportion of trial moves (i) and (ii) was chosen according to physical time scales as in our earlier works [12,24]. In one MC cycle, on average, all of the membrane patches were attempted to be vertically displaced ten times, whereas all of the ligands were attempted to be horizontally shifted once. Each of the MC simulation runs comprised MC cycles, where the initial cycles were used for equilibration and the subsequent cycles for data acquisition.
In the fluorescence experiments [9], the area concentration of receptor–ligand complexes was in the range between about 35 and 85 CD47–SIRP complexes per . We performed the MC simulations in the same range of concentrations. The membrane in the MC simulations was composed of square patches, corresponding to an area of the adhesion zone of 36 . We thus performed a series of simulations with the total number of ligands , and 3240, corresponding to an area concentration of ligands between 30 and 90 per .
Molecular modeling shows that the complex of surface-immobilized GST-tagged SIRP and the membrane-anchored GFP-labeled CD47 has an average length nm [9]. The thickness of the BSA layer has not been determined. Thus, the exact value of parameter is unknown. Therefore, we performed a series of simulations with and nm.
In summary, we performed the MC simulations with the following values of the model parameters: nm, , , , nm, nm, and . We determined the average area concentration of free ligands, the average area concentration of receptor–ligand complexes, and, hence, the two-dimensional binding affinity
We also measured the average distance between the membrane and the BSA-coated surface as well as the membrane roughness
where the angular brackets denote the ensemble average. The roughness is caused by the thermal fluctuations of the membrane.
2.2. Analysis of Binding Kinetics
We adapt the maximum likelihood method developed by Hu et al. [6] for extracting the binding kinetics from DPD trajectories [6]. We apply this method to the MC trajectories of the CD47–SIRP system under study. The receptor–ligand binding and unbinding events divide any trajectory into different states with different numbers of receptor–ligand complexes. A system with receptors and ligands has states in total, where is the maximum number of receptor–ligand complexes. Thus, a given trajectory can be mapped to a Markov model
where the transition rates and are related, respectively, to the on- and off-rate constants and via
and
where A denotes the area of the contact zone.
The on- and off-rate constants and in Equations (7) and (8) can be determined from the observed number of transitions between the states and from the overall dwell times in the states. The binding and unbinding events divide a given trajectory into time windows j of length in state , which are followed by a transition into state with or . The probability for staying in state for a dwell time is . The probability for the time window j with the observed transition is for and for . The likelihood function is the probability of the whole trajectory and takes the form
where is the total number of transitions from state n to , the total number of transitions from state n to , and the total dwell time in state n.
Maximizing the likelihood function L in Equation (9) with respect to the rate constants and leads to the maximum likelihood estimators
and
In each simulation, we record the numbers of transitions and as well as the overall dwell times in each state , and then we estimate the binding rate constants in each state according to Equations (10) and (11).
For n around the average number of receptor–ligand complexes, , the values of and hardly change with n. We thus define the association rate constants and the dissociation rate constants . The binding affinity given by Equation (4) is then consistent with .
The dwell times in Equations (10) and (11) are in units of the number of MC steps. To relate one MC step to the physical time, , we follow Weikl and Lipowsky [19] and use the two-dimensional diffusion relation , where D is the diffusion coefficient of membrane proteins. Taking /s and nm, we obtain , which implies that each of the simulations comprising MC cycles corresponds to the physical time of about one hour.
3. Results
We performed MC simulations of the model introduced in Section 2. We measured the area concentration of receptor–ligand complexes and the two-dimensional binding affinity as given by Equation (4). The results of these simulations with , and nm are shown in Figure 2 as points in blue, orange, and purple, respectively. Importantly, the MC simulation results are in quantitative agreement with experimental FRAP data taken from Reference [9], which validates our computational model.
