
A Novel Membrane-like 2D A’-MoS2 as Anode for Lithium- and Sodium-Ion Batteries

 ,
,  ,
,
Abstract
1. Introduction
2. Methods
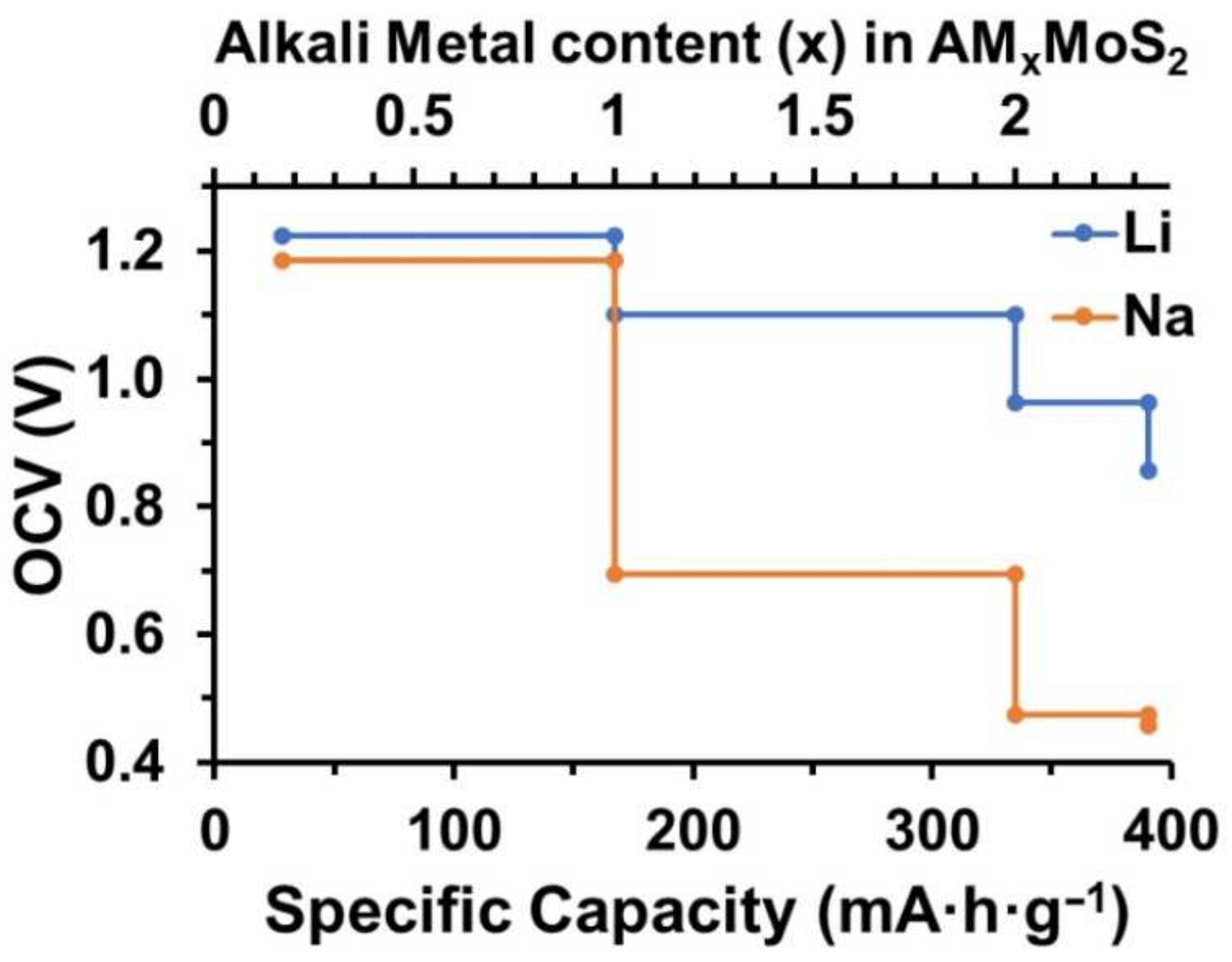
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, Y.; Dong, H.; Aurbach, D.; Yao, Y. Current Status and Future Directions of Multivalent Metal-Ion Batteries. Nat. Energy 2020, 5, 646–656. [Google Scholar] [CrossRef]
- Shea, J.J.; Luo, C. Organic Electrode Materials for Metal Ion Batteries. ACS Appl. Mater. Interfaces 2020, 12, 5361–5380. [Google Scholar] [CrossRef] [PubMed]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-Ion Battery Materials: Present and Future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Lee, H.-M.; Ghovanloo, M. Energy management integrated circuits for wireless power transmission. In Implantable Biomedical Microsystems; Bhunia, S., Majerus, S., Sawan, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 87–111. ISBN 978-0-323-26208-8. [Google Scholar]
- Tarascon, J.-M. Key Challenges in Future Li-Battery Research. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2010, 368, 3227–3241. [Google Scholar] [CrossRef] [PubMed]
- Nayak, P.K.; Yang, L.; Brehm, W.; Adelhelm, P. From Lithium-Ion to Sodium-Ion Batteries: Advantages, Challenges, and Surprises. Angew. Chem. Int. Ed. 2018, 57, 102–120. [Google Scholar] [CrossRef]
- Slater, M.D.; Kim, D.; Lee, E.; Johnson, C.S. Sodium-Ion Batteries. Adv. Funct. Mater. 2013, 23, 947–958. [Google Scholar] [CrossRef]
- Ellis, B.L.; Nazar, L.F. Sodium and Sodium-Ion Energy Storage Batteries. Curr. Opin. Solid State Mater. Sci. 2012, 16, 168–177. [Google Scholar] [CrossRef]
- Eftekhari, A.; Kim, D.-W. Sodium-Ion Batteries: New Opportunities beyond Energy Storage by Lithium. J. Power Sources 2018, 395, 336–348. [Google Scholar] [CrossRef]
- Peng, L.; Zhu, Y.; Chen, D.; Ruoff, R.S.; Yu, G. Two-Dimensional Materials for Beyond-Lithium-Ion Batteries. Adv. Energy Mater. 2016, 6, 1600025. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Zhu, Z.; Zhong, Y.; Bando, Y.; Golberg, D.; Yao, J.; Wang, X. The Role of Geometric Sites in 2D Materials for Energy Storage. Joule 2018, 2, 1075–1094. [Google Scholar] [CrossRef]
- Tang, X.; Ye, H.; Liu, W.; Liu, Y.; Guo, Z.; Wang, M. Lattice-Distorted Lithiation Behavior of a Square Phase Janus MoSSe Monolayer for Electrode Applications. Nanoscale Adv. 2021, 3, 2902–2910. [Google Scholar] [CrossRef]
