
Synergistic Integration of HDAC Inhibitors and Individualized Neoantigen Therapy (INT): A Next-Generation Combinatorial Approach for Cancer Immunotherapy

 , ,
, ,
Abstract
1. Introduction
2. Potential Coordination Mechanisms
2.1. Synergizing with DCs
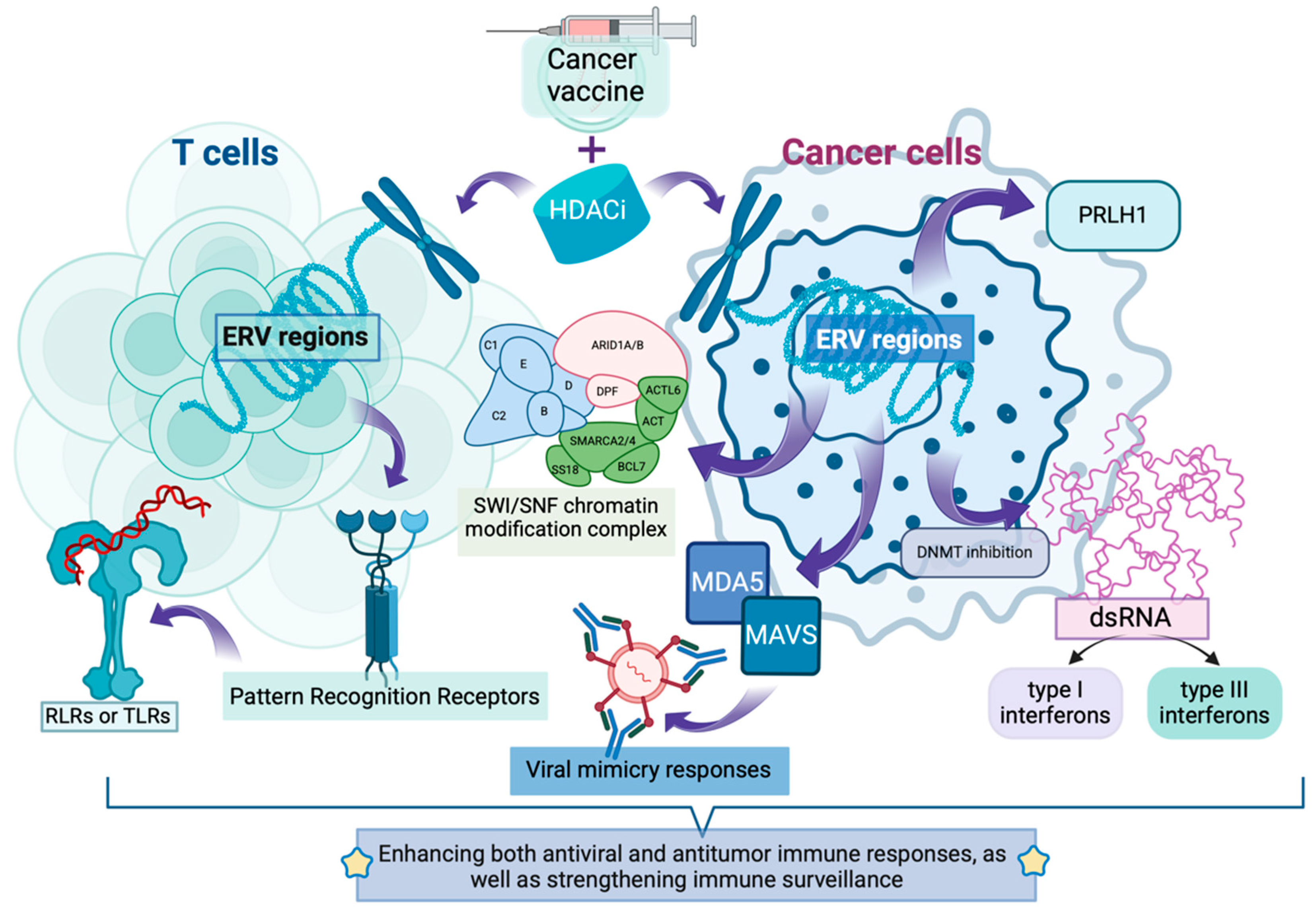
2.2. Activation of Endogenous Retroelements: Expanding the “Antigen Repository”
2.3. Enhancement of Antigen Processing Machinery
2.4. Upregulation of MHC Class I Molecule Expression
2.5. Improvement of MHC-I Complex Stability
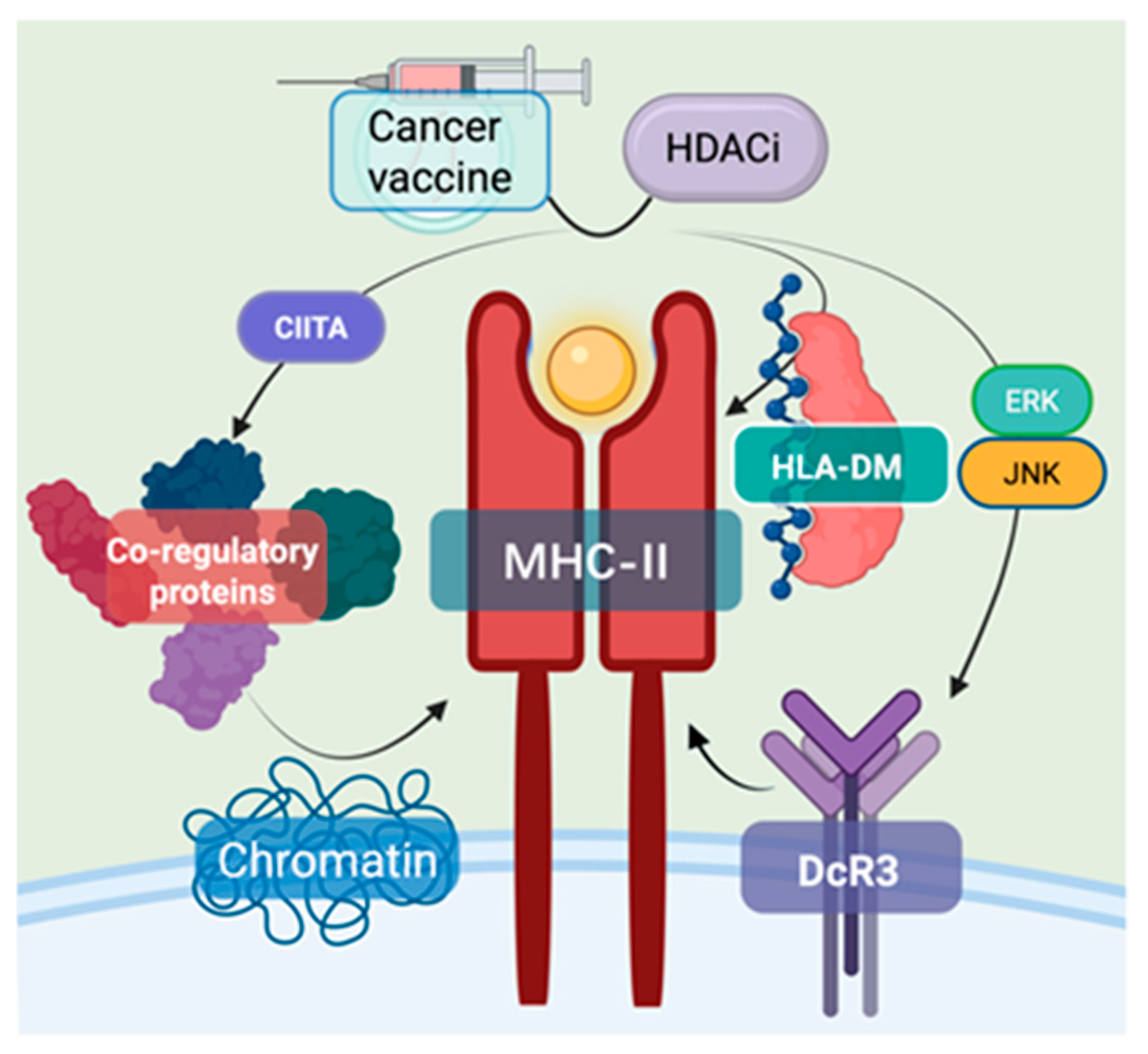
2.6. Enhancement of MHC Class II-Mediated Antitumor Immunity
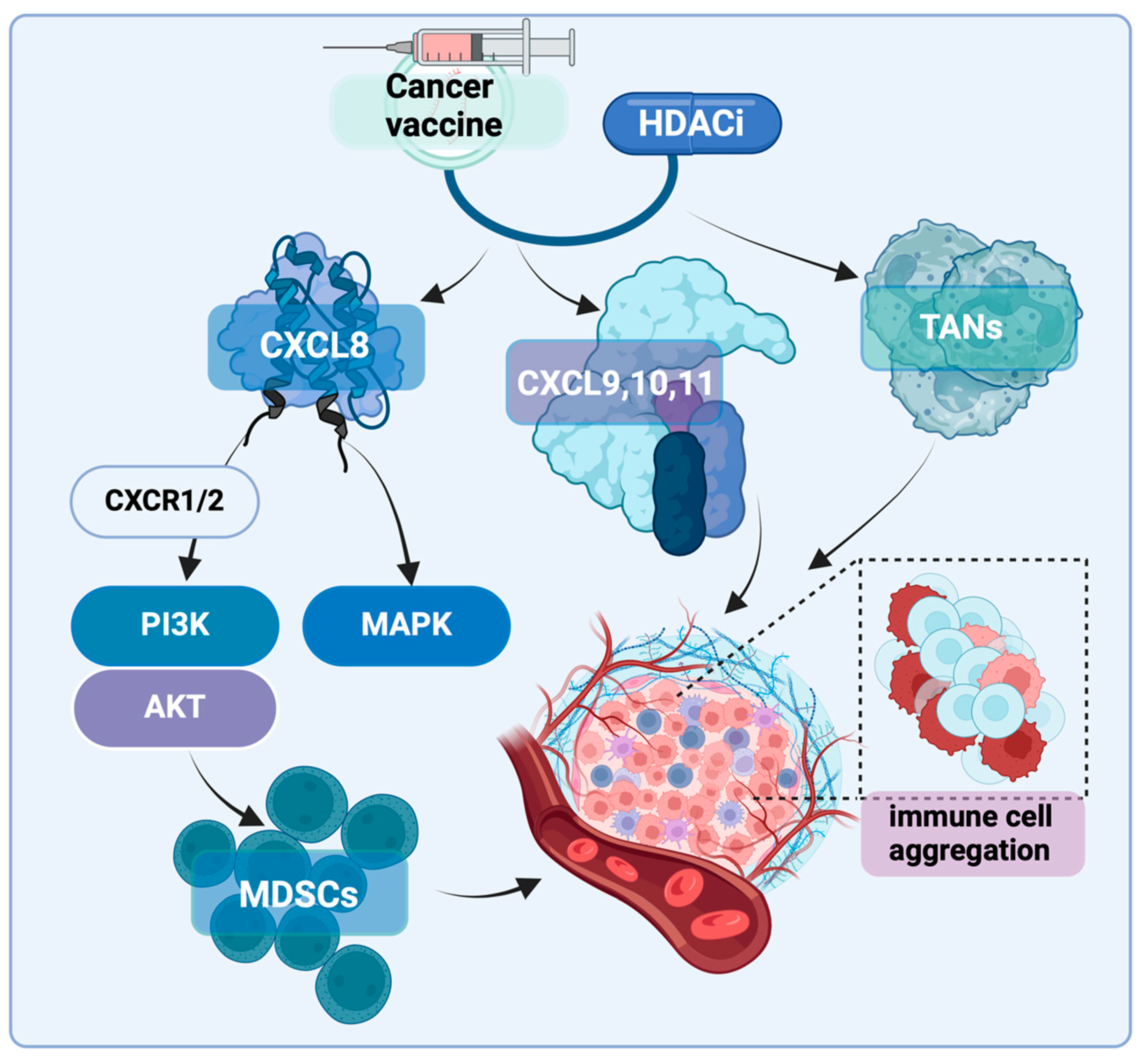
2.7. Other Impacts on the Immune Microenvironment
3. Downregulation of HDACs Using Genome Editing Mechanisms
3.1. CRISPR-Cas9 Mediated Gene Editing
3.2. Transcriptional Repression Mediated by Deactivated Cas9 (dCas9)
3.3. Prime Editing Technology
3.4. TALEN Technology
3.5. RNA Interference Technology and CRISPR-Cas13 System
3.6. Switchable Cas12a-Based System
4. Clinical Applications of HDACi Combined with Tumor Vaccines
4.1. Polypeptide Vaccine PVX-410 Combined with Citarinostat
4.2. Multi-Drug Combination Regimens Beyond Dual Therapy
5. Current Issues and Challenges
5.1. Impact on PD-L1 Expression
5.2. Optimization of Safety and Toxicity Management Strategies
5.3. Investigation of Optimal Sequencing in Combination Therapy
5.4. Incorporating a Third Agent
5.5. Development of Biomarkers for Patient Stratification
5.6. Mechanistic Studies of Long-Term Immune Memory Formation
5.7. Development of Novel Delivery Systems
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lang, F.; Schrörs, B.; Löwer, M.; Türeci, Ö.; Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Carlino, M.S.; Khattak, A.; Meniawy, T.; Ansstas, G.; Taylor, M.H.; Kim, K.B.; McKean, M.; Long, G.V.; Sullivan, R.J.; et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): A randomised, phase 2b study. Lancet 2024, 403, 632–644. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Wang, L.; He, Y.; He, T.; Liu, G.; Lin, C.; Li, K.; Lu, L.; Cai, K. Lymph node-targeted immune-activation mediated by imiquimod-loaded mesoporous polydopamine based-nanocarriers. Biomaterials 2020, 255, 120208. