Safety and Immunogenicity of a Novel Recombinant Simian Adenovirus ChAdOx2 as a Vectored Vaccine

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design And Participants
2.2. Design and Construction of ChAdOx2 HAV Vaccine
2.3. IFN-γ ELISpot
2.4. Statistical Analysis
2.5. Anti-Vector Neutralising Antibodies (NAb)
3. Results
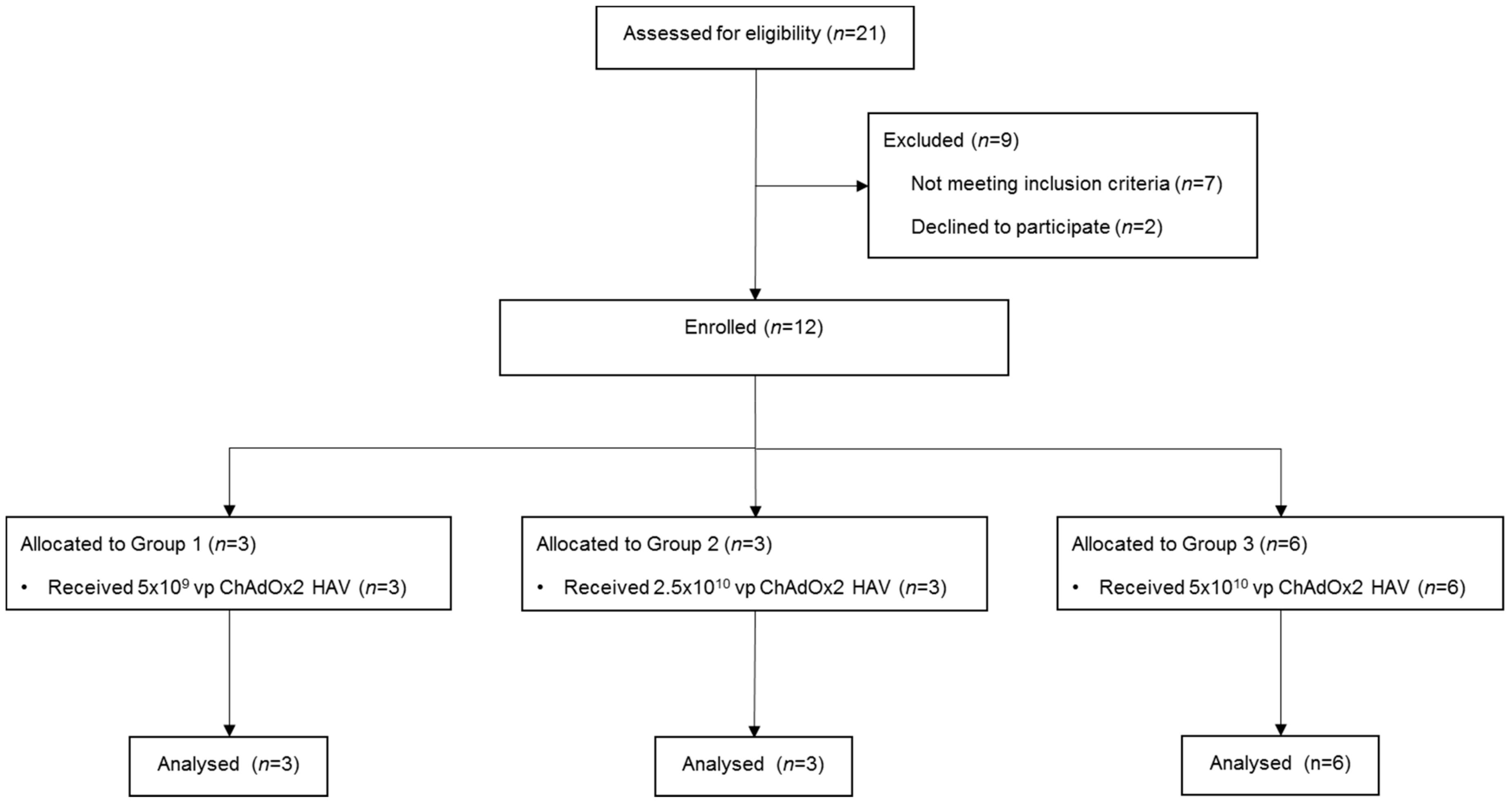
3.1. Study Population
3.2. Vaccine Safety
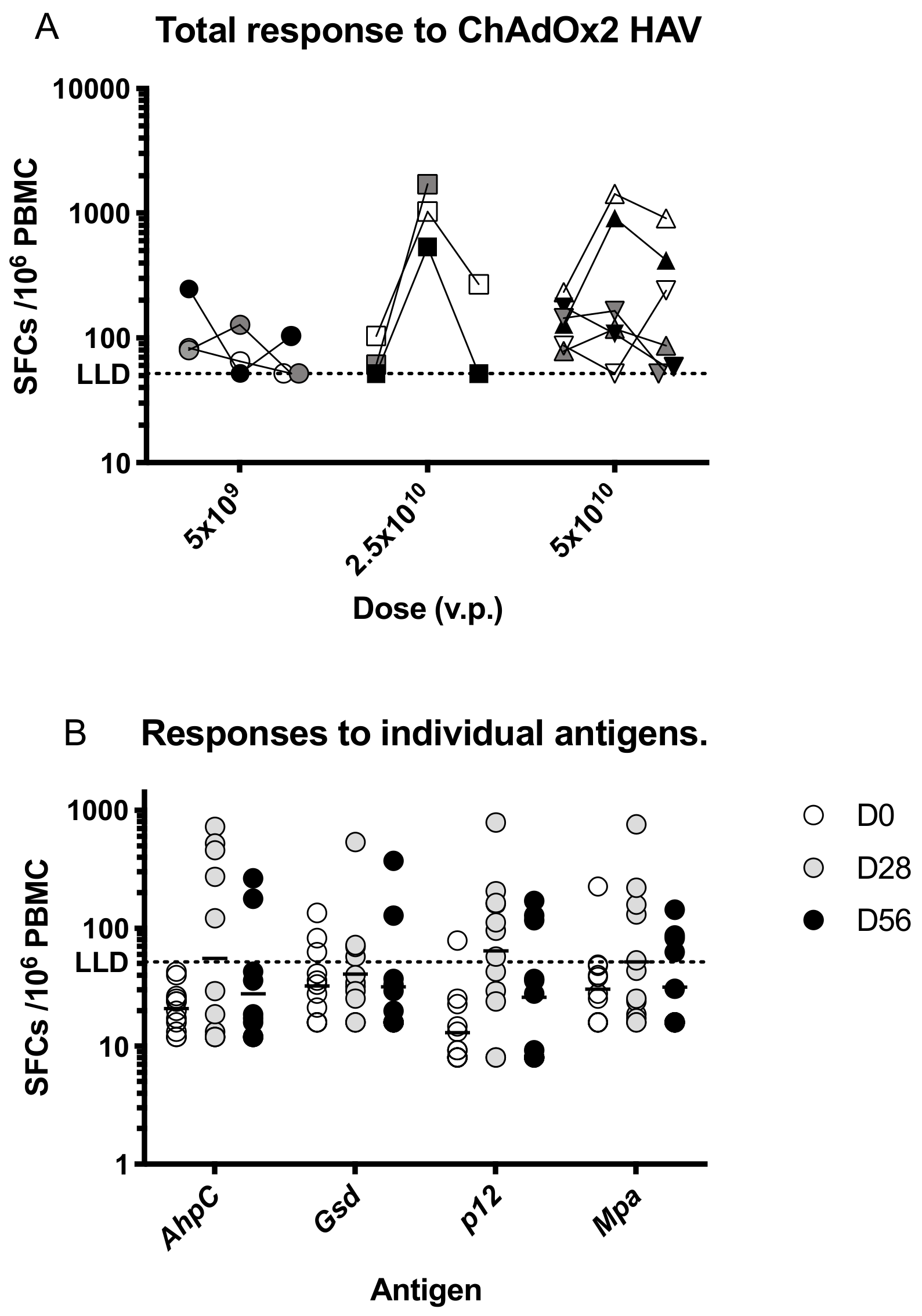
3.3. Immunogenicity
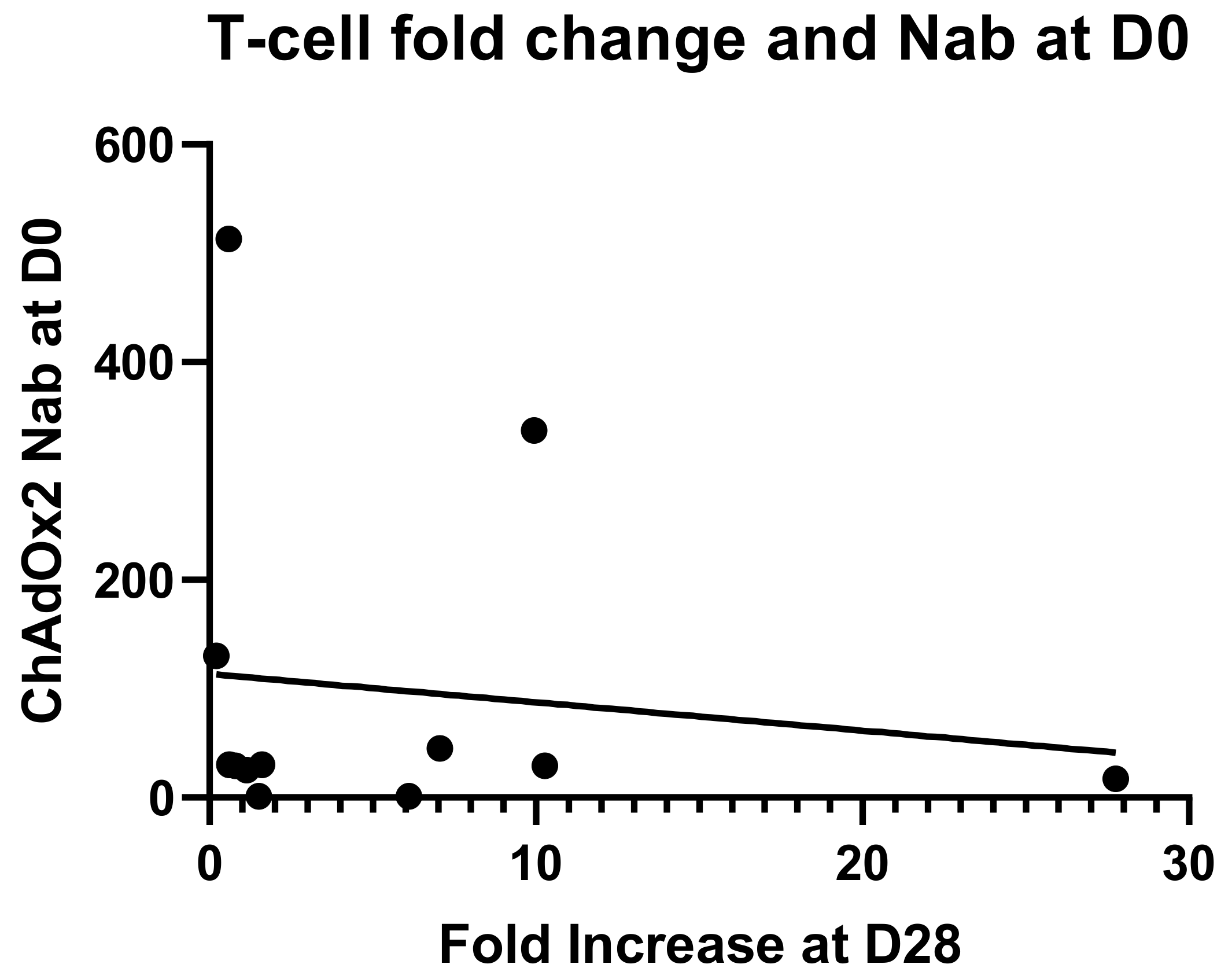
3.4. Anti-Vector NAbs
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Draper, S.J.; Heeney, J.L. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol. 2010, 8, 62–73. [Google Scholar] [CrossRef]
- Rocha, C.D.; Caetano, B.C.; Machado, A.V.; Bruna-Romero, O. Recombinant viruses as tools to induce protective cellular immunity against infectious diseases. Int. Microbiol. 2004, 7, 83–94. [Google Scholar] [PubMed]
- Randrianarison-Jewtoukoff, V.; Perricaudet, M. Recombinant adenoviruses as vaccines. Biologicals 1995, 23, 145–157. [Google Scholar] [CrossRef]
- Catanzaro, A.T.; Koup, R.A.; Roederer, M.; Bailer, R.T.; Enama, M.E.; Moodie, Z.; Gu, L.; Martin, J.E.; Novik, L.; Chakrabarti, B.K.; et al. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J. Infect. Dis. 2006, 194, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Priddy, F.H.; Brown, D.; Kublin, J.; Monahan, K.; Wright, D.P.; Lalezari, J.; Santiago, S.; Marmor, M.; Lally, M.; Novak, R.M.; et al. Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clin. Infect. Dis. 2008, 46, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Harro, C.D.; Robertson, M.N.; Lally, M.A.; O’Neill, L.D.; Edupuganti, S.; Goepfert, P.A.; Mulligan, M.J.; Priddy, F.H.; Dubey, S.A.; Kierstead, L.S.; et al. Safety and immunogenicity of adenovirus-vectored near-consensus HIV type 1 clade B gag vaccines in healthy adults. AIDS Res. Human Retrovirus. 