- Bahari, Y.; Mortazavi, B.; Rajabpour, A.; Zhuang, X.; Rabczuk, T. Application of Two-Dimensional Materials as Anodes for Rechargeable Metal-Ion Batteries: A Comprehensive Perspective from Density Functional Theory Simulations. Energy Storage Mater. 2021, 35, 203–282. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, Y.; Ren, D.; Wang, L.; He, X. Graphite as Anode Materials: Fundamental Mechanism, Recent Progress and Advances. Energy Storage Mater. 2021, 36, 147–170. [Google Scholar] [CrossRef]
- Tarascon, J.-M.; Armand, M. Issues and Challenges Facing Rechargeable Lithium Batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, H.; Tang, X.; Fan, W.; Peng, G.; Qu, M. Graphite/Graphene Oxide Composite as High Capacity and Binder-Free Anode Material for Lithium Ion Batteries. J. Power Sources 2013, 241, 619–626. [Google Scholar] [CrossRef]
- Senyshyn, A.; Mühlbauer, M.J.; Dolotko, O.; Ehrenberg, H. Low-Temperature Performance of Li-Ion Batteries: The Behavior of Lithiated Graphite. J. Power Sources 2015, 282, 235–240. [Google Scholar] [CrossRef]
- Wen, Y.; He, K.; Zhu, Y.; Han, F.; Xu, Y.; Matsuda, I.; Ishii, Y.; Cumings, J.; Wang, C. Expanded Graphite as Superior Anode for Sodium-Ion Batteries. Nat. Commun. 2014, 5, 4033. [Google Scholar] [CrossRef]
- Stevens, D.A.; Dahn, J.R. The Mechanisms of Lithium and Sodium Insertion in Carbon Materials. J. Electrochem. Soc. 2001, 148, A803. [Google Scholar] [CrossRef]
- Cao, Y.; Xiao, L.; Sushko, M.L.; Wang, W.; Schwenzer, B.; Xiao, J.; Nie, Z.; Saraf, L.V.; Yang, Z.; Liu, J. Sodium Ion Insertion in Hollow Carbon Nanowires for Battery Applications. Nano Lett. 2012, 12, 3783–3787. [Google Scholar] [CrossRef]
- Moriwake, H.; Kuwabara, A.; Fisher, C.A.J.; Ikuhara, Y. Why Is Sodium-Intercalated Graphite Unstable? RSC Adv. 2017, 7, 36550–36554. [Google Scholar] [CrossRef]
- Liu, Y.; Merinov, B.V.; Goddard, W.A. Origin of Low Sodium Capacity in Graphite and Generally Weak Substrate Binding of Na and Mg among Alkali and Alkaline Earth Metals. Proc. Natl. Acad. Sci. USA 2016, 113, 3735–3739. [Google Scholar] [CrossRef] [PubMed]
- Chepkasov, I.V.; Ghorbani-Asl, M.; Popov, Z.I.; Smet, J.H.; Krasheninnikov, A.V. Alkali Metals inside Bi-Layer Graphene and MoS2: Insights from First-Principles Calculations. Nano Energy 2020, 75, 104927. [Google Scholar] [CrossRef]
