1. Introduction
Human epidermal growth factor receptor-2 (HER2) is overexpressed in a number of different cancer types, including carcinomas of the bladder, ovary, endometrium, pancreas, colon, stomach, gallbladder, esophagus, and breast [
1]. HER2 overexpression occurs in 20% to 30% of invasive breast carcinomas and correlates with an aggressive disease phenotype and a poor prognosis [
2]. To date, a significant improvement in overall survival in early-stage and metastatic HER2+ breast cancer patients has been achieved due to passive immunization therapy using Trastuzumab, a humanized monoclonal antibody directed against the extracellular domain of HER2, in addition to chemotherapy [
3,
4,
5]. However, despite the benefits displayed by the combined immune–chemotherapy, long-term follow-up data showed that 15% to 24% of the patients experienced tumor recurrence and disease progression, mainly due to the development of resistance to Trastuzumab [
6,
7]. Hence, there is an urgent need for alternative HER2-targeted immune–therapies that could promote robust and persistent antitumor immune responses without the emergence of resistance. Recent anti-HER2 vaccines focused on triggering a patient’s own immune system to induce tumor-specific antibodies (Abs) have shown promising outcomes [
8,
9,
10]. Among the different vaccine strategies, the use of virus-like particles (VLPs) has shown superiority over monomeric protein antigens or DNA vaccines [
11,
12,
13,
14]. VLPs’ advantageous features include higher immunogenicity due to their virus-resembling surface, straightforward antigen delivery, and more effective stimulation of B- and CD4+ T-cell responses due to the high avidity of epitopes on their surface [
15,
16,
17]. Moreover, internalization by antigen-presenting cells has been shown to activate CD8+ T-cells to elicit a specific cytotoxic T-lymphocyte response [
18].
Recently, we developed a new VLP platform for complex antigen display based on the baculovirus–insect cell expression vector system (BEVS). The BEVS has been proven to be well suited to the expression of eukaryotic proteins and the production of versatile VLPs [
19,
20,
21]. Although baculovirus-derived VLPs have shown higher immunogenicity in comparison to mammalian cell-based VLPs in vivo, one of the limitations of BEVS lies in the inability to perform the mammalian-like glycosylation required for the production of therapeutic glycoproteins. To address this limitation, we applied SweetBac technology [
22] to generate mammalian-like
N-glycan structures on the antigen. Overall, our VLP technology allowed us to (I) provide HER2 in an authentic conformation and as a complete molecule [
23] and to (II) investigate the impact of different glycosylation patterns on the immunogenic effect of HER2-displaying VLPs.
In the present study, we hypothesized that chimeric antigen-displaying VLPs, produced in
Spodoptera frugiperda Sf9 insect cells, are suitable as cancer vaccines and that differences in glycosylation structures have an impact on antigenicity and antitumor activity in vivo. We generated HER2-displaying VLPs exhibiting insect cell
N-linked glycosylation composition [
23] and novel VLPs, presenting HER2 with a mammalian-like N-linked glycosylation pattern, and investigated immunogenicity and antitumor activity in conjunction with AddaVax (an MF59-like squalene-based oil-in-water nanoemulsion) and Poly (I:C) (polyinosinic–polycytidylic acid, a synthetic analog of double-stranded RNA) adjuvants. Overall, our study sheds a light on future challenges in developing active cancer vaccination strategies, including a determination of the glycosylation pattern on the antigen surface and an assessment of the optimal vaccine adjuvant to generate the most effective antitumor immune response.
2. Materials and Methods
2.1. Ethics Statement
The animal studies were approved by the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai Hospital (IACUC-2014-0234). All research staff were trained in animal care and handling. All efforts were made to minimize animal suffering.
2.2. Animals and Cells
Six to eight-week-old female BALB/c mice (albino in-bred mice) were used for the vaccination and tumor challenge studies and were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were maintained in microisolator cages, kept on a 12-h light and 12-h dark cycle, and had free access to food and water.
The
Sf9 insect cells (ATCC CRL-1711) [
24] used for the production of recombinant baculoviruses (rBVs) and VLPs were maintained in serum-free HyClone SFM4 Insect cell medium (GE Healthcare, Little Chalfont, UK) supplemented with 0.1% Kolliphor P188 (Sigma-Aldrich, St. Louis, MO, USA) at 27 °C. Suspension cultures were grown in 500-mL shaker flasks at 100 rpm.
An SK-BR-3 (ATCC HTB-30) human mammary gland cancer cell line expressing human HER2 was grown and propagated in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) containing a penicillin–streptomycin antibiotics mix (100 U/ml penicillin, 100 µg/mL streptomycin; Gibco) and 10% fetal bovine serum (PAN BioTech, P170203), resulting in complete DMEM (cDMEM).
The TUBO cells (mouse mammary tumor cells) used in our in vivo mammalian cancer syngeneic tumor model were a cloned cell line established in vitro from a lobular carcinoma that arose spontaneously in a BALB-neuT mouse [
25]. The TUBO cells were kindly provided by Dr. Federica Carvallo and Dr. Pier-Luigi Lollini (Department of Molecular Biotechnology and Health Science, University of Torino, Italy) and were maintained in cDMEM medium.
2.3. Generation of rBV
Here, rBVs encoding full-length human HER2 (GenBank accession No. X03363.1) and the HIV-1 Gag matrix protein (GenBank accession No. K03455.1) were generated using a MultiBac (EMBL, Grenoble, France) expression system, as previously described [