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.C.; Araya, R.E.; Huang, A.; Chen, Q.; Di Modica, M.; Rodrigues, R.R.; Lopès, A.; Johnson, S.B.; Schwarz, B.; Bohrnsen, E.; et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 2021, 184, 5338–5356.e21. [Google Scholar] [CrossRef] [PubMed]
- Ingelfinger, F.; De Feo, D.; Becher, B. GM-CSF: Master regulator of the T cell-phagocyte interface during inflammation. Semin. Immunol. 2021, 54, 101518. [Google Scholar] [CrossRef]
- Moran, B.; Davern, M.; Reynolds, J.V.; Donlon, N.E.; Lysaght, J. The impact of histone deacetylase inhibitors on immune cells and implications for cancer therapy. Cancer Lett. 2023, 559, 216121. [Google Scholar] [CrossRef]
- Buchwald, M.; Krämer, O.H.; Heinzel, T. HDACi—targets beyond chromatin. Cancer Lett. 2009, 280, 160–167. [Google Scholar] [CrossRef]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef]
- Kim, Y.H.; Bagot, M.; Pinter-Brown, L.; Rook, A.H.; Porcu, P.; Horwitz, S.M.; Whittaker, S.; Tokura, Y.; Vermeer, M.; Zinzani, P.L.; et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): An international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2018, 19, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, S.M.; White, L.A.; Kolesar, J.M. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am. J. Health Syst. Pharm. 2010, 67, 793–797. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Janne, P.A.; Opyrchal, M.; Hafez, N.; Raez, L.E.; Gabrilovich, D.I.; Wang, F.; Trepel, J.B.; Lee, M.J.; Yuno, A.; et al. Entinostat plus Pembrolizumab in Patients with Metastatic NSCLC Previously Treated with Anti-PD-(L)1 Therapy. Clin. Cancer Res. 2021, 27, 1019–1028. [Google Scholar] [CrossRef]
- Wozniak, M.B.; Villuendas, R.; Bischoff, J.R.; Aparicio, C.B.; Martinez Leal, J.F.; de La Cueva, P.; Rodriguez, M.E.; Herreros, B.; Martin-Perez, D.; Longo, M.I.; et al. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica 2010, 95, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Shariat-Madar, Z.; Walker, L.A.; Tekwani, B.L. Mechanism for neurotropic action of vorinostat, a pan histone deacetylase inhibitor. Mol. Cell. Neurosci. 2016, 77, 11–20. [Google Scholar] [CrossRef]