2009, 25, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Harro, C.; Sun, X.; Stek, J.E.; Leavitt, R.Y.; Mehrotra, D.V.; Wang, F.; Bett, A.J.; Casimiro, D.R.; Shiver, J.W.; DiNubile, M.J.; et al. Safety and immunogenicity of the Merck adenovirus serotype 5 (MRKAd5) and MRKAd6 human immunodeficiency virus type 1 trigene vaccines alone and in combination in healthy adults. Clin. Vacc. Immunol. 2009, 16, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Kibuuka, H.; Kimutai, R.; Maboko, L.; Sawe, F.; Schunk, M.S.; Kroidl, A.; Shaffer, D.; Eller, L.A.; Kibaya, R.; Eller, M.A.; et al. A phase 1/2 study of a multiclade HIV-1 DNA plasmid prime and recombinant adenovirus serotype 5 boost vaccine in HIV-Uninfected East Africans (RV 172). J. Infect. Dis. 2010, 201, 600–607. [Google Scholar] [CrossRef]
- Schooley, R.T.; Spritzler, J.; Wang, H.; Lederman, M.M.; Havlir, D.; Kuritzkes, D.R.; Pollard, R.; Battaglia, C.; Robertson, M.; Mehrotra, D.; et al. AIDS clinical trials group 5197: A placebo-controlled trial of immunization of HIV-1-infected persons with a replication-deficient adenovirus type 5 vaccine expressing the HIV-1 core protein. J. Infect. Dis. 2010, 202, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Jaoko, W.; Karita, E.; Kayitenkore, K.; Omosa-Manyonyi, G.; Allen, S.; Than, S.; Adams, E.M.; Graham, B.S.; Koup, R.A.; Bailer, R.T.; et al. Safety and immunogenicity study of Multiclade HIV-1 adenoviral vector vaccine alone or as boost following a multiclade HIV-1 DNA vaccine in Africa. PLoS ONE 2010, 5, e12873. [Google Scholar] [CrossRef]
- Peiperl, L.; Morgan, C.; Moodie, Z.; Li, H.; Russell, N.; Graham, B.S.; Tomaras, G.D.; De Rosa, S.C.; McElrath, M.J. Safety and immunogenicity of a replication-defective adenovirus type 5 HIV vaccine in Ad5-seronegative persons: A randomized clinical trial (HVTN 054). PLoS ONE 2010, 5, e13579. [Google Scholar] [CrossRef] [PubMed]
- Churchyard, G.J.; Morgan, C.; Adams, E.; Hural, J.; Graham, B.S.; Moodie, Z.; Grove, D.; Gray, G.; Bekker, L.G.; McElrath, M.J.; et al. A phase IIA randomized clinical trial of a multiclade HIV-1 DNA prime followed by a multiclade rAd5 HIV-1 vaccine boost in healthy adults (HVTN204). PLoS ONE 2011, 6, e21225. [Google Scholar] [CrossRef] [PubMed]