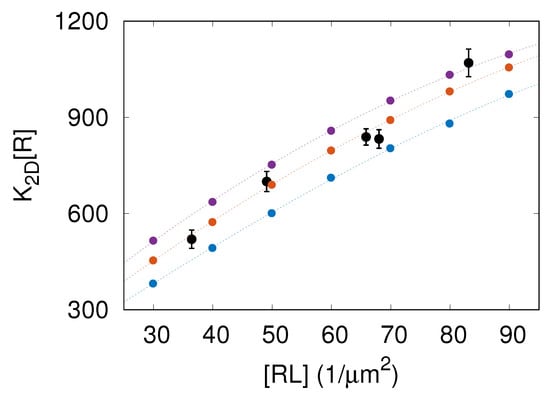
Figure 2.
Two-dimensional binding affinity times the area concentration of receptors, , versus the average area concentration of receptor–ligand complexes. The data points in black correspond to the experimental FRAP data taken from Figure 2A in Reference [9]. The points in blue, orange, and purple represent the results of MC simulations with , and nm, respectively. The dashed lines are to guide the eye.
The data presented in Figure 2 clearly demonstrate that the binding affinity is not constant but rather increases with the area concentration of CD47–SIRP complexes. Thus, the more CD47–SIRP complexes are formed, the larger the CD47–SIRP binding affinity gets, which implies that increasing the amount of CD47–SIRP complexes facilitates the formation of extra CD47–SIRP complexes. Therefore, CD47–SIRP binding is a cooperative process. The cause of this binding cooperativity is that the formation of CD47–SIRP complexes smoothens membrane fluctuations, which, in turn, facilitates the formation of additional CD47–SIRP complexes [6,8,9].
In the MC simulations, we also measured the thermal roughness of the membrane. This roughness results from thermally excited undulations of the flexible membrane. Figure 3 shows that the results of MC simulations with nm overlie on a master curve with . Since [9] and the dissociation constant of the soluble variants of CD47 and SIRP is [1], the latter relation is equivalent to with a dimensionless coefficient . The relation has been observed previously in DPD simulations of a generic, coarse-grained molecular model [6].
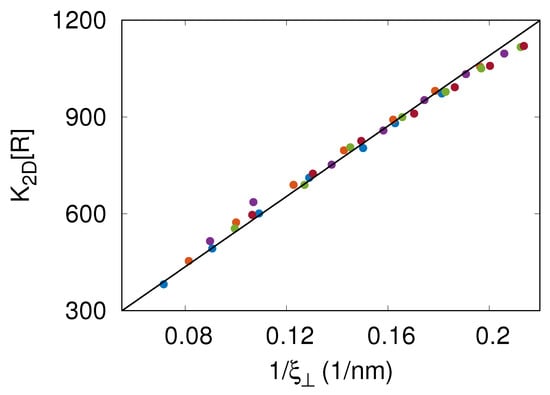
Figure 3.
Two-dimensional binding affinity times the area concentration of receptors, , versus the thermal roughness of the membrane. The different colors represent the results of MC simulations with nm. The solid line in black shows the relation with being a fitting parameter. This relation is equivalent to with , where is the dissociation constant of the soluble variants of CD47 and SIRP.
MC simulations with local moves can be used to study membrane dynamics in the overdamped limit [23,25]. Here, we keep track of receptor–ligand binding and unbinding events in the course of the MC simulations. The maximum likelihood method used to extract the binding rate constants and from the MC simulation trajectories is detailed in Section 2.2. Figure 4A shows that the values obtained from the simulations are in the range between and s. These values indicate that the off-rate is reaction-limited because , where is the simulation step time and is the depth of the receptor–ligand binding potential given by Equation (2). Importantly, the values obtained from the simulations compare well with s measured in surface plasmon resonance experiments [14], which additionally validates our computational model because no kinetic information is incorporated into the construction of the model.
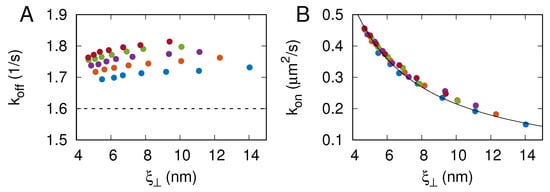
Figure 4.