23]. In brief, the sequence coding for human HER2 was ligated into the MultiBac acceptor vector pACEBac-1 (EMBL, Grenoble, France), and the sequence coding for the HIV-1 matrix protein was ligated into the pIDC donor vector (EMBL, Grenoble, France). Acceptor–donor fusions were generated by Cre-LoxP recombination and were inserted into the MultiBac genome using competent DH10MultiBac cells according to Fitzgerald et al. [
26]. The rBVs were generated by transfecting
Sf9 cells with the bacmid DNA in conjunction with FuGENE HD transfection reagent (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The viruses were amplified, and viral titers were determined via a tissue culture infectious dose of 50% (TCID
50): they are given as plaque-forming units per milliliter (PFU/mL).
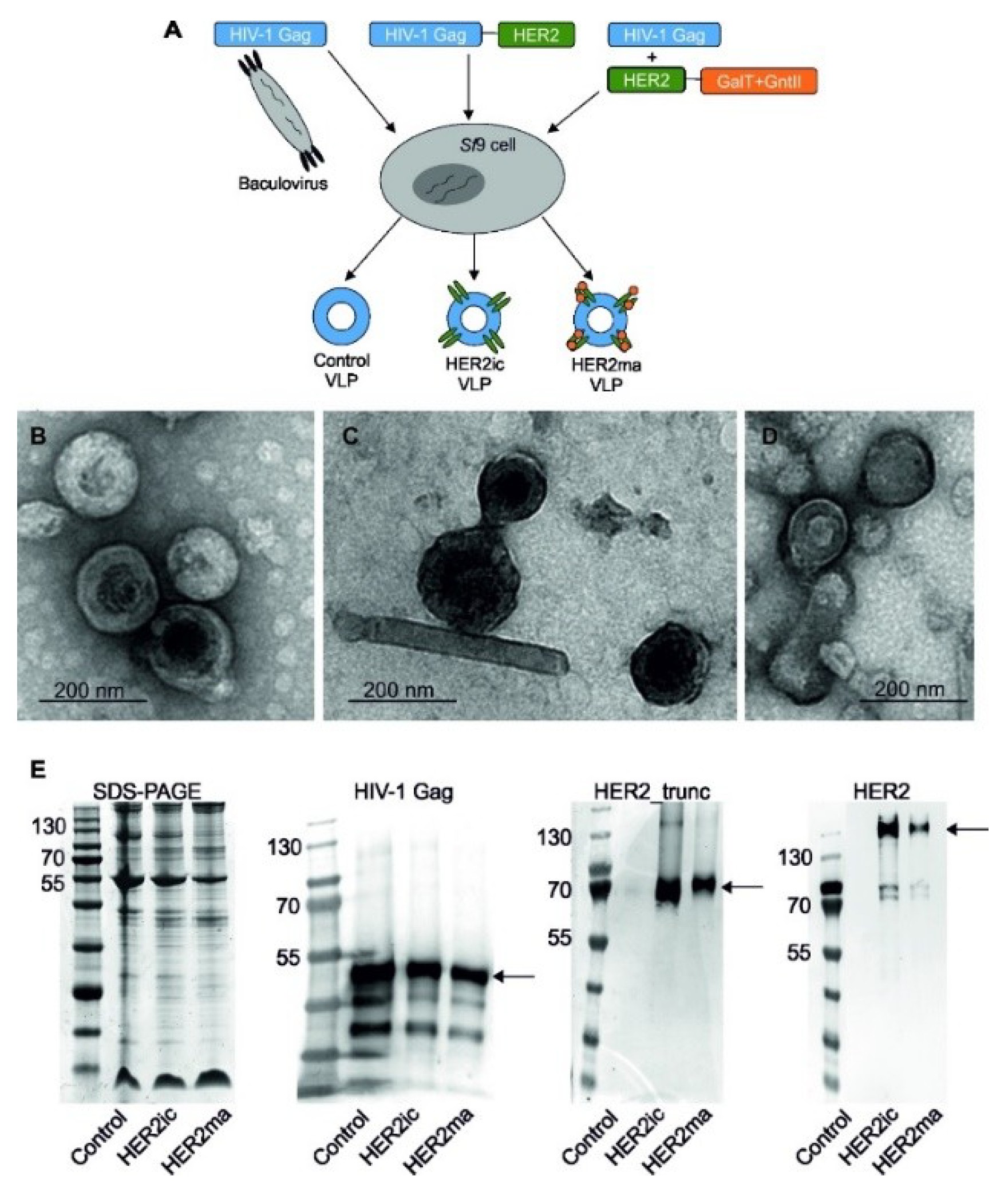
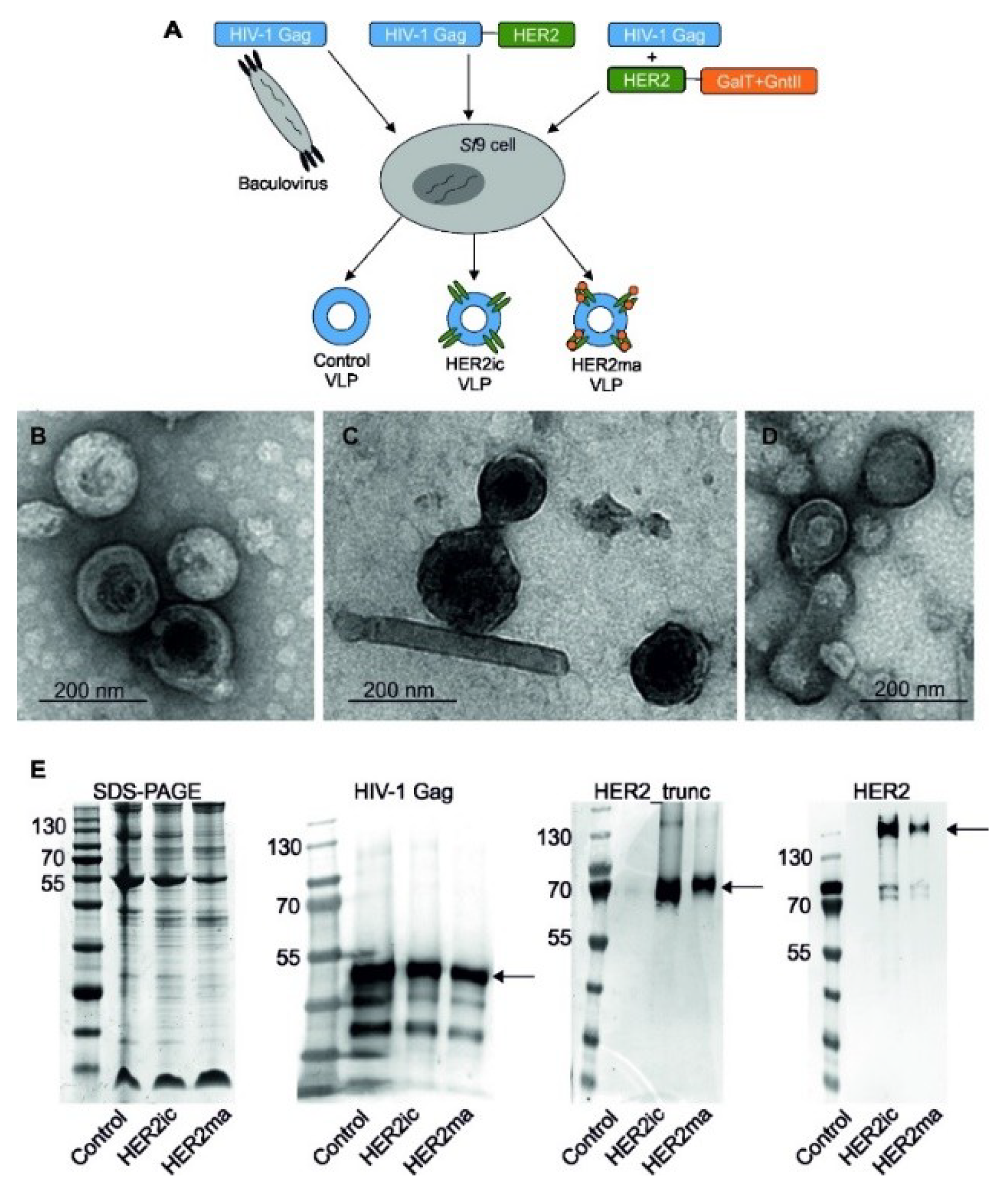
2.4. Generation and Purification of VLPs
The generation of VLPs, displaying full-length human HER2 with insect cell-specific glycosylation patterns (HER2ic) on the surface, was conducted as previously described [
23]. Briefly, rBVs coding for the HIV-1 Gag matrix protein and the full-length human HER2 were used to infect
Sf9 cells. After harvesting the cell supernatant, VLPs were purified via sucrose gradient ultracentrifugation. Different sucrose gradient fractions were aliquoted and investigated for their HIV-1 Gag and HER2 protein content via western blot analysis. Fractions containing the most target proteins were pooled and investigated by transmission electron microscopy (TEM) and enzyme-linked immunosorbent assay (ELISA) binding studies to confirm the presence of HER2 on the VLP surface in its native conformation.
Moreover, VLPs displaying HER2 with a modified, mammalianized N-linked glycosylation pattern on the surface (HER2ma) were produced. For this, a glycomodule consisting of open reading frames coding for the
Caenorhabditis elegans N-acetylglucosaminyltransferase II and the bovine β1,4 galactosyltransferase I (controlled by polyhedrin and p10 promoters, respectively) was introduced in the LoxP site of the MultiBac baculovirus genome, as previously described [
22]. The resulting backbone was subsequently used as a shuttle vector for the expression of full-length human HER2 integrated into the standard Tn7 site. The generation of VLPs was achieved through coinfection of
Sf9 cells with rBVs coding for HER2 and glycosyltransferases and rBVs coding for the Gag matrix protein from HIV-1. The parallel expression of target genes and glycosyltransferases modifies the glycan pattern of the proteins expressed in insect cells. VLPs solely comprising of the HIV-1 Gag matrix protein and the host cell surface (Control VLPs) were applied as controls in the following experiments.
2.5. Characterization and Quantification of VLPs
Sucrose-gradient purified VLP samples were mixed with a 4× NuPage LDS sample buffer (Invitrogen, Carlsbad, CA, USA) containing lithium dodecyl sulfate (pH 8.4) and were heated to 70 °C for 10 min. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and were electroblotted onto a polyvinylidene difluoride (PVDF) transfer membrane (GE healthcare Life Sciences, Vienna, Austria). Membranes were probed with primary antihuman HIV-1 Gag rabbit antibody (E63H00201, Enogene, Atlanta, GA, USA), followed by incubation with a secondary antibody (antirabbit IgG alkaline phosphatase-labeled goat antibody; A9919, Sigma-Aldrich, St. Louis, MO, USA). The detection of truncated human HER2 was conducted using Trastuzumab (BioVision, A1046-100) and a secondary antihuman IgG alkaline phosphatase-labeled goat antibody (abcam, ab6858). Full-length human HER2 was detected using anti-HER2 mAb (3B5; Thermo Fisher, Waltham, MA, USA, MA5-13675) and anti-mouse IgG alkaline phosphatase-labeled antibody (Sigma, A3438). The morphology of negatively stained VLPs was determined using TEM (FEI Tecnai G2 200 kV, FEI, Hillsboro, OR, USA) at various magnifications. The VLP concentration was determined by nanoparticle tracking analysis (NTA) using a NanoSight LM-10 (Malvern Instruments Ltd., Worcestershire, UK) [
27]. The particle number is given as total particles per mL. The VLPs’ mean diameter and the homogeneity were analyzed by dynamic light scattering (DLS) using a Zetasizer Nano-ZS with software 7.03 (Malvern Instruments, Malvern, UK) at 25 °C. For this, sucrose-gradient purified VLP suspensions were diluted 1:100 in particle-free phosphate buffered saline (PBS) and were measured in five replicates. The size distribution was calculated by the intensity report. The homogeneity of the VLP samples was determined by the polydispersity index (PDI). The total protein concentration of the VLP samples was quantified using Quickstart Bradford Dye Reagent (Bio-Rad, 5000201) using a bovine serum albumin standard curve. To define the HER2 protein content in the VLP samples, a quantification ELISA was performed. Therefore, 96-well plates (Maxisorp; Nunc Thermo Scientific, Waltham, MA, USA) were coated with VLPs and different concentrations of recombinant human HER2 protein overnight at 4 °C. Wells were blocked using PBS containing 0.1% Tween20 (PBS-T) and 0.3% BSA (bovine serum albumin) at room temperature for 1 h. HER2 molecules were stained with 5 µg/mL of Trastuzumab (BioVision, Milpitas, CA, USA, A1046-100) for 1 h at room temperature. Detection was accomplished using an antihuman IgG-horseradish peroxidase (HRP)-conjugated antibody (628420, Invitrogen, Carlsbad, CA, USA), 1:2000 diluted. After washing six times with PBS-T, wells were developed using a 100-µL/well HRP substrate, 3,30,50-tetramethylbenzidine (TMB Stabilized chromogen, Invitrogen, Carlsbad, CA, USA). The reaction was stopped with 100 µL of 2 N H
2SO
4, and optical density was measured at 450 nm using an Infinite M1000 plate reader (Tecan Group, Maennedorf, Switzerland). Raw data were analyzed in Microsoft Excel spreadsheets (Version 2003, Microsoft, Redmond, WA, USA). A standard curve for HER2 at known concentrations was established. The relative HER2 protein concentration of the VLP samples was determined using the linear relationship between the absorbance and the HER2 standard.