- Keefer, M.C.; Gilmour, J.; Hayes, P.; Gill, D.; Kopycinski, J.; Cheeseman, H.; Cashin-Cox, M.; Naarding, M.; Clark, L.; Fernandez, N.; et al. A phase I double blind, placebo-controlled, randomized study of a multigenic HIV-1 adenovirus subtype 35 vector vaccine in healthy uninfected adults. PLoS ONE 2012, 7, e41936. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; Walsh, S.R.; Seaman, M.S.; Tucker, R.P.; Krause, K.H.; Patel, A.; Johnson, J.A.; Kleinjan, J.; Yanosick, K.E.; Perry, J.; et al. First-in-human evaluation of the safety and immunogenicity of a recombinant adenovirus serotype 26 HIV-1 Env vaccine (IPCAVD 001). J. Infect. Dis. 2013, 207, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.M.; Sobieszczyk, M.E.; Janes, H.; Karuna, S.T.; Mulligan, M.J.; Grove, D.; Koblin, B.A.; Buchbinder, S.P.; Keefer, M.C.; Tomaras, G.D.; et al. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N. Engl. J. Med. 2013, 369, 2083–2092. [Google Scholar] [CrossRef]
- Gray, G.E.; Moodie, Z.; Metch, B.; Gilbert, P.B.; Bekker, L.G.; Churchyard, G.; Nchabeleng, M.; Mlisana, K.; Laher, F.; Roux, S.; et al. Recombinant adenovirus type 5 HIV gag/pol/nef vaccine in South Africa: unblinded, long-term follow-up of the phase 2b HVTN 503/Phambili study. Lancet Infect. Dis. 2014, 14, 388–396. [Google Scholar] [CrossRef]
- Omosa-Manyonyi, G.; Mpendo, J.; Ruzagira, E.; Kilembe, W.; Chomba, E.; Roman, F.; Bourguignon, P.; Koutsoukos, M.; Collard, A.; Voss, G.; et al. A Phase I Double Blind, Placebo-Controlled, Randomized Study of the Safety and Immunogenicity of an Adjuvanted HIV-1 Gag-Pol-Nef Fusion Protein and Adenovirus 35 Gag-RT-Int-Nef Vaccine in Healthy HIV-Uninfected African Adults. PLoS ONE 2015, 10, e0125954. [Google Scholar] [CrossRef]