CD47–SIRP binding rate constants (A) and (B) as a function of membrane roughness . The different colors represent the results of MC simulations with nm. The dashed line in panel (A) indicates s, determined in surface plasmon resonance experiments [14]. The solid line in panel (B) shows with being a fit parameter.
As can be seen in Figure 4A, the CD47–SIRP dissociation rate constant does not exhibit any particular dependence on the membrane roughness and varies only very weakly with . In contrast, the CD47–SIRP association rate constant decreases monotonically with the membrane roughness , as can be seen in Figure 4B. In fact, the data points obtained from the simulations with nm overlie on a master curve with nm/s. The relation is not fully consistent with the results reported by Hu et al. [6], probably because the relatively fast off-rates in the DPD simulations were not reaction-limited.
In the MC simulations, we also computed the two-dimensional pair correlation function for receptor–ligand complexes and the corresponding potential of mean force . Panels A and B in Figure 5 show the potential of mean force for nm and nm, respectively. The lines in orange, red, purple, and blue correspond to , and 90 per . Importantly, and in all of the cases studied here, which means that the receptor–ligand complexes are always effectively attracted one to another. This effective attraction between the receptor–ligand complexes is rather weak () and has a very long range, as it vanishes on the length scale of a micrometer.
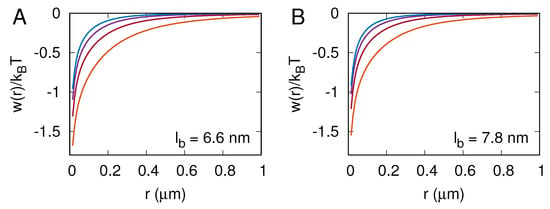
Figure 5.
Potential of mean force , where is the two-dimensional pair correlation function for receptor–ligand complexes. Panels (A,B) correspond to nm and nm, respectively. The lines in orange, red, purple, and blue correspond to 30, 50, 70, and 90 receptor–ligand complexes per square micron.
The membrane-mediated attraction between receptor–ligand complexes, as quantified here with the potential of mean force , is entropic in nature and originates from the suppression of conformational fluctuations of the membrane by receptor–ligand complexes [26,27,28]. It can be seen in Figure 5 that this attraction is strongest at the lowest receptor–ligand concentration, , which makes sense because conformational fluctuations of the membrane are largest in that case. The graphs of in Figure 5 also show that both the magnitude and the range of the effective attraction decrease with increasing the area concentration of receptor–ligand complexes, which is reasonable because the more receptor–ligand complexes are formed, the weaker the membrane fluctuates.
Based on the potential of mean force, , we computed the two-dimensional second virial coefficient [29]
Figure 6 shows the computed values of versus for , and nm. The color code is as in Figure 2. Importantly, in the whole range of parameters studied here. The negative values of mean that the receptor–ligand complexes are effectively attracted one to another. More negative values of imply stronger effective attraction between the receptor–ligand complexes.
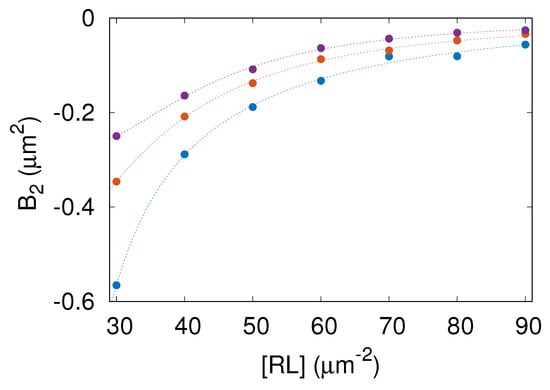
Figure 6.
Second virial coefficient, , as a function of the area concentration of the receptor–ligand complexes, , computed for nm (blue), (orange), and (purple). The color code and symbols are as in Figure 2.