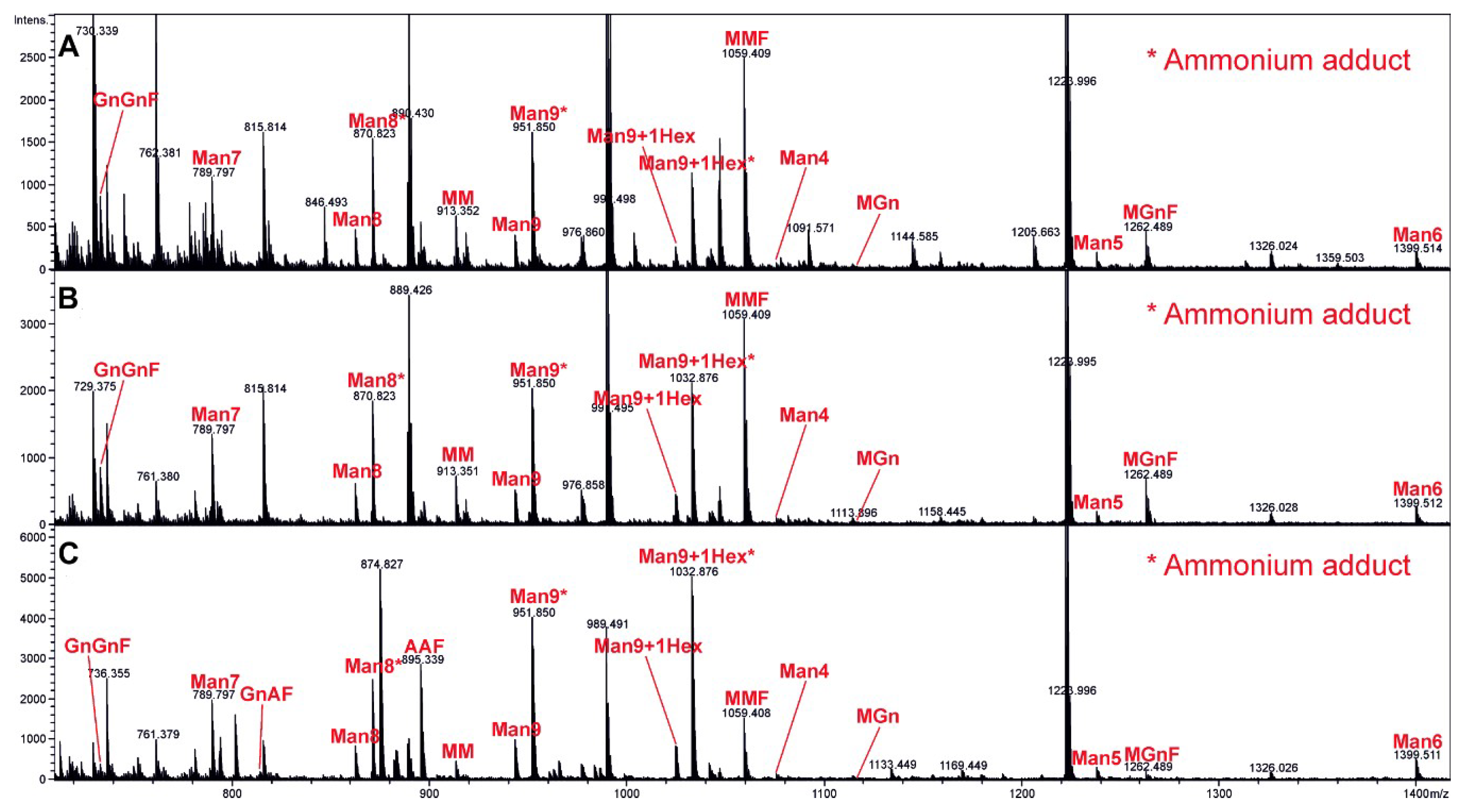
2.6. Mass Spectrometry Analysis of Complete Glycan Pattern on VLP Surface
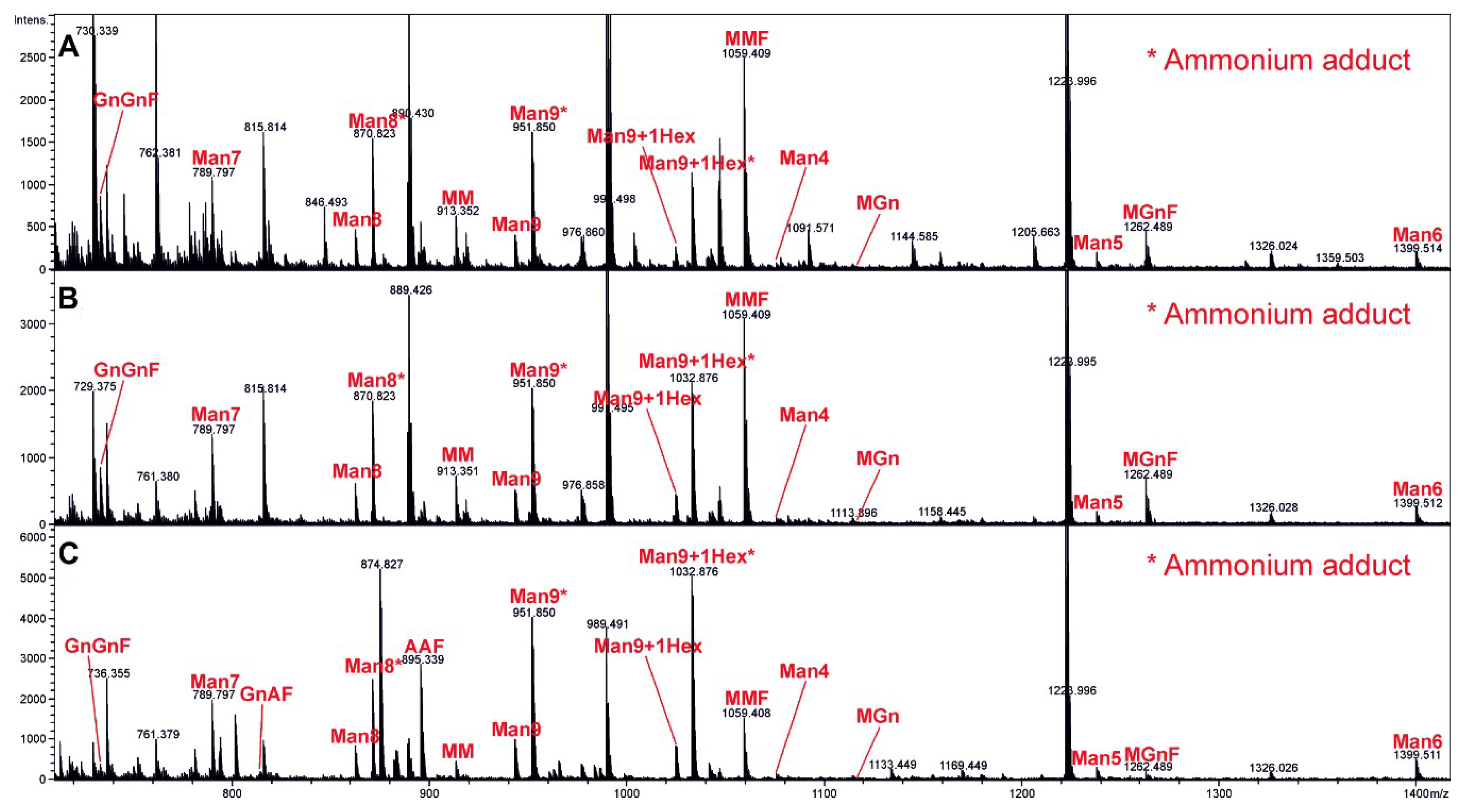
Each VLP sample (Control, HER2ic, HER2ma) was transferred into a fresh 1.5-mL screw cap microtube, and cysteine bridges were reduced by the addition of 15 mM of dithiothreitol (Sigma Aldrich, CAS: 3483-12-3) in 100 mM of ammonium bicarbonate buffer and subsequent incubation for 45 min at 56 °C. Samples were then treated with 55 mM of iodoacetamide (Sigma, CAS: 144-48-9) in 100 mM of ammonium bicarbonate (Acros Organics, 393215500) and incubated for 30 min at room temperature in the dark. Proteins were precipitated with acetone (4-fold volume added, 20 min storage at −20 °C) and dried in a SpeedVac (Sorvall). The samples were dissolved in 30 µL of 100 mM ammonium bicarbonate and digested with trypsin (Promega, V511A 20 µg) at 37 °C overnight (enzyme to substrate ratio of 1:50). To destroy any residual protease activity, the samples were incubated at 95 °C for 10 min. One-hundred micro-litres of Peptide N-Glycosidase A (PNGase) A buffer (citric acid monohydrate 244, Merck Darmstadt, Germany, sodium dihydrogen phosphate, J.T. Baker, 303; pH 5) and 0.75 µL (=11.25 mU) of PNGase A (Proglycan) per sample were then added, and the samples were incubated at 37 °C overnight. The released glycans were dried in a SpeedVac (Sorvall) and reconstituted in 90 µL of 100-mM ammonium bicarbonate buffer. Ten micro-litres of 10% sodium borohydride solution (Sigma Aldrich, 452882) were added and incubated overnight at room temperature. The digested glycans were purified using HyperSep Hypercarb solid phase extraction (SPE) 10-mg cartridges (Thermo Scientific, 60302606). The elution of bound glycans was performed using 500-µL SPE of 60% acetonitrile. The samples were dried in a SpeedVac (Sorvall), redissolved in 20 µL of high-quality water, and subjected to liquid chromatography-electrospray ionization mass spectrometry (LC-ESI-MS). The digested samples were loaded on a porous graphitic carbon column (100 × 0.32 mm, Thermo Scientific) using 80 mM of ammonium formiate buffer (Ammonia solution, VWR, CAS: 1336-21-6; Formic acid, Acros Organics, CAS: 64-18-6) as aqueous solvent and acetonitrile (VWR, CAS: 75-05-8) as solvent B. A gradient from 2% to 42% solvent B (98%–58% solvent A) was developed over 20 min at a flow rate of 8 μL/min using an Ultimate 3000 capillary flow LC (Dionex, Thermo