- Barouch, D.H.; Kik, S.V.; Weverling, G.J.; Dilan, R.; King, S.L.; Maxfield, L.F.; Clark, S.; Ng’ang’a, D.; Brandariz, K.L.; Abbink, P.; et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine 2011, 29, 5203–5209. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J. Viral vectors as vaccine carriers. Curr. Opin. Virol. 2016, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gordo, E.; Podgorski, I.I.; Downes, N.; Alemany, R. Circumventing antivector immunity: Potential use of nonhuman adenoviral vectors. Human Gene Ther. 2014, 25, 285–300. [Google Scholar] [CrossRef]
- Gilbert, S.C. Adenovirus-vectored Ebola vaccines. Expert Rev. Vacc. 2015, 14, 1347–1357. [Google Scholar] [CrossRef]
- Cocito, C.; Gilot, P.; Coene, M.; de Kesel, M.; Poupart, P.; Vannuffel, P. Paratuberculosis. Clin. Microbiol. Rev. 1994, 7, 328–345. [Google Scholar] [CrossRef]
- Waddell, L.A.; Rajic, A.; Stark, K.D.; Mc, E.S. The zoonotic potential of Mycobacterium avium ssp. paratuberculosis: A systematic review and meta-analyses of the evidence. Epidemiol. Infect. 2015, 143, 3135–3157. [Google Scholar] [CrossRef]
- Dalziel, T.K. Chronic Interstitial Enteritis. Brit. Med. J. 1913, 2, 1068–1070. [Google Scholar]
- Atreya, R.; Bulte, M.; Gerlach, G.F.; Goethe, R.; Hornef, M.W.; Kohler, H.; Meens, J.; Mobius, P.; Roeb, E.; Weiss, S. Facts, myths and hypotheses on the zoonotic nature of Mycobacterium avium subspecies paratuberculosis. Int. J. Med. Microbiol. IJMM 2014, 304, 858–867. [Google Scholar] [CrossRef]
- Pickup, R.W.; Rhodes, G.; Arnott, S.; Sidi-Boumedine, K.; Bull, T.J.; Weightman, A.; Hurley, M.; Hermon-Taylor, J. Mycobacterium avium subsp. paratuberculosis in the catchment area and water of the River Taff in South Wales, United Kingdom, and its potential relationship to clustering of Crohn’s disease cases in the city of Cardiff. Appl. Environ. Microbiol. 2005, 71, 2130–2139. [Google Scholar] [CrossRef] [PubMed]
- Grant, I.R.; Ball, H.J.; Rowe, M.T. Incidence of Mycobacterium paratuberculosis in bulk raw and commercially pasteurized cows’ milk from approved dairy processing establishments in the United Kingdom. Appl. Environ. Microbiol. 2002, 68, 2428–2435. [Google Scholar] [CrossRef]