The lowest value of found in the parameter range studied here is about . As can be seen in Figure 6, increases with both and . Consequently, the membrane-mediated attraction between receptor–ligand complexes is strongest for nm and gets weaker as is increased. This result is understandable because conformational fluctuations of the membrane are suppressed to a larger extent by receptor–ligand complexes when is smaller.
The effective, fluctuation-induced, membrane-mediated attraction between receptor–ligand complexes is not strong enough to induce phase separation within the membrane. Generally, if the adhesion of tensionless membranes is mediated by only one type of receptor–ligand complex, as in the system studied here, additional interactions (such as direct attraction between adhesion molecules [21] or generic repulsion between the apposing membranes [22] or the association of adhesion molecules with lipid rafts [11,12]) are necessary for separation between a phase depleted of receptors and a phase enriched in receptor–ligand complexes. However, the negative values of the second virial coefficient found in this study reveal a propensity of CD47–SIRP complexes to form transient clusters.
4. Discussion
The simulations presented here allowed us to capture processes occurring at various length scales, ranging from the specific receptor–ligand binding at the distance nm all the way up to membrane elastic deformations at the length comparable to the simulation box size . These processes were simulated on the time scale of about one hour. Importantly, the simulations not only reproduced the dependence of on obtained in the fluorescence microscopy experiments [9] (Figure 2) but also yielded the CD47–SIRP dissociation rate constant consistent with the value determined via surface plasmon resonance [14] (Figure 4A). The simulation results also complied with the general relationship derived by Hu et al. [6] (Figure 3) and demonstrated that the CD47–SIRP association rate constant (Figure 4B).
The CD47–SIRP complex concentration and binding affinity are found to be positively correlated (Figure 2). This means that the more CD47–SIRP complexes are formed in the adhesion zone, the larger the CD47–SIRP binding affinity gets, which implies that increasing the number of CD47–SIRP complexes leads to the formation of extra CD47–SIRP complexes. Therefore, CD47–SIRP binding is a cooperative process.
The CD47–SIRP binding cooperativity is due to thermal fluctuations and elastic properties of the membrane. Namely, the GPMV membrane is rather soft (its bending rigidity modulus ) and undergoes thermally excited fluctuations, which is reflected in the membrane roughness up to 14 nm (Figure 3). As CD47–SIRP complexes are formed, fluctuations in the local distance between the membrane and the planar surface are suppressed and, thus, the membrane roughness decreases. Then, the CD47 molecules anchored in the membrane are more likely to be present in the binding distance from the surface-immobilized SIRP molecules, which facilitates formation of additional CD47–SIRP complexes. Indeed, the binding affinity is found to increase as the membrane roughness is suppressed due to increasing the CD47–SIRP complex concentration (Figure 3).
Our analysis of the simulation data revealed long-ranged, membrane-mediated, entropic attraction between CD47–SIRP complexes. To explain the origin of this attraction, we note that the number of membrane conformations is larger when many CD47–SIRP complexes form one cluster and act effectively as one constraint on the local distance between the membrane and the surface than when CD47–SIRP complexes are distributed more-or-less uniformly and act as multiple constraints on the local distance between the membrane and the surface. Thus, clustering CD47–SIRP complexes decreases the entropy of the adhered membrane, which is the cause of the membrane-mediated, entropic attraction between CD47–SIRP complexes. This type of effect has been studied theoretically using generic models [27] and demonstrated in experiments on cadherin-mediated adhesion [28]. To the best of our knowledge, however, membrane-mediated interactions between CD47–SIRP complexes have not been determined so far. Here, we quantified the membrane-mediated, effective attraction between CD47–SIRP complexes in terms of the potential of mean force (Figure 5), which adds to the novelty of our study. Moreover, we determined the second virial coefficient as a function of the area concentration of CD47–SIRP complexes (Figure 6). Apparently, such detailed information on indirect interactions between CD47–SIRP complexes cannot currently be obtained from experiments alone. Our approach combining physics-based computer simulations with available experimental data is unique and provides new insights into the interactions between CD47 and SIRP.