Fisher, Waltham, MA, USA). With this steep gradient, all glycans eluted within a time window of a few minutes, which facilitated depiction of the whole glycan profile. Detection was performed using quadrupole time of flight (QTOF) MS (Bruker maXis 4G) equipped with a standard ESI source in positive ion data dependent acquisition (DDA) mode (=switching to tandem mass spectrometry mode for eluting peaks). MS-scans were recorded in a range from 150 to 2200 Da. Data interpretation and quantification was performed with DataAnalysis 4.0 (Bruker, Billerica, MA, USA) and QuantAnalysis 2.0 (Bruker). The extracted ion chromatograms of the first four isotopic mass peaks of every detected glycoform were integrated. Glycoforms were quantified by comparing peak areas. As shown by Grünwald-Gruber et al. [
28], signal responses of detected glycan variants are in a similar range, and thus comparing peak areas is valid.
2.7. Immunization and In Vivo Tumor Challenge
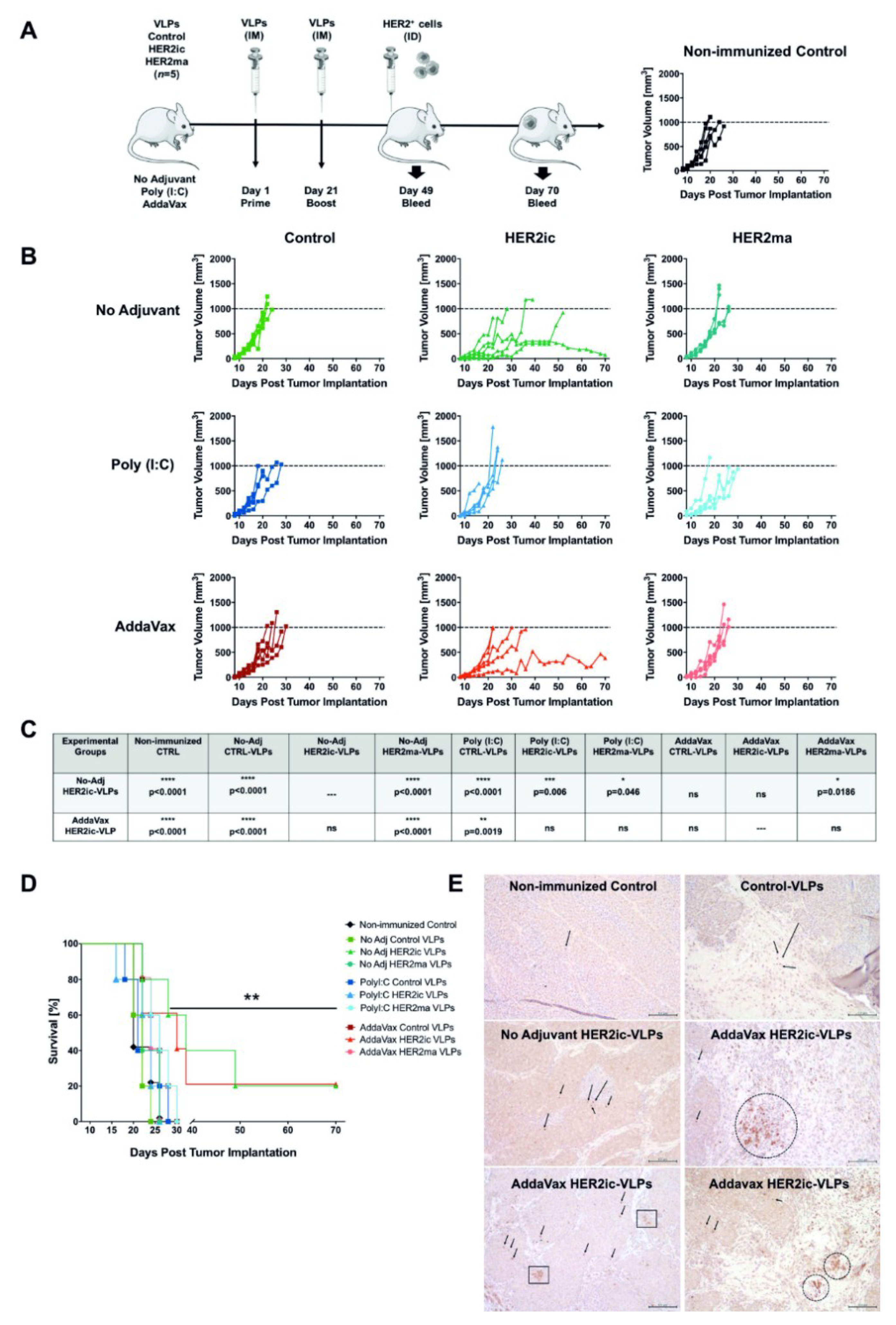
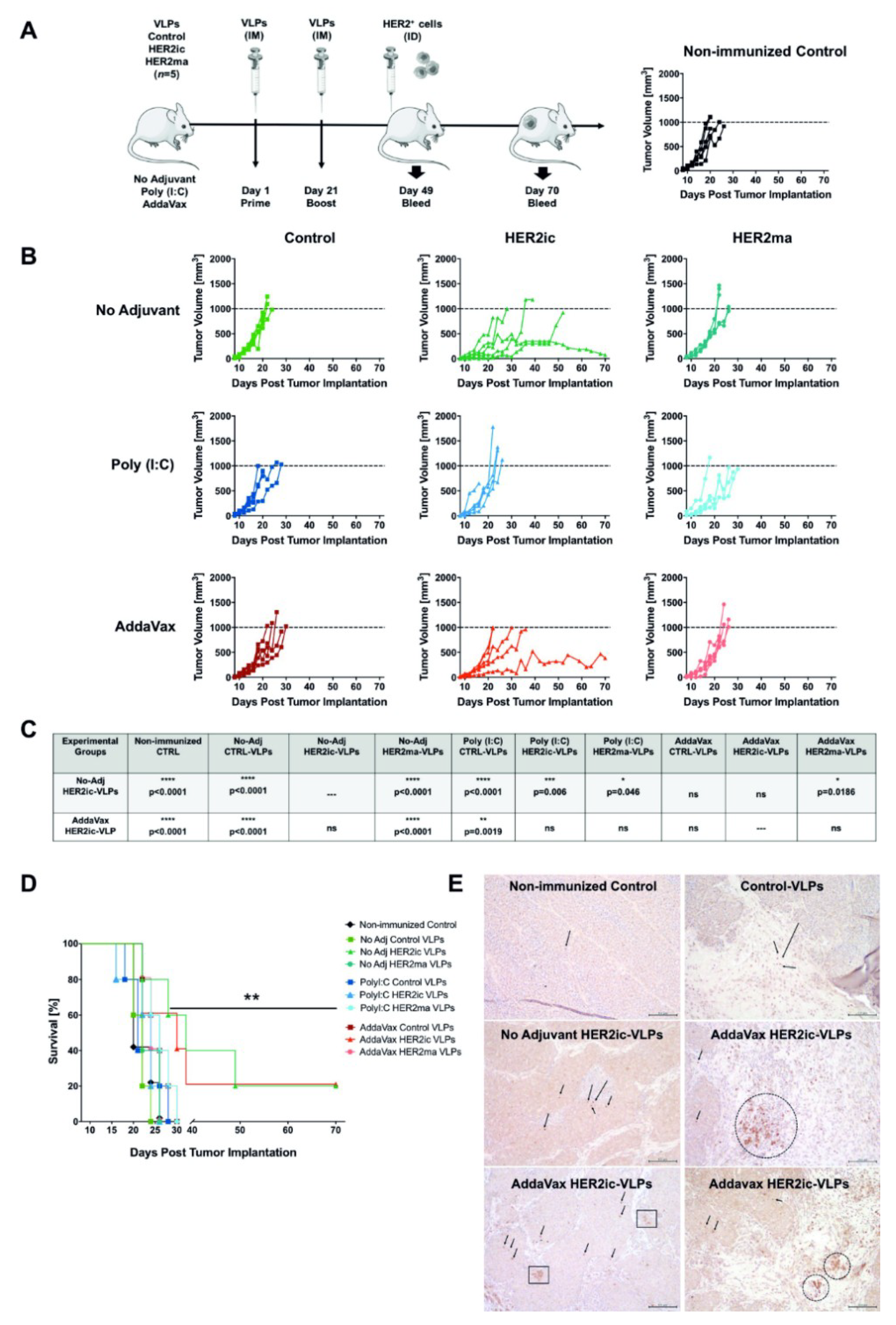
For the immunization studies, 6–8-week-old female BALB/c mice (n = 5 mice/group) were vaccinated intramuscularly (IM) with 5 µg total protein of Control VLPs, HER2ic VLPs, and HER2ma VLPs in a prime-boost regimen at day 1 and day 21. For each immunization, vaccines were adjuvant with 5 µg Poly (I:C) (InvivoGen, tlrl-pic) or 25 µL AddaVax (InvivoGen, vac-adx-10) per dose or were left nonadjuvant, resulting in nine different vaccination groups. Four weeks after the boost vaccination, 2.5 × 105 TUBO cells were intradermally (ID) implanted into the flank of the right posterior leg of each mouse and under light anesthesia. Starting from the day when tumors were first palpable, tumor growth was monitored every 48 h for the extent of the study. Tumor measurement was determined using a digital caliper, and total volume was calculated using the formula tumor volume (V) = L × W × W/2, where L, or tumor length, is the larger diameter, and W, or tumor width, is the smallest. Animals that bore a tumor larger than 1000 mm3 or displayed an open necrotic lesion were humanely euthanized by inhalation of 2% CO2. Growth curves were plotted for each vaccination group. Blood was collected the day before tumor implantation (4 weeks after the booster vaccination) and 3 weeks after the tumor challenge by submandibular bleeding. Blood samples were centrifuged for 10 min at 2000× g in a microcentrifuge, and the serum was stored at −20 °C and was used for ELISA, an antibody-dependent cellular cytotoxicity (ADCC) reporter assay, and IgG subtype determination.