- De Souza, H.S.P. Etiopathogenesis of inflammatory bowel disease: Today and tomorrow. Curr. Opin. Gastroenterol. 2017, 33, 222–229. [Google Scholar] [CrossRef]
- Kim, D.H.; Cheon, J.H. Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies. Immune Netw. 2017, 17, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, R.J.; Chamberlin, W.M.; Sarosiek, J.; McCallum, R.W. Crohn’s disease and the mycobacterioses: A quarter century later. Causation or simple association? Crit. Rev. Microbiol. 2012, 38, 52–93. [Google Scholar] [CrossRef] [PubMed]
- Feller, M.; Huwiler, K.; Stephan, R.; Altpeter, E.; Shang, A.; Furrer, H.; Pfyffer, G.E.; Jemmi, T.; Baumgartner, A.; Egger, M. Mycobacterium avium subspecies paratuberculosis and Crohn’s disease: A systematic review and meta-analysis. Lancet Infect. Dis. 2007, 7, 607–613. [Google Scholar] [CrossRef]
- Songini, M.; Mannu, C.; Targhetta, C.; Bruno, G. Type 1 diabetes in Sardinia: Facts and hypotheses in the context of worldwide epidemiological data. Acta Diabetol. 2017, 54, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Cossu, D.; Yokoyama, K.; Hattori, N. Conflicting Role of Mycobacterium Species in Multiple Sclerosis. Front. Neurol. 2017, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Bull, T.J.; Gilbert, S.C.; Sridhar, S.; Linedale, R.; Dierkes, N.; Sidi-Boumedine, K.; Hermon-Taylor, J. A novel multi-antigen virally vectored vaccine against Mycobacterium avium subspecies paratuberculosis. PLoS ONE 2007, 2, e1229. [Google Scholar] [CrossRef]
- Antrobus, R.D.; Coughlan, L.; Berthoud, T.K.; Dicks, M.D.; Hill, A.V.; Lambe, T.; Gilbert, S.C. Clinical assessment of a novel recombinant simian adenovirus ChAdOx1 as a vectored vaccine expressing conserved Influenza A antigens. Mol. Ther. 2014, 22, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.J.; Sebastian, S.; Spencer, A.J.; Gilbert, S.C. Simian adenoviruses as vaccine vectors. Fut. Virol. 2016, 11, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Chartier, C.; Degryse, E.; Gantzer, M.; Dieterle, A.; Pavirani, A.; Mehtali, M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 1996, 70, 4805–4810. [Google Scholar] [PubMed]