The CD47–SIRP innate immune checkpoint has been in the focus of biomedical research [30,31,32,33,34]. The binding of CD47 to SIRP has been found to play important roles in phagocytosis, auto-immunity, and host defense [1,2]. As such, the CD47–SIRP protein complex has been recognized as a potential therapeutic target in cancer [3,4,30,32,33,35] and inflammation [5]. Our simulation results extend the present understanding of cooperative effects in CD47–SIRP interactions and thus can influence advancements of new cancer treatments [4,35].
It is important to note that the lattice-based mesoscale model employed in this study has several limitations. First of all, as CD47 and SIRP molecules are represented by single particles with no internal structure, the conformational and rotational entropy of these proteins is not included in the model. Secondly, the discretization of the membrane into square patches can impose artifacts in the distribution of the ligands within the membrane. It also limits the spatial resolution in the membrane lateral directions to nm and the temporal resolution to about 60 s. In principle, all of the aforementioned limitations can be overcome by using coarse-grained molecular dynamics simulations [6,36,37]. However, the computational costs still prohibit coarse-grained molecular dynamics simulations from exploring the length and time scales investigated in the present study using a lattice-based mesoscale model.
The major advantage of our present work over previous studies on the cooperative binding of CD47 with SIRP [9] is that, here, we captured and quantified the long-ranged, membrane-mediated, entropic attraction between CD47–SIRP complexes (Figure 5 and Figure 6). We also established how membrane fluctuations affect the CD47–SIRP binding rate constants and (Figure 4). This progress was possible because we carefully parameterized a lattice-based mesoscale model, performed extensive simulations of a sufficiently large adhesion zone with the linear size , and analyzed the simulation data in detail to determine various physical quantities. Further insights into the dynamics of the CD47–SIRP checkpoint can be gained in the future using coarse-grained molecular dynamics simulations [36,37].
Author Contributions
Conceptualization, B.R.; methodology, L.L., J.H. and B.R.; software, L.L., J.H., and B.R.; validation, L.L., C.G., J.H. and B.R.; formal analysis, L.L., C.G., J.H. and B.R.; investigation, L.L., J.H. and B.R.; resources, L.L., J.H. and B.R.; data curation, L.L. and B.R.; writing—original draft preparation, J.H. and B.R.; writing—review and editing, B.R.; visualization, L.L., C.G. and B.R.; supervision, J.H. and B.R.; project administration, J.H. and B.R.; funding acquisition, J.H. and B.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Centre of Poland (grant number 2021/40/Q/NZ1/00017), the National Natural Science Foundation of China (grant numbers 12232019, 12272388, 21973040, and 22161132012), and the Youth Innovation Promotion Association of the Chinese Academy of Sciences.
Data Availability Statement
The data presented in this study are openly available in [Monte Carlo Simulations of the Membrane-Mediated Cooperative Interactions between CD47 and SIRP] at [https://doi.org/10.18150/RLZZSW].