- Okabe, S.; Tauchi, T.; Tanaka, Y.; Kimura, S.; Maekawa, T.; Ohyashiki, K. Activity of histone deacetylase inhibitors and an Aurora kinase inhibitor in BCR-ABL-expressing leukemia cells: Combination of HDAC and Aurora inhibitors in BCR-ABL-expressing cells. Cancer Cell Int. 2013, 13, 32. [Google Scholar] [CrossRef]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Reddy, P. HDAC inhibition and graft versus host disease. Mol. Med. 2011, 17, 404–416. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, S.; Song, T.; Guan, X.; Zhang, R.; Chen, X. Trichostatin a Protects Dendritic Cells Against Oxygen-Glucose Deprivation via the SRSF3/PKM2/Glycolytic Pathway. Front. Pharmacol. 2018, 9, 612. [Google Scholar] [CrossRef]
- Nencioni, A.; Beck, J.; Werth, D.; Grünebach, F.; Patrone, F.; Ballestrero, A.; Brossart, P. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin. Cancer Res. 2007, 13, 3933–3941. [Google Scholar] [CrossRef]
- Ge, Z.; Da, Y.; Xue, Z.; Zhang, K.; Zhuang, H.; Peng, M.; Li, Y.; Li, W.; Simard, A.; Hao, J.; et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp. Neurol. 2013, 241, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Cornel, A.M.; Dunnebach, E.; Hofman, D.A.; Das, S.; Sengupta, S.; van den Ham, F.; Wienke, J.; Strijker, J.G.M.; van den Beemt, D.; Essing, A.H.W.; et al. Epigenetic modulation of neuroblastoma enhances T cell and NK cell immunogenicity by inducing a tumor-cell lineage switch. J. Immunother. Cancer 2022, 10, e005002. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.D.; Tomasi, T.B. Spatial distribution of histone methylation during MHC class II expression. Mol. Immunol. 2008, 45, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, A.; Scholtka, B.; Gerecke, C.; Kleuser, B. Epigenetic histone modulation contributes to improvements in inflammatory bowel disease via EBI3. Cell. Mol. Life Sci. 2020, 77, 5017–5030. [Google Scholar] [CrossRef]
- Guimarães, L.D.; Webber, L.P.; Gaio, E.J.; Junior, D.S.; Gonçalves, P.; Wick, M.J.; Burr, N.S.; Squarize, C.H.; Castilho, R.M. Using PDX animal models to identify and stratify adenoid cystic carcinoma patients presenting an enhanced response to HDAC inhibitors. Am. J. Cancer Res. 2023, 13, 143–160. [Google Scholar]