- Jian, Z.; Luo, W.; Ji, X. Carbon Electrodes for K-Ion Batteries. J. Am. Chem. Soc. 2015, 137, 11566–11569. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Ruiz, N.; Armstrong, A.R.; Alptekin, H.; Amores, M.A.; Au, H.; Barker, J.; Boston, R.; Brant, W.R.; Brittain, J.M.; Chen, Y.; et al. 2021 Roadmap for Sodium-Ion Batteries. J. Phys. Energy 2021, 3, 031503. [Google Scholar] [CrossRef]
- Xu, X.; Li, F.; Zhang, D.; Liu, Z.; Zuo, S.; Zeng, Z.; Liu, J. Self-Sacrifice Template Construction of Uniform Yolk–Shell ZnS@C for Superior Alkali-Ion Storage. Adv. Sci. 2022, 9, 2200247. [Google Scholar] [CrossRef]
- Yuan, J.; Mu, M.; Xu, X.; Gan, Y.; He, H.; Zhang, X.; Li, X.; Kuang, F.; Li, H.; Liu, J. Three-Dimensional Porous FeS@ N Doped Carbon Nanosheets for High-Rate and High-Stable Sodium/Potassium Storage. Compos. Part B Eng. 2022, 247, 110300. [Google Scholar] [CrossRef]
- Chen, B.; Chao, D.; Liu, E.; Jaroniec, M.; Zhao, N.; Qiao, S.-Z. Transition Metal Dichalcogenides for Alkali Metal Ion Batteries: Engineering Strategies at the Atomic Level. Energy Environ. Sci. 2020, 13, 1096–1131. [Google Scholar] [CrossRef]
- Yun, Q.; Li, L.; Hu, Z.; Lu, Q.; Chen, B.; Zhang, H. Layered Transition Metal Dichalcogenide-Based Nanomaterials for Electrochemical Energy Storage. Adv. Mater. 2020, 32, 1903826. [Google Scholar] [CrossRef]
- He, H.; Lu, P.; Wu, L.; Zhang, C.; Song, Y.; Guan, P.; Wang, S. Structural Properties and Phase Transition of Na Adsorption on Monolayer MoS2. Nanoscale Res. Lett. 2016, 11, 330. [Google Scholar] [CrossRef]
- Wang, L.; Shih, E.-M.; Ghiotto, A.; Xian, L.; Rhodes, D.A.; Tan, C.; Claassen, M.; Kennes, D.M.; Bai, Y.; Kim, B.; et al. Correlated Electronic Phases in Twisted Bilayer Transition Metal Dichalcogenides. Nat. Mater. 2020, 19, 861–866. [Google Scholar] [CrossRef]
- David, L.; Bhandavat, R.; Barrera, U.; Singh, G. Polymer-Derived Ceramic Functionalized MoS2 Composite Paper as a Stable Lithium-Ion Battery Electrode. Sci. Rep. 2015, 5, 9792. [Google Scholar] [CrossRef]
- Liu, T.; Jin, Z.; Liu, D.-X.; Du, C.; Wang, L.; Lin, H.; Li, Y. A Density Functional Theory Study of High-Performance Pre-Lithiated MS2 (M = Mo, W, V) Monolayers as the Anode Material of Lithium Ion Batteries. Sci. Rep. 2020, 10, 6897. [Google Scholar] [CrossRef]