Acknowledgments
The Monte Carlo simulations were performed using the computer resources at the Centre of Informatics—Tricity Academic Supercomputer and Network (CI TASK) in Gdańsk, Poland, and the computing facilities in the High-Performance Computing Center (HPCC) of Nanjing University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Barclay, A.N.; Van den Berg, T.K. The interaction between signal regulatory protein alpha (SIRPα) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Andrechak, J.C.; Dooling, L.J.; Discher, D.E. The macrophage checkpoint CD47: SIRPα for recognition of ‘self’ cells: From clinical trials of blocking antibodies to mechanobiological fundamentals. Philos. Trans. R. Soc. B 2019, 374, 20180217. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody–mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Zhou, X.; Li, Y.; Li, B.; Cheng, L. The CD47-SIRPα axis is a promising target for cancer immunotherapies. Int. Immunopharmacol. 2023, 120, 110255. [Google Scholar] [CrossRef]
- Rodriguez, P.L.; Harada, T.; Christian, D.A.; Pantano, D.A.; Tsai, R.K.; Discher, D.E. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science 2013, 339, 971–975. [Google Scholar] [CrossRef]
- Hu, J.; Lipowsky, R.; Weikl, T.R. Binding constants of membrane-anchored receptors and ligands depend strongly on the nanoscale roughness of membranes. Proc. Natl. Acad. Sci. USA 2013, 110, 15283–15288. [Google Scholar] [CrossRef]
- Xu, G.K.; Hu, J.; Lipowsky, R.; Weikl, T.R. Binding constants of membrane-anchored receptors and ligands: A general theory corroborated by Monte Carlo simulations. J. Chem. Phys. 2015, 143, 243136. [Google Scholar] [CrossRef]
- Krobath, H.; Różycki, B.; Lipowsky, R.; Weikl, T.R. Binding cooperativity of membrane adhesion receptors. Soft Matter 2009, 5, 3354–3361. [Google Scholar] [CrossRef]
- Steinkühler, J.; Różycki, B.; Alvey, C.; Lipowsky, R.; Weikl, T.R.; Dimova, R.; Discher, D.E. Membrane fluctuations and acidosis regulate cooperative binding of ‘marker of self’ protein CD47 with the macrophage checkpoint receptor SIRPα. J. Cell Sci. 2019, 132, jcs216770. [Google Scholar]
- Weikl, T.R.; Hu, J.; Kav, B.; Różycki, B. Binding and segregation of proteins in membrane adhesion: Theory, modeling, and simulations. In Advances in Biomembranes and Lipid Self-Assembly; Elsevier: Amsterdam, The Netherlands, 2019; Volume 30, pp. 159–194. [Google Scholar]
- Li, L.; Hu, J.; Różycki, B.; Song, F. Intercellular receptor–ligand binding and thermal fluctuations facilitate receptor aggregation in adhering membranes. Nano Lett. 2019, 20, 722–728. [Google Scholar] [CrossRef]
- Li, L.; Hu, J.; Shi, X.; Różycki, B.; Song, F. Interplay between cooperativity of intercellular receptor–ligand binding and coalescence of nanoscale lipid clusters in adhering membranes. Soft Matter 2021, 17, 1912–1920. [Google Scholar] [CrossRef]
- Różycki, B.; Weikl, T.R. Cooperative stabilization of close-contact zones leads to sensitivity and selectivity in T-cell recognition. Cells 2021, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Brooke, G.; Holbrook, J.D.; Brown, M.H.; Barclay, A.N. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J. Immunol. 2004, 173, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- Różycki, B.; Lipowsky, R.; Weikl, T.R. Segregation of receptor-ligand complexes in cell adhesion zones: Phase diagrams and the role of thermal membrane roughness. New J. Phys. 2010, 12, 095003. [Google Scholar] [CrossRef]
- Weikl, T.R. Membrane-mediated cooperativity of proteins. Annu. Rev. Phys. Chem. 2018, 69, 521–539. [Google Scholar] [CrossRef]
- Goetz, R.; Gompper, G.; Lipowsky, R. Mobility and elasticity of self-assembled membranes. Phys. Rev. Lett. 1999, 82, 221. [Google Scholar] [CrossRef]