- Wang, J.H.; Shih, K.S.; Wu, Y.W.; Wang, A.W.; Yang, C.R. Histone deacetylase inhibitors increase microRNA-146a expression and enhance negative regulation of interleukin-1β signaling in osteoarthritis fibroblast-like synoviocytes. Osteoarthr. Cartil. 2013, 21, 1987–1996. [Google Scholar] [CrossRef]
- Salmon, J.M.; Todorovski, I.; Stanley, K.L.; Bruedigam, C.; Kearney, C.J.; Martelotto, L.G.; Rossello, F.; Semple, T.; Arnau, G.M.; Zethoven, M.; et al. Epigenetic Activation of Plasmacytoid DCs Drives IFNAR-Dependent Therapeutic Differentiation of AML. Cancer Discov. 2022, 12, 1560–1579. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Biery, M.C.; Myers, C.; Ferguson, E.; Zheng, Y.; Girard, E.J.; Przystal, J.M.; Park, G.; Noll, A.; Pakiam, F.; et al. Optimal therapeutic targeting by HDAC inhibition in biopsy-derived treatment-naïve diffuse midline glioma models. Neuro. Oncol. 2021, 23, 376–386. [Google Scholar] [CrossRef]
- Jarosz, A.S.; Halo, J.V. Transcription of Endogenous Retroviruses: Broad and Precise Mechanisms of Control. Viruses 2024, 16, 1312. [Google Scholar] [CrossRef]
- Goyal, A.; Bauer, J.; Hey, J.; Papageorgiou, D.N.; Stepanova, E.; Daskalakis, M.; Scheid, J.; Dubbelaar, M.; Klimovich, B.; Schwarz, D.; et al. DNMT and HDAC inhibition induces immunogenic neoantigens from human endogenous retroviral element-derived transcripts. Nat. Commun. 2023, 14, 6731. [Google Scholar] [CrossRef]
- Krchlikova, V.; Lu, Y.; Sauter, D. Viral influencers: Deciphering the role of endogenous retroviral LTR12 repeats in cellular gene expression. J. Virol. 2025, 99, e0135124. [Google Scholar] [CrossRef] [PubMed]
- Kronung, S.K.; Beyer, U.; Chiaramonte, M.L.; Dolfini, D.; Mantovani, R.; Dobbelstein, M. LTR12 promoter activation in a broad range of human tumor cells by HDAC inhibition. Oncotarget 2016, 7, 33484–33497. [Google Scholar] [CrossRef]
- Karttunen, K.; Patel, D.; Xia, J.; Fei, L.; Palin, K.; Aaltonen, L.; Sahu, B. Transposable elements as tissue-specific enhancers in cancers of endodermal lineage. Nat. Commun. 2023, 14, 5313. [Google Scholar] [CrossRef]