- Mikhaleva, N.S.; Visotin, M.A.; Kuzubov, A.A.; Popov, Z.I. VS2/Graphene Heterostructures as Promising Anode Material for Li-Ion Batteries. J. Phys. Chem. C 2017, 121, 24179–24184. [Google Scholar] [CrossRef]
- Yang, F.; Feng, X.; Glans, P.-A.; Guo, J. MoS2 for beyond Lithium-Ion Batteries. APL Mater. 2021, 9, 050903. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Liu, L.; Min, C.; Liang, S.; Xu, Z.; Xue, Y.; Hong, C.; Cai, Z. Construction of MoS2/Mxene Heterostructure on Stress-Modulated Kapok Fiber for High-Rate Sodium-Ion Batteries. J. Colloid Interface Sci. 2022, 605, 472–482. [Google Scholar] [CrossRef]
- Kulka, A.; Hanc, A.; Walczak, K.; Płotek, J.; Sun, J.; Lu, L.; Borca, C.; Huthwelker, T. Direct Evidence of an Unanticipated Crystalline Phase Responsible for the High Performance of Few-Layered-MoS2 Anodes for Na-Ion Batteries. Energy Storage Mater. 2022, 48, 314–324. [Google Scholar] [CrossRef]
- Barik, G.; Pal, S. 2D Square Octagonal Molybdenum Disulfide: An Effective Anode Material for LIB/SIB Applications. Adv. Theory Simul. 2020, 3, 2000157. [Google Scholar] [CrossRef]
- Gavryushkin, P.; Sagatov, N.; Sukhanova, E.; Medrish, I.; Popov, Z. Janus Structures of SMoSe and SVSe Compositions with Low Enthalpy and Unusual Crystal Chemistry. J. Appl. Cryst. 2022, 55, 1324–1335. [Google Scholar] [CrossRef]
- Zhang, J.; Xia, Y.; Wang, B.; Jin, Y.; Tian, H.; Ho, W.k.; Xu, H.; Jin, C.; Xie, M. Single-Layer Mo5Te8—A New Polymorph of Layered Transition-Metal Chalcogenide. 2D Mater. 2020, 8, 015006. [Google Scholar] [CrossRef]
- Wang, X.; Guan, X.; Ren, X.; Liu, T.; Huang, W.; Cao, J.; Jin, C. Deriving 2D M2X3 (M = Mo, W, X = S, Se) by Periodic Assembly of Chalcogen Vacancy Lines in Their MX2 Counterparts. Nanoscale 2020, 12, 8285–8293. [Google Scholar] [CrossRef]
- Chepkasov, I.V.; Sukhanova, E.V.; Kvashnin, A.G.; Zakaryan, H.A.; Aghamalyan, M.A.; Mamasakhlisov, Y.S.; Manakhov, A.M.; Popov, Z.I.; Kvashnin, D.G. Computational Design of Gas Sensors Based on V3S4 Monolayer. Nanomaterials 2022, 12, 774. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, E.V.; Kvashnin, A.G.; Agamalyan, M.A.; Zakaryan, H.A.; Popov, Z.I. Map of Two-Dimensional Tungsten Chalcogenide Compounds (W–S, W–Se, W–Te) Based on USPEX Evolutionary Search. JETP Lett. 2022, 115, 292–296. [Google Scholar] [CrossRef]
- Sukhanova, E.; Kvashnin, A.; Bereznikova, L.; Zakaryan, H.; Aghamalyan, M.; Kvashnin, D.; Popov, Z. 2D-Mo3S4 Phase as Promising Contact for MoS2. Appl. Surf. Sci. 2022, 589, 152971. [Google Scholar] [CrossRef]