- Coughlan, L.; Sridhar, S.; Payne, R.; Edmans, M.; Milicic, A.; Venkatraman, N.; Lugonja, B.; Clifton, L.; Qi, C.; Folegatti, P.M.; et al. Heterologous Two-Dose Vaccination with Simian Adenovirus and Poxvirus Vectors Elicits Long-Lasting Cellular Immunity to Influenza Virus A in Healthy Adults. EBioMedicine 2018, 29, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Dicks, M.D.; Spencer, A.J.; Edwards, N.J.; Wadell, G.; Bojang, K.; Gilbert, S.C.; Hill, A.V.; Cottingham, M.G. A novel chimpanzee adenovirus vector with low human seroprevalence: Improved systems for vector derivation and comparative immunogenicity. PLoS ONE 2012, 7, e40385. [Google Scholar] [CrossRef] [PubMed]
- Warimwe, G.M.; Lorenzo, G.; Lopez-Gil, E.; Reyes-Sandoval, A.; Cottingham, M.G.; Spencer, A.J.; Collins, K.A.; Dicks, M.D.; Milicic, A.; Lall, A.; et al. Immunogenicity and efficacy of a chimpanzee adenovirus-vectored Rift Valley fever vaccine in mice. Virol. J. 2013, 10, 349. [Google Scholar] [CrossRef]
- Stylianou, E.; Griffiths, K.L.; Poyntz, H.C.; Harrington-Kandt, R.; Dicks, M.D.; Stockdale, L.; Betts, G.; McShane, H. Improvement of BCG protective efficacy with a novel chimpanzee adenovirus and a modified vaccinia Ankara virus both expressing Ag85A. Vaccine 2015, 33, 6800–6808. [Google Scholar] [CrossRef] [PubMed]
- McElrath, M.J.; De Rosa, S.C.; Moodie, Z.; Dubey, S.; Kierstead, L.; Janes, H.; Defawe, O.D.; Carter, D.K.; Hural, J.; Akondy, R.; et al. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: A case-cohort analysis. Lancet 2008, 372, 1894–1905. [Google Scholar] [CrossRef]