- Hashimoto, K.; Suzuki, A.M.; Dos Santos, A.; Desterke, C.; Collino, A.; Ghisletti, S.; Braun, E.; Bonetti, A.; Fort, A.; Qin, X.Y.; et al. CAGE profiling of ncRNAs in hepatocellular carcinoma reveals widespread activation of retroviral LTR promoters in virus-induced tumors. Genome Res. 2015, 25, 1812–1824. [Google Scholar] [CrossRef]
- Sherrill-Mix, S.; Ocwieja, K.E.; Bushman, F.D. Gene activity in primary T cells infected with HIV89.6: Intron retention and induction of genomic repeats. Retrovirology 2015, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Srinivasachar Badarinarayan, S.; Shcherbakova, I.; Langer, S.; Koepke, L.; Preising, A.; Hotter, D.; Kirchhoff, F.; Sparrer, K.M.J.; Schotta, G.; Sauter, D. HIV-1 infection activates endogenous retroviral promoters regulating antiviral gene expression. Nucleic Acids Res. 2020, 48, 10890–10908. [Google Scholar] [CrossRef] [PubMed]
- Vincendeau, M.; Gottesdorfer, I.; Schreml, J.M.; Wetie, A.G.; Mayer, J.; Greenwood, A.D.; Helfer, M.; Kramer, S.; Seifarth, W.; Hadian, K.; et al. Modulation of human endogenous retrovirus (HERV) transcription during persistent and de novo HIV-1 infection. Retrovirology 2015, 12, 27. [Google Scholar] [CrossRef]
- Yang, Z.; Chu, B.; Tu, Y.; Li, L.; Chen, D.; Huang, S.; Huang, W.; Fan, W.; Li, Q.; Zhang, C.; et al. Dual inhibitors of DNMT and HDAC remodels the immune microenvironment of colorectal cancer and enhances the efficacy of anti-PD-L1 therapy. Pharmacol. Res. 2024, 206, 107271. [Google Scholar] [CrossRef]
- Wang, R.; Dong, X.; Zhang, X.; Liao, J.; Cui, W.; Li, W. Exploring viral mimicry combined with epigenetics and tumor immunity: New perspectives in cancer therapy. Int. J. Biol. Sci. 2025, 21, 958–973. [Google Scholar] [CrossRef]
- Fan, W.; Li, W.; Li, L.; Qin, M.; Mao, C.; Yuan, Z.; Wang, P.; Chu, B.; Jiang, Y. Bifunctional HDAC and DNMT inhibitor induces viral mimicry activates the innate immune response in triple-negative breast cancer. Eur. J. Pharm. Sci. 2024, 197, 106767. [Google Scholar] [CrossRef]