- Kung, C.-W.; Han, P.-C.; Chuang, C.-H.; Wu, K.C.-W. Electronically Conductive Metal–Organic Framework-Based Materials. APL Mater. 2019, 7, 110902. [Google Scholar] [CrossRef]
- Zou, G.; Hou, H.; Ge, P.; Huang, Z.; Zhao, G.; Yin, D.; Ji, X. Metal-Organic Framework-Derived Materials for Sodium Energy Storage. Small 2018, 14, 1702648. [Google Scholar] [CrossRef]
- Dang, S.; Zhu, Q.-L.; Xu, Q. Nanomaterials Derived from Metal–Organic Frameworks. Nat. Rev. Mater. 2018, 3, 17075. [Google Scholar] [CrossRef]
- Li, X.; He, C.; Zheng, J.; Ye, W.; Yin, W.; Tang, B.; Rui, Y. Preparation of Promising Anode Materials with Sn-MOF as Precursors for Superior Lithium and Sodium Storage. J. Alloy. Compd. 2020, 842, 155605. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Hafner, J. Ab-Initio Simulations of Materials Using VASP: Density-Functional Theory and Beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J.; Hafner, J. Theory of the Crystal Structures of Selenium and Tellurium: The Effect of Generalized-Gradient Corrections to the Local-Density Approximation. Phys. Rev. B 1994, 50, 13181–13185. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First Principles Phonon Calculations in Materials Science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Eriksson, F.; Fransson, E.; Erhart, P. The Hiphive Package for the Extraction of High-Order Force Constants by Machine Learning. Adv. Theory Simul. 2019, 2, 1800184. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Kumar, A.; Ahluwalia, P.K. Electronic Structure of Transition Metal Dichalcogenides Monolayers 1H-MX2 (M = Mo, W; X = S, Se, Te) from Ab-Initio Theory: New Direct Band Gap Semiconductors. Eur. Phys. J. B 2012, 85, 186. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Lin, G.; Ju, Q.; Guo, X.; Zhao, W.; Adimi, S.; Ye, J.; Bi, Q.; Wang, J.; Yang, M.; Huang, F. Intrinsic Electron Localization of Metastable MoS2 Boosts Electrocatalytic Nitrogen Reduction to Ammonia. Adv. Mater. 2021, 33, 2007509. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.-L.; Araujo, C.M.; Luo, W.; Ahuja, R. Single-Layer MoS2 as an Efficient Photocatalyst. Catal. Sci. Technol. 2013, 3, 2214–2220. [Google Scholar] [CrossRef]
- Barik, G.; Pal, S. Defect Induced Performance Enhancement of Monolayer MoS2 for Li- and Na-Ion Batteries. J. Phys. Chem. C 2019, 123, 21852–21865. [Google Scholar] [CrossRef]