2.8. Histological Analysis of Tumor Biopsies
Representatives of each experimental group were humanely euthanized once the tumors reached the experimental end point of 1000 mm3. Long-term survivors were euthanized at day 70 post-tumor implantation. Excision biopsies were preserved by formalin fixation and paraffin embedding for immunohistochemistry analysis (IHC). A histological evaluation was carried out at the Comparative Pathology Laboratory, Center of Comparative Medicine and Surgery at Icahn School of Medicine at Mount Sinai, New York (US). IHC staining of CD8+ lymphocytes was performed on 5-µm-thick tumor sections. A polyclonal, mouse-specific anti-CD8 antibody (Abcam, Cambridge, UK; ab203035) was used following the manufacturer’s instructions. The final counterstaining was performed using hematoxylin.
2.9. Serum Antibody Responses
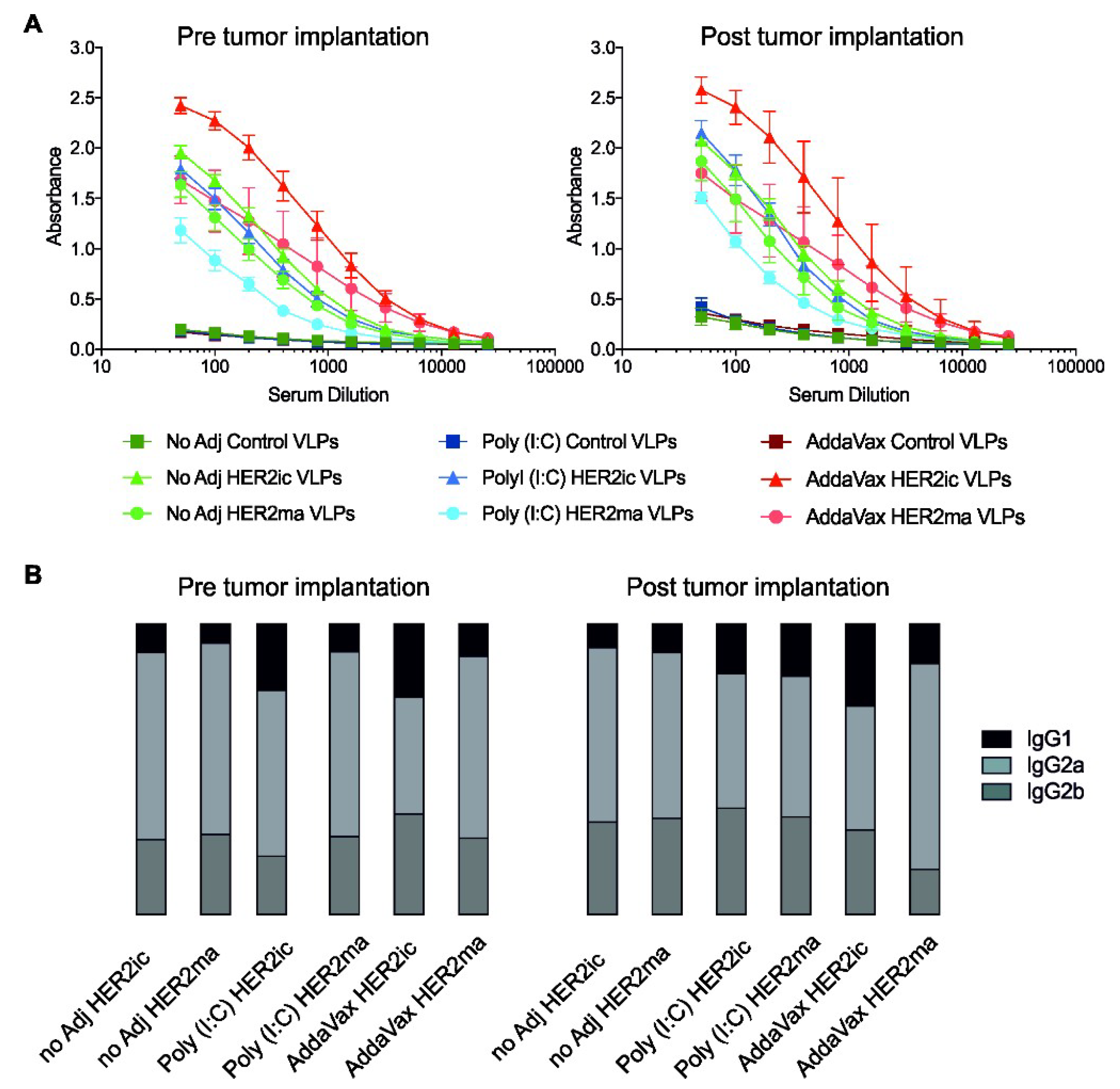
To measure the serum antibody responses of vaccinated mice, ELISAs were conducted. Ninety-six-well flat-bottomed immunoplates (Thermo Fisher) were coated with 50 µL of recombinant human HER2 protein (expressed in High Five insect cells) at a concentration of 2 µg/mL in phosphate buffered saline (PBS, Gibco) at 4 °C overnight. The following day, 220 µL of blocking solution, PBS supplemented with 0.1% Tween-20 (PBS-T, Fisher Scientific), and 0.3% low-fat milk powder (American-Bio, Canton, MA, USA) were added to all wells of the microtiter plate and were incubated for 1 h at room temperature. Serum Abs were serially diluted 1:2, starting with an initial dilution of 1:50 in the first well, and were incubated for 2 h at room temperature. After washing three times with PBS-T, 50 µL per well of antimouse IgG (whole molecule)-HRP conjugated antibody (produced in rabbit, Sigma, #A9044) diluted 1:3000 in blocking buffer was added and incubated for 1 h at room temperature. The plates were washed four times with PBS-T and were developed by adding 100 µL/well of SigmaFast o-phenylenediamine dihydrochloride (OPD, Sigma). The reaction was stopped using 50 µL/well of 3 M hydrochloric acid (Thermo Fischer), and plates were read at 490 nm using a microtiter plate reader (Bio-Tek, Winooski, VT, USA). Raw data were analyzed in Microsoft Excel (Version 2003) and GraphPad Prism 7 and were normalized to blank.