- White, C.H.; Beliakova-Bethell, N.; Lada, S.M.; Breen, M.S.; Hurst, T.P.; Spina, C.A.; Richman, D.D.; Frater, J.; Magiorkinis, G.; Woelk, C.H. Transcriptional Modulation of Human Endogenous Retroviruses in Primary CD4+ T Cells Following Vorinostat Treatment. Front. Immunol. 2018, 9, 603. [Google Scholar] [CrossRef] [PubMed]
- Curty, G.; Iñiguez, L.P.; Nixon, D.F.; Soares, M.A.; de Mulder Rougvie, M. Hallmarks of Retroelement Expression in T-Cells Treated With HDAC Inhibitors. Front. Virol. 2021, 1, 756635. [Google Scholar] [CrossRef]
- Russ, E.; Iordanskiy, S. Endogenous Retroviruses as Modulators of Innate Immunity. Pathogens 2023, 12, 162. [Google Scholar] [CrossRef]
- Wang, F.; Jin, Y.; Wang, M.; Luo, H.Y.; Fang, W.J.; Wang, Y.N.; Chen, Y.X.; Huang, R.J.; Guan, W.L.; Li, J.B.; et al. Combined anti-PD-1, HDAC inhibitor and anti-VEGF for MSS/pMMR colorectal cancer: A randomized phase 2 trial. Nat. Med. 2024, 30, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Gregorie, C.J.; Tomasi, T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. 2008, 57, 647–654. [Google Scholar] [CrossRef]
- Ritter, C.; Fan, K.; Paschen, A.; Reker Hardrup, S.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 2290. [Google Scholar] [CrossRef]
- Basler, M.; Groettrup, M. On the Role of the Immunoproteasome in Protein Homeostasis. Cells 2021, 10, 3216. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.G.; Driscoll, J.; Monaco, J.J. Structural and serological similarity of MHC-linked LMP and proteasome (multicatalytic proteinase) complexes. Nature 1991, 353, 355–357. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef]
- Kelly, A.; Trowsdale, J. Genetics of antigen processing and presentation. Immunogenetics 2019, 71, 161–170. [Google Scholar] [CrossRef]
- Zaitoua, A.J.; Kaur, A.; Raghavan, M. Variations in MHC class I antigen presentation and immunopeptidome selection pathways. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The regulatory network behind MHC class I expression. Mol. Immunol. 2019, 113, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Gomez, S.; Tabernacki, T.; Kobyra, J.; Roberts, P.; Chiappinelli, K.B. Combining epigenetic and immune therapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 65, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef]