- Ewer, K.J.; Lambe, T.; Rollier, C.S.; Spencer, A.J.; Hill, A.V.S.; Dorrell, L. Viral vectors as vaccine platforms: from immunogenicity to impact. Curr. Opin. Immunol. 2016, 41, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, S.; Harris, S.A.; Satti, I.; Hokey, D.A.; Dheenadhayalan, V.; Stockdale, L.; Manjaly Thomas, Z.-R.; Minhinnick, A.; Wilkie, M.; Vermaak, S.; et al. A Phase I, Open-Label Trial, Evaluating the Safety and Immunogenicity of Candidate Tuberculosis Vaccines AERAS-402 and MVA85A, Administered by Prime-Boost Regime in BCG-Vaccinated Healthy Adults. PLoS ONE 2015, 10, e0141687. [Google Scholar] [CrossRef]
- Janes, H.E.; Cohen, K.W.; Frahm, N.; De Rosa, S.C.; Sanchez, B.; Hural, J.; Magaret, C.A.; Karuna, S.; Bentley, C.; Gottardo, R.; et al. Higher T-Cell Responses Induced by DNA/rAd5 HIV-1 Preventive Vaccine Are Associated With Lower HIV-1 Infection Risk in an Efficacy Trial. J. Infect. Dis. 2017, 215, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.; Rampling, T.; Venkatraman, N.; Bowyer, G.; Wright, D.; Lambe, T.; Imoukhuede, E.B.; Payne, R.; Fehling, S.K.; Strecker, T.; et al. A Monovalent Chimpanzee Adenovirus Ebola Vaccine Boosted with MVA. N. Engl. J. Med. 2016, 374, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Mensah, V.A.; Gueye, A.; Ndiaye, M.; Edwards, N.J.; Wright, D.; Anagnostou, N.A.; Syll, M.; Ndaw, A.; Abiola, A.; Bliss, C.; et al. Safety, Immunogenicity and Efficacy of Prime-Boost Vaccination with ChAd63 and MVA Encoding ME-TRAP against Plasmodium falciparum Infection in Adults in Senegal. PLoS ONE 2016, 11, e0167951. [Google Scholar] [CrossRef] [PubMed]
- Modjarrad, K. MERS-CoV vaccine candidates in development: The current landscape. Vaccine 2016, 34, 2982–2987. [Google Scholar] [CrossRef]
- O’Hara, G.A.; Duncan, C.J.A.; Ewer, K.J.; Collins, K.A.; Elias, S.C.; Halstead, F.D.; Goodman, A.L.; Edwards, N.J.; Reyes-Sandoval, A.; Bird, P.; et al. Clinical Assessment of a Recombinant Simian Adenovirus ChAd63: A Potent New Vaccine Vector. J. Infect. Dis. 2012, 205, 772–781. [Google Scholar] [CrossRef]