- Tsourkas, P.K.; Longo, M.L.; Raychaudhuri, S. Monte Carlo study of single molecule diffusion can elucidate the mechanism of B cell synapse formation. Biophys. J. 2008, 95, 1118–1125. [Google Scholar] [CrossRef]
- Weikl, T.R.; Lipowsky, R. Pattern formation during T-cell adhesion. Biophys. J. 2004, 87, 3665–3678. [Google Scholar] [CrossRef]
- Helfrich, W. Elastic properties of lipid bilayers: Theory and possible experiments. Z. Naturforschung C 1973, 28, 693–703. [Google Scholar] [CrossRef]
- Weikl, T.R.; Lipowsky, R. Adhesion-induced phase behavior of multicomponent membranes. Phys. Rev. E 2001, 64, 011903. [Google Scholar] [CrossRef]
- Weikl, T.R.; Andelman, D.; Komura, S.; Lipowsky, R. Adhesion of membranes with competing specific and generic interactions. Eur. Phys. J. E 2002, 8, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Różycki, B.; Weikl, T.R.; Lipowsky, R. Adhesion of membranes via switchable molecules. Phys. Rev. E 2006, 73, 061908. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, J.; Shi, X.; Shao, Y.; Song, F. Lipid rafts enhance the binding constant of membrane-anchored receptors and ligands. Soft Matter 2017, 13, 4294–4304. [Google Scholar] [CrossRef]
- Lipowsky, R.; Zielinska, B. Binding and unbinding of lipid membranes: A Monte Carlo study. Phys. Rev. Lett. 1989, 62, 1572. [Google Scholar] [CrossRef] [PubMed]
- Weikl, T.R.; Asfaw, M.; Krobath, H.; Różycki, B.; Lipowsky, R. Adhesion of membranes via receptor–ligand complexes: Domain formation, binding cooperativity, and active processes. Soft Matter 2009, 5, 3213–3224. [Google Scholar] [CrossRef]
- Speck, T.; Reister, E.; Seifert, U. Specific adhesion of membranes: Mapping to an effective bond lattice gas. Phys. Rev. E 2010, 82, 021923. [Google Scholar] [CrossRef]
- Fenz, S.F.; Bihr, T.; Schmidt, D.; Merkel, R.; Seifert, U.; Sengupta, K.; Smith, A.S. Membrane fluctuations mediate lateral interaction between cadherin bonds. Nat. Phys. 2017, 13, 906–913. [Google Scholar] [CrossRef]
- Neal, B.; Asthagiri, D.; Lenhoff, A. Molecular origins of osmotic second virial coefficients of proteins. Biophys. J. 1998, 75, 2469–2477. [Google Scholar] [CrossRef]
- Sugimura-Nagata, A.; Koshino, A.; Inoue, S.; Matsuo-Nagano, A.; Komura, M.; Riku, M.; Ito, H.; Inoko, A.; Murakami, H.; Ebi, M.; et al. Expression and prognostic significance of CD47–SIRPA macrophage checkpoint molecules in colorectal cancer. Int. J. Mol. Sci. 2021, 22, 2690. [Google Scholar] [CrossRef]
- Martínez-Sanz, P.; Hoogendijk, A.J.; Verkuijlen, P.J.; Schornagel, K.; van Bruggen, R.; van den Berg, T.K.; Tytgat, G.A.; Franke, K.; Kuijpers, T.W.; Matlung, H.L. CD47-SIRPα checkpoint inhibition enhances neutrophil-mediated killing of dinutuximab-opsonized neuroblastoma cells. Cancers 2021, 13, 4261. [Google Scholar] [CrossRef]
- Dizman, N.; Buchbinder, E.I. Cancer therapy targeting CD47/SIRPα. Cancers 2021, 13, 6229. [Google Scholar] [CrossRef] [PubMed]
- Behrens, L.M.; van den Berg, T.K.; van Egmond, M. Targeting the CD47-SIRPα innate immune checkpoint to potentiate antibody therapy in cancer by neutrophils. Cancers 2022, 14, 3366. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Akilov, O.E. Targeting the CD47-SIRPα Axis: Present Therapies and the Future for Cutaneous T-cell Lymphoma. Cells 2022, 11, 3591. [Google Scholar] [CrossRef] [PubMed]
- Olaoba, O.T.; Ayinde, K.S.; Lateef, O.M.; Akintubosun, M.O.; Lawal, K.A.; Adelusi, T.I. Is the new angel better than the old devil? Challenges and opportunities in CD47-SIRPα-based cancer therapy. Crit. Rev. Oncol. 2023, 184, 103939. [Google Scholar] [CrossRef]
- Reynwar, B.J.; Illya, G.; Harmandaris, V.A.; Müller, M.M.; Kremer, K.; Deserno, M. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature 2007, 447, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.C.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).