2.10. Determination of Induced Anti-HER2 IgG Subtypes
To investigate the IgG subtypes of the induced anti-HER2 antibodies in mouse serum, an ELISA was performed as described previously (see
Section 2.9). In brief, recombinant human HER2 (2 µg/mL in PBS) was coated on 96-well MaxiSorp plates (Thermo Fisher) overnight at 4 °C. Afterwards, the plates were blocked using PBS-T supplemented with 0.3% milk powder (AmericanBio) for 1 h at room temperature. Serum samples were two-fold serially diluted and incubated for 2 h at room temperature. After washing three times with PBS-T, anti-mIgG1-HRP (abcam, ab97240), anti-mIgG2a-HRP (abcam, ab97245), and anti-mIgG2b-HRP (abcam, ab97250) antibodies (1 mg/mL) were diluted 1:3000 in blocking buffer and were added for 1 h at room temperature. Plates were washed four times and were developed using OPD substrate (Sigma). The enzymatic reaction was stopped by adding 100 µL of 3-M hydrochloric acid (Thermo Fischer), and optical density was measured at 490 nm using a plate reader (BioTek). Raw data were analyzed in Microsoft Excel (Version 2003) and GraphPad Prism 7. Results were normalized to blank and are plotted as IgG1:IgG2a:IgG2b ratios of total induced anti-HER2 Abs.
2.11. ADCC Reporter Assay
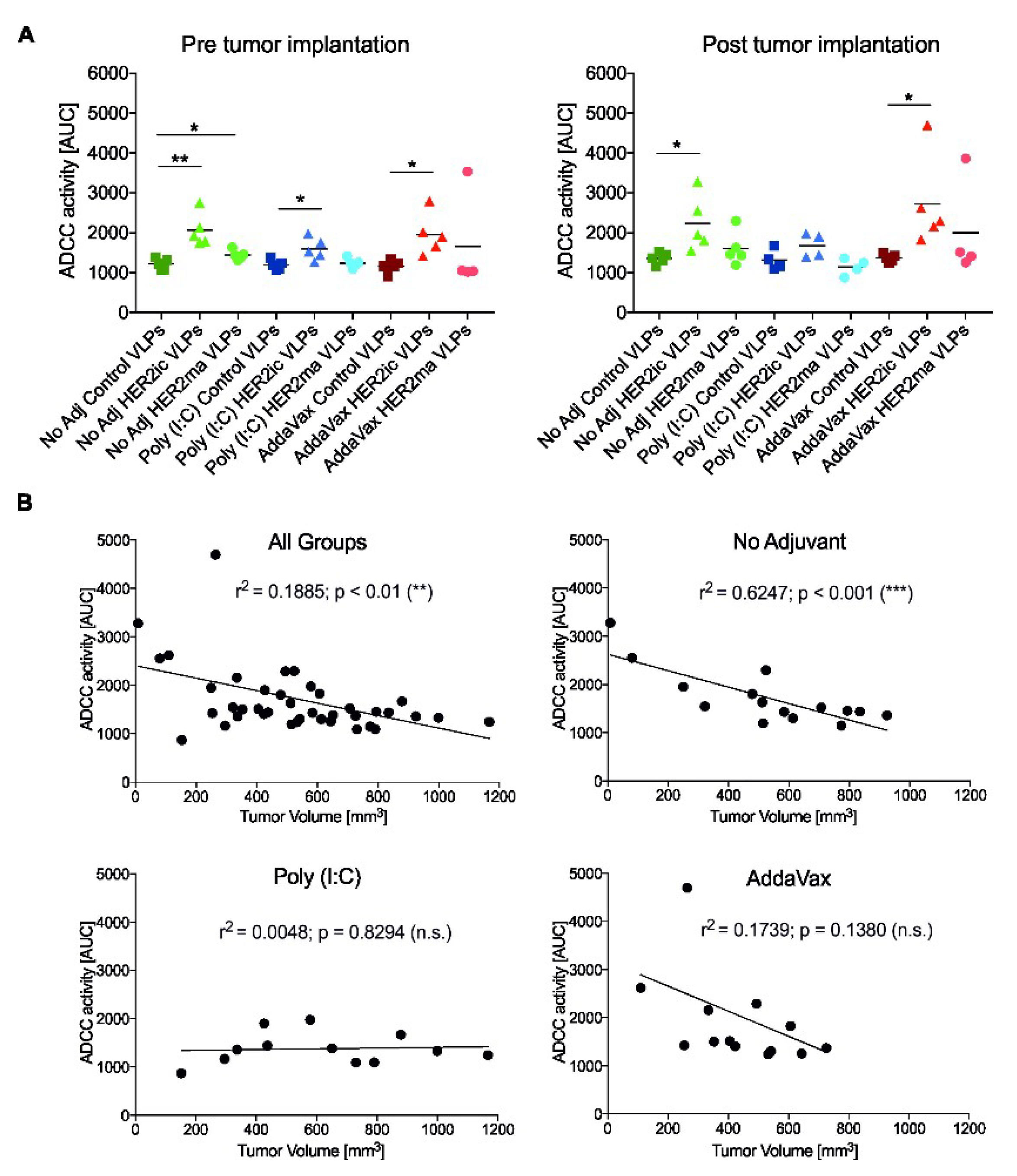
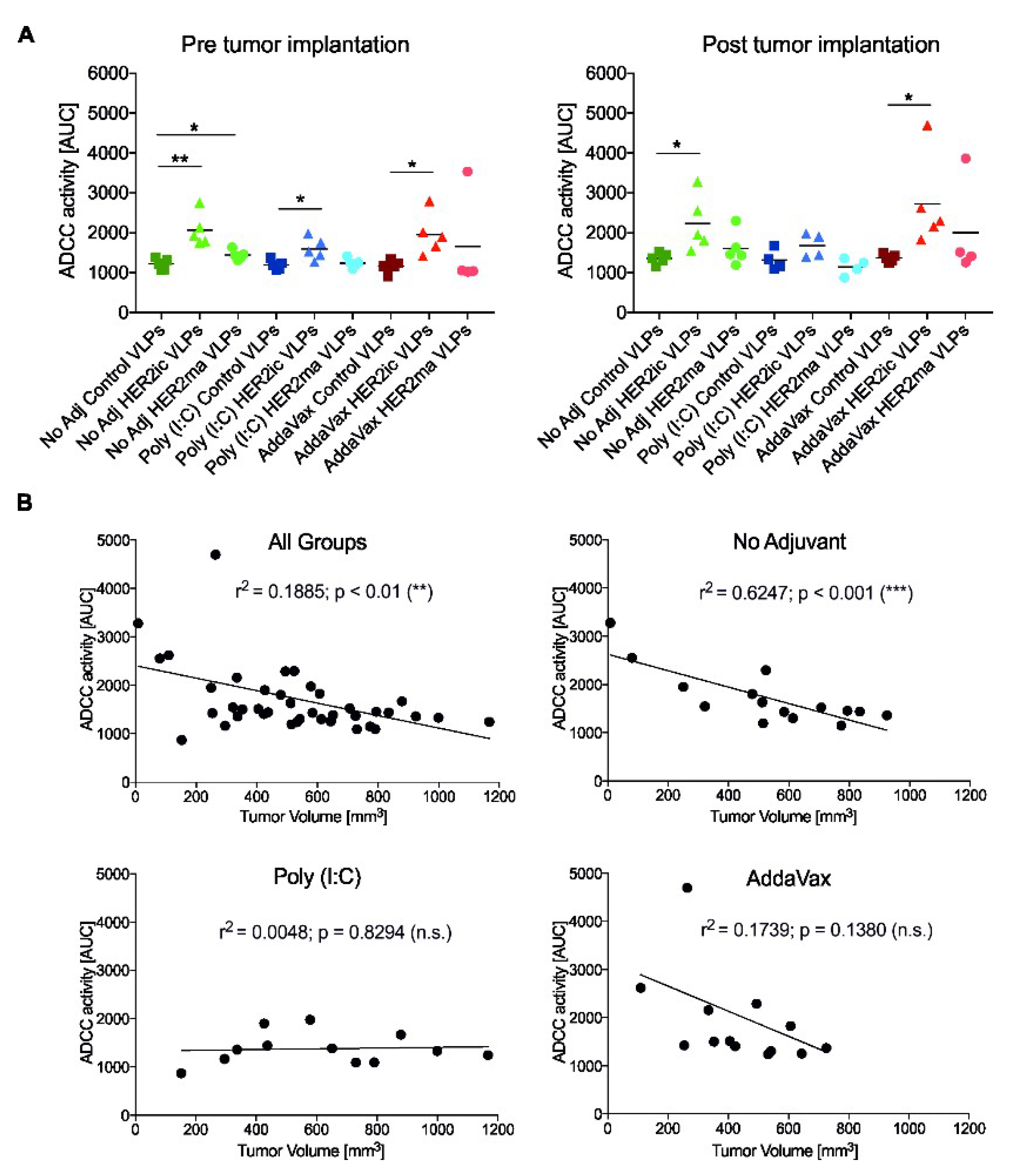
An ADCC reporter bioassay kit (BioGlo Luciferase Assay System, Promega, G720B, G7198) was applied to assess whether any of the anti-HER2 serum Abs elicited ADCC activity. The assay was carried out according to the manufacturer’s instructions. In brief, SK-BR-3 (ATCC HTB-30) cells (5 × 104/well) were seeded onto white 96-well cell culture plates (Corning Costar, 3917) and were incubated for 48 h at 37 °C, 5% CO2. Twofold serial dilutions of each mouse serum Ab sample were added to the cells, starting from an overall dilution of 1:50. Seventy-five-thousand effector cells, expressing the murine Fcγ receptor IV (mFcγRIV), were subsequently added to every 96-well containing the SK-BR-3 cells and the antibody dilutions. Cells were incubated at 37 °C for 6 h. Luciferase substrate was added, and luminescence was measured after 5 min incubation using a Synergy Hybrid Reader (BioTek). Raw data were processed using a Microsoft Excel spreadsheet (Version 2003) and GraphPad Prism 7 and are shown as the area under the curve (AUC). Significant differences between groups were analyzed using a standard t-test.