- Su, J.; Pei, Y.; Yang, Z.; Wang, X. Ab Initio Study of Graphene-like Monolayer Molybdenum Disulfide as a Promising Anode Material for Rechargeable Sodium Ion Batteries. RSC Adv. 2014, 4, 43183–43188. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Position | Relative Energy (eV) | |||
|---|---|---|---|---|
| Li | Na | Li | Na | |
| P1 | −1.22 | −1.18 | 0.00 | 0.00 |
| P2 | −1.00 | −0.87 | 0.25 | 0.35 |
| P3 | −1.01 | −0.93 | 0.21 | 0.25 |
| P4 | −0.91 | −0.80 | 0.31 | 0.39 |
| P5 | −0.99 | −0.93 | 0.24 | 0.26 |
| P6 | −1.04 | −0.86 | 0.19 | 0.34 |
| P7 | −0.79 | −0.16 | 0.43 | 0.48 |
| P8 | −1.03 | −0.43 | 0.19 | 0.71 |
| Transition Path | Li | Na | ||||
|---|---|---|---|---|---|---|
| Direct | Reverse | Direct | Reverse | |||
| On the surface | A | P2→P5 | 0.16 | 0.16 | 0.06 | 0.13 |
| P5→P3 | 0.11 | 0.13 | 0.06 | 0.05 | ||
| A’ | P6→P4 | 0.19 | 0.07 | 0.08 | 0.02 | |
| P4→P7 | 0.21 | 0.09 | 0.14 | 0.04 | ||
| B | P4→P1 | 0.14 | 0.46 | 0.11 | 0.50 | |
| P1→P5 | 0.40 | 0.17 | 0.35 | 0.10 | ||
| P5→P2 | 0.16 | 0.16 | 0.13 | 0.06 | ||
| P2→P6 | 0.21 | 0.26 | 0.16 | 0.15 | ||
| P6→P4 | 0.19 | 0.07 | 0.08 | 0.02 | ||
| Through the slab | C | P3→P8 | 0.22 | 0.38 | 1.05 | 0.55 |
| P8→P1 | 0.22 | 0.28 | 0.07 | 0.82 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukhanova, E.V.; Bereznikova, L.A.; Manakhov, A.M.; Al Qahtani, H.S.; Popov, Z.I. A Novel Membrane-like 2D A’-MoS2 as Anode for Lithium- and Sodium-Ion Batteries. Membranes 2022, 12, 1156. https://doi.org/10.3390/membranes12111156
Sukhanova EV, Bereznikova LA, Manakhov AM, Al Qahtani HS, Popov ZI. A Novel Membrane-like 2D A’-MoS2 as Anode for Lithium- and Sodium-Ion Batteries. Membranes. 2022; 12(11):1156. https://doi.org/10.3390/membranes12111156
Chicago/Turabian StyleSukhanova, Ekaterina V., Liudmila A. Bereznikova, Anton M. Manakhov, Hassan S. Al Qahtani, and Zakhar I. Popov. 2022. "A Novel Membrane-like 2D A’-MoS2 as Anode for Lithium- and Sodium-Ion Batteries" Membranes 12, no. 11: 1156. https://doi.org/10.3390/membranes12111156
APA StyleSukhanova, E. V., Bereznikova, L. A., Manakhov, A. M., Al Qahtani, H. S., & Popov, Z. I. (2022). A Novel Membrane-like 2D A’-MoS2 as Anode for Lithium- and Sodium-Ion Batteries. Membranes, 12(11), 1156. https://doi.org/10.3390/membranes12111156