- Thorner, A.R.; Lemckert, A.A.; Goudsmit, J.; Lynch, D.M.; Ewald, B.A.; Denholtz, M.; Havenga, M.J.; Barouch, D.H. Immunogenicity of heterologous recombinant adenovirus prime-boost vaccine regimens is enhanced by circumventing vector cross-reactivity. J. Virol. 2006, 80, 12009–12016. [Google Scholar] [CrossRef] [PubMed]
- Frahm, N.; DeCamp, A.C.; Friedrich, D.P.; Carter, D.K.; Defawe, O.D.; Kublin, J.G.; Casimiro, D.R.; Duerr, A.; Robertson, M.N.; Buchbinder, S.P.; et al. Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J. Clin. Invest. 2012, 122, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Kardani, K.; Bolhassani, A.; Shahbazi, S. Prime-boost vaccine strategy against viral infections: Mechanisms and benefits. Vaccine 2016, 34, 413–423. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folegatti, P.M.; Bellamy, D.; Roberts, R.; Powlson, J.; Edwards, N.J.; Mair, C.F.; Bowyer, G.; Poulton, I.; Mitton, C.H.; Green, N.; et al. Safety and Immunogenicity of a Novel Recombinant Simian Adenovirus ChAdOx2 as a Vectored Vaccine. Vaccines 2019, 7, 40. https://doi.org/10.3390/vaccines7020040
Folegatti PM, Bellamy D, Roberts R, Powlson J, Edwards NJ, Mair CF, Bowyer G, Poulton I, Mitton CH, Green N, et al. Safety and Immunogenicity of a Novel Recombinant Simian Adenovirus ChAdOx2 as a Vectored Vaccine. Vaccines. 2019; 7(2):40. https://doi.org/10.3390/vaccines7020040
Chicago/Turabian StyleFolegatti, Pedro M., Duncan Bellamy, Rachel Roberts, Jonathan Powlson, Nick J. Edwards, Catherine F. Mair, Georgina Bowyer, Ian Poulton, Celia H. Mitton, Nicky Green, and et al. 2019. "Safety and Immunogenicity of a Novel Recombinant Simian Adenovirus ChAdOx2 as a Vectored Vaccine" Vaccines 7, no. 2: 40. https://doi.org/10.3390/vaccines7020040
APA StyleFolegatti, P. M., Bellamy, D., Roberts, R., Powlson, J., Edwards, N. J., Mair, C. F., Bowyer, G., Poulton, I., Mitton, C. H., Green, N., Berrie, E., Lawrie, A. M., Hill, A. V. S., Ewer, K. J., Hermon-Taylor, J., & Gilbert, S. C. (2019). Safety and Immunogenicity of a Novel Recombinant Simian Adenovirus ChAdOx2 as a Vectored Vaccine. Vaccines, 7(2), 40. https://doi.org/10.3390/vaccines7020040