To assess whether there was a correlation between ADCC activity in vitro and tumor growth in vivo, the ADCC activity, as the AUC, was plotted over the tumor volume on the day the serum samples were taken. Correlations are indicated by the Pearson correlation coefficient (r2) and significance values.
2.12. Assessment of T-Cell Activation
2.12.1. Processing of Spleens
Mice were immunized as previously described (see
Section 2.7). Eighteen days after the boost vaccination, spleens were harvested from the mice and were pushed through a 70-µM strainer (Foxx Life Sciences, Salem, NH, USA). The splenocytes were then centrifuged at 1600 rpm for 5 min (HERAEUS Megafuge16, Thermo Scientific, Waltham, MA, USA), and red blood cells were lysed in ammonium-chloride-potassium (ACK) lysis buffer comprised of 0.15 M NH
4Cl (Sigma A-4514, MW = 53.49), 10 mM KHCO
3 (Sigma P-9144), and Na
2EDTA (Sigma ED2SS), pH 7.2. Splenocytes were then centrifuged at 1600 rpm for 5 min and were resuspended in complete α-medium (MEM α-modification; Sigma M4526, 10% FCS; Sigma F-2442, 100 U penicillin/100 µg streptomycin mix; Sigma P-0781, 4 mM
l-glutamine; Sigma G-7513, 50 µM 2-Mercaptoethanol; Gibco BRL 31350-010) at a cell concentration of 2.5 × 10
6 cells/mL.
2.12.2. Enzyme-Linked Immunospot (ELISpot) Assay
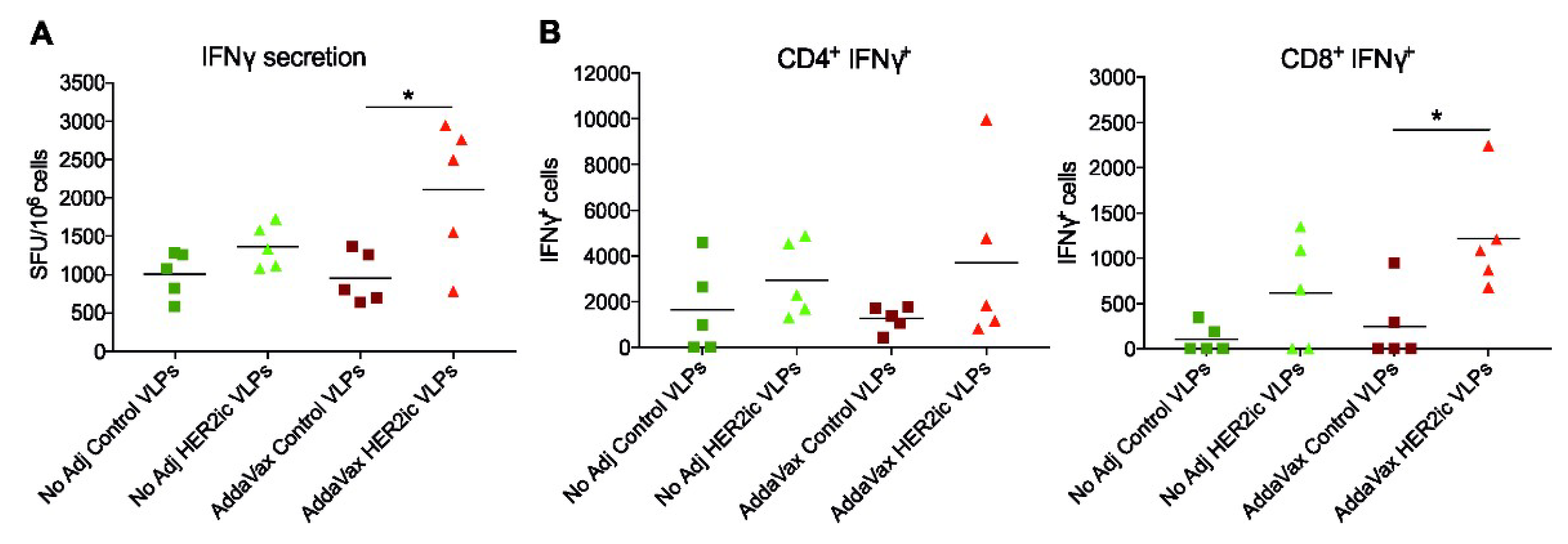
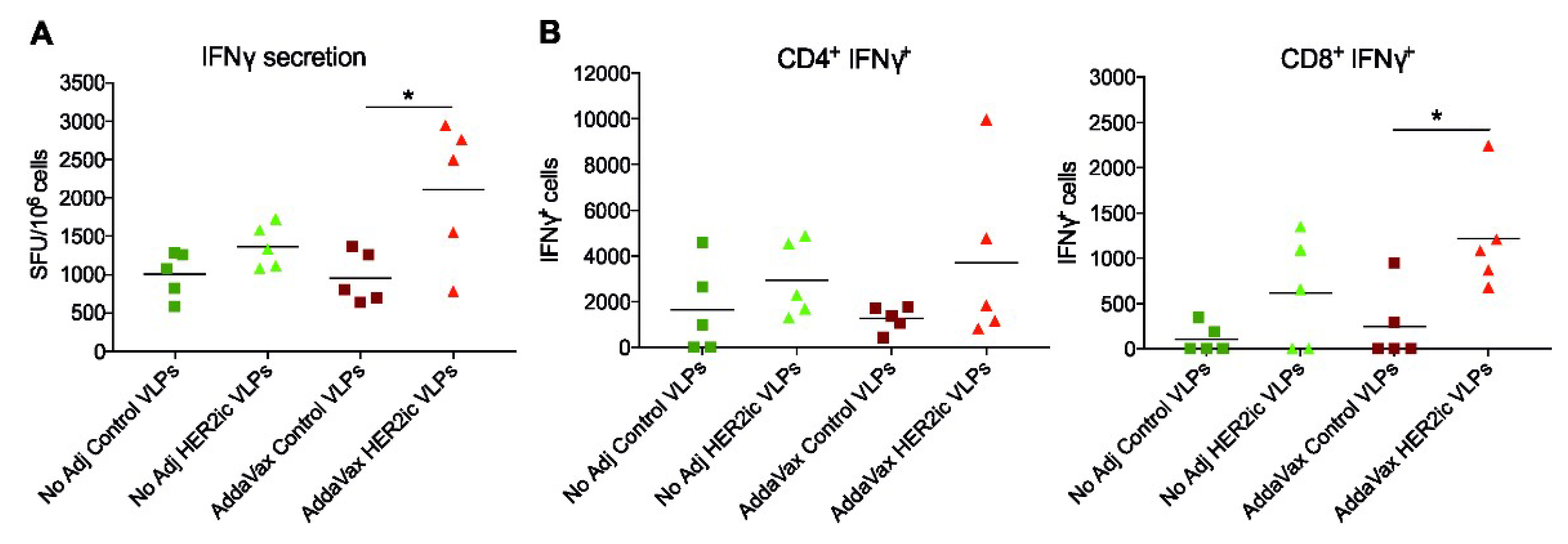
An interferon (IFN)γ ELISpot assay was performed according to the manufacturer’s instructions (MabTech, 3321-2A). Briefly, ELISpot plates (EMD Millipore, MAIPS4510) were coated with 15 µg/mL antimouse IFNγ capture antibody. The following day, splenocytes (2.5 × 106 cells/mL) were added to the plate and were incubated at 37 °C for 48 h in the presence or absence of 1 µg/mL full-length recombinant human HER2 protein. After 48 h, the plates were washed and incubated with the biotinylated mouse anti-IFNγ monoclonal detection antibody (R4-6A2, 1mg/mL) diluted in PBS containing 0.5% FBS (PBS-0.5% FBS) for 2 h at room temperature. Following the secondary incubation, plates were stained with HRP-conjugated streptavidin (1 mg/mL in PBS-0.5% FBS) for 1 h at room temperature. Visualization of IFNγ-producing cells occurred by using a BioRad AP substrate kit (BioRad, 1706432) per the manufacturer’s instructions, and spots were counted in an ImmunoSpot Analyzer 5.0.3 Pro DC (Cellular Technology, LLC). Raw data were processed using Microsoft Excel (2003) and GraphPad Prism 7. Significant differences between groups were analyzed using a standard t-test.
2.12.3. Intracellular Cytokine Staining (ICS)
One-hundred micro-litres of cells (2.5 × 106 cells/mL) per sample were stimulated for 5 h at 37 °C using 1 µg/mL full-length recombinant human HER2 protein in the presence of Brefeldin A Solution (BioLegend, San Diego, CA, 420601). Unstimulated samples were included as controls. After stimulation, splenocytes were washed in fluorescence-activated cell sorting (FACS) staining buffer (PBS supplemented with 0.1% bovine serum albumin) and stained for anti-CD4 (Clone RM4.4, 116013) and anti-CD8 (Clone 53-6.7; BioLegend, 140414) T-cell cell surface markers. Antibodies were incubated with cells for 30 min at 4 °C and were subsequently washed in FACS buffer and fixed using fixation buffer (BioLegend, 420801) according to the manufacturer’s instructions. Following fixation, splenocytes were washed in permeabilization buffer (BioLegend, 421002) and stained for IFNγ (Clone XMG1.2, BioLegend, 505805) expression for 30 min at 4 °C. Afterwards, cells were sequentially washed with permeabilization buffer, resuspended in 200 µL FACS buffer, and immediately analyzed on a modified LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed using FlowJo v10 and were presented using GraphPad Prism 7.0.
2.13. Statistical Analysis
Data analysis was performed using Excel (Version 2003) and GraphPad Prism 7.0 software (GraphPad Software Inc., San Diego, CA, USA). The unpaired nonparametric t-test was used to assess differences between two groups. A p-value <0.05 was considered statistically significant (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). A two-way Anova–Tukey multiple comparisons test was used for the analysis of tumor growth over time. A survival curves analysis was performed using the long-rank (Mantel–Cox) test (** p = 0.0061). Linear correlations between ADCC activity and tumor volumes are given by the Pearson correlation coefficient (r2).
4. Discussion
Passive immunotherapy with anti-HER2 monoclonal antibodies (such as Trastuzumab) alongside chemotherapy is the standard of care for HER2-positive breast cancer patients. However, resistance to these treatments and a restricted duration of action due to the short half-life and elimination rate of the applied antibodies remain an issue to be overcome. Taking into account these limitations, active anti-HER2 vaccination strategies have become an attractive therapeutic approach. Such strategies could potentially trigger tumor-specific immune responses, induce long-lasting immunity against tumor antigens, and reduce severe side effects [
8,
9].
In the present study, we tested antigen-displaying enveloped VLPs expressed in
Sf9 insect cells as vaccine candidates. It has been shown that the composition of glycans attached to therapeutic proteins potentially influences the efficacy and immunogenicity of the drug [
32]. However, the impact of the glycosylation in the case of HER2 studies remains poorly explored. We generated chimeric HIV-1 Gag VLPs expressing full-length HER2 with insect cell-specific glycosylation on the surface [
23] as well as a variant displaying HER2 with mammalianized glycan structures by deploying SweetBac technology [
22]. To investigate the glycan composition of our VLP preparations, we applied LC-ESI-MS. The major glycoforms detected in the HER2ic VLPs were oligomannosidic and paucimannosidic structures. In the glycoengineered strain (HER2ma VLPs), a major part of the insect cell-type MMF was modified to the more complex-type mammalian structure AAF (G2F).
For our in vivo tumor challenge, we used the TUBO cell line [
25], a HER2+ cloned cell line established in vitro from a spontaneous mammary adenocarcinoma developed by a BALB/c-neuT mouse, which is transgenic for the rat
HER2-neu oncogene [
33]. TUBO-based in vivo syngeneic tumor models have been extensively used in the field of anti-HER2 DNA vaccine development, allowing HER2+ tumors to arise in the receptor animal without rejection and facilitating structural and functional anti-HER2 in vivo studies without off-target effects [
25,
33]. Although rat HER2 displays 95% of the amino acidic sequence homology of mouse HER2 (85% in the case of the human protein), and cross-reactivity of induced anti-HER2 antibodies was confirmed, this slight difference in amino acid composition between proteins constituted a limitation in our model and should be taken into consideration in order to evaluate the positive and future potential of our prophylactic strategy, especially when it pursues antigen-specific responses.
In order to achieve an adaptive, long-term, protecting antitumor response in vivo, our prophylactic strategy must be able to induce at first an effective innate immune response that could lead to an efficient tumor-specific antigen presentation. In that matter, the relatively high residual baculovirus contamination present in our VLP preparations could stimulate innate immune responses via the toll-like receptor 9 (TLR9), hence contributing to building up the antigen-specific antitumor response [
34].
When we compared the antitumor activity of HER2ic and HER2ma VLP immunizations in our in vivo model, higher protection was induced by insect cell glycosylated HER2ic VLPs compared to mammalian-like glycosylated HER2ma VLPs. HER2ic VLP vaccination was able to delay tumor emergence, control tumor growth, and extend survival significantly, with 20% of nonadjuvant and AddaVax-treated mice displaying stable disease. Strikingly, no protection was achieved with mammalian-like glycosylated VLP vaccinations. It has been previously described that expression of HER2 in
Drosophila S2 insect cells has the potential advantage of improving antigen uptake by antigen presenting cells (APCs) due to the addition of insect glycans. Specifically, the paucimannose insect glycan structure has been reported to induce improved uptake by dendritic cells and other APCs due to the expression of membrane-bound mannose receptors in these cells [
14,
35,
36,
37]. In addition, in our study, the presence of high levels of paucimannose insect cell glycans could be beneficial for antigen uptake by APCs and therefore enhance the antitumor effect elicited by HER2ic VLPs. In general, we clearly showed that there was a significant difference in the antitumor response induced by the VLP vaccines that displayed insect cell or mammalian-like glycosylated antigens. The Control VLPs did not show any antitumor effects.
We have also revealed that the use of Poly (I:C) as an adjuvant did not contribute to achieving an efficient antitumor response in our VLP-based prophylactic strategy. The synthetic double-stranded RNA analog is known to exert a strong inflammatory response by interacting with toll-like receptor 3 (TLR3). TLR3 is expressed in the membranes of B-cells, macrophages, and dendritic cells, all three of them with very well-established APC capacity. Although triggering TLR3 signaling induces the activation of multiple proinflammatory pathways (as NFκB and IRF3 signals) [
38], it is likely that in our system the baculovirus component in combination with Poly (I:C) strongly drove the innate response toward a favorable recognition of viral components rather than toward the tumor antigen, due to the synergetic effect of the activation of TLR9 and TLR3. Thus, the antigen-specific antitumor response mediated by HER2+ VLPs could be diminished when administered with Poly (I:C). On the other hand, AddaVax is a squalene-based oil-in-water emulsion that elicits both cellular (Th1) and humoral (Th2) immune responses. AddaVax is believed to act through recruitment and activation of APCs and through stimulation of cytokine production by macrophages and granulocytes [
39]. It has been previously shown that cytokines such as IL-12 are required for the establishment of long-term protection and a tumor-specific memory against mammary adenocarcinomas, which are mediated by CD8+ lymphocytes [
33]. As we have previously noted, HER2ic-AddaVax-immunized tumor biopsies have shown an increased presence of tumor-infiltrating CD8+ lymphocytes that could contribute to the long-term response observed in this group.
When we investigated the underlying tumor-specific immune responses, we observed that HER2ic VLP immunizations induced higher anti-HER2 Ab titers than the mammalianized counterparts. In any case, the induced antibodies were biased toward the more active IgG2a and IgG2b subtypes. These murine IgG2 subtypes have been described as having higher biological activity than IgG1 or IgG3 antibodies [
30]. Accordingly, the induced anti-HER2 Abs mediated ADCC effector functions. The highest activity was detected in HER2ic VLP-immunized mice that were given AddaVax or were left nonadjuvant. In addition, both of these vaccination groups showed the highest tumor protection, and hence showed significant correlations between the ADCC activity in vitro and reduced tumor volumes in vivo. In the Poly (I:C) cohort, no correlation was observed. This strongly suggests that the antitumor effect was partly driven by effector functions of the induced HER2-specific Abs.
Since it is known that IgG2a and IgG2b Ab subtypes mostly drive ADCC effector functions [
30], we anticipated higher IgG2a and IgG2b levels than IgG1 levels in the vaccination groups that showed high ADCC activity. As we did not observe any obvious changes in the subtype ratios among the different vaccination groups, we suggest that the induced anti-HER2 antibodies might display different glycosylation patterns on their Fc regions that lead to differences in binding to Fc
γ receptors on natural killer (NK) cells [
40], therefore leading to differences in the intensity of the ADCC activity.
Moreover, T-cell activity was clearly increased in HER2 VLP-immunized mice when compared to Control VLP-immunized mice, indicating that cell-mediated immune responses were also involved in the tumor protective effects. However, more research needs to be addressed to confirm that.
We are also aware of the fact that using the HIV-1 Gag matrix protein in a human vaccine might be problematic, since it could trigger nonspecific immune responses and cause side effects. Yet, there exist a variety of alternative matrix proteins, such as M1 from the influenza virus [
41], that form VLPs and that could be used when given to patients.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}