- Zhu, M.; Han, Y.; Gu, T.; Wang, R.; Si, X.; Kong, D.; Zhao, P.; Wang, X.; Li, J.; Zhai, X.; et al. Class I HDAC inhibitors enhance antitumor efficacy and persistence of CAR-T cells by activation of the Wnt pathway. Cell Rep. 2024, 43, 114065. [Google Scholar] [CrossRef]
- Dong, W.; He, B.; Cao, Y.; Yang, R.; Zhang, S.; Kong, Y.; Lu, D.; Zheng, X.; Hou, Y.; Zhu, M.; et al. Low-dose SAHA enhances CD8+ T cell-mediated antitumor immunity by boosting MHC I expression in non-small cell lung cancer. Cell. Oncol. 2025, 48, 249–264. [Google Scholar] [CrossRef]
- Sun, T.; Li, Y.; Yang, W.; Wu, H.; Li, X.; Huang, Y.; Zhou, Y.; Du, Z. Histone deacetylase inhibition up-regulates MHC class I to facilitate cytotoxic T lymphocyte-mediated tumor cell killing in glioma cells. J. Cancer 2019, 10, 5638–5645. [Google Scholar] [CrossRef]
- Shi, M.-Q.; Xu, Y.; Fu, X.; Pan, D.-S.; Lu, X.-P.; Xiao, Y.; Jiang, Y.-Z. Advances in targeting histone deacetylase for treatment of solid tumors. J. Hematol. Oncol. 2024, 17, 37. [Google Scholar] [CrossRef]
- Liang, T.; Wang, F.; Elhassan, R.M.; Cheng, Y.; Tang, X.; Chen, W.; Fang, H.; Hou, X. Targeting histone deacetylases for cancer therapy: Trends and challenges. Acta Pharm. Sin. B 2023, 13, 2425–2463. [Google Scholar] [CrossRef]
- Narukawa, T.; Yasuda, S.; Horinaka, M.; Taniguchi, K.; Tsujikawa, T.; Morita, M.; Ukimura, O.; Sakai, T. The Novel HDAC Inhibitor OBP-801 Promotes MHC Class I Presentation Through LMP2 Upregulation, Enhancing the PD-1-Targeting Therapy in Clear Cell Renal Cell Carcinoma. Cancers 2024, 16, 4058. [Google Scholar] [CrossRef]
- Ramaiah, M.J.; Tangutur, A.D.; Manyam, R.R. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021, 277, 119504. [Google Scholar] [CrossRef] [PubMed]
- Kurdistani, S.K.; Tavazoie, S.; Grunstein, M. Mapping global histone acetylation patterns to gene expression. Cell 2004, 117, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, K.P.; Gendreizig, S.; White, D.A.; Bradbury, C.; Hollfelder, F.; Turner, B.M. Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J. Biol. Chem. 2007, 282, 4408–4416. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Kallingal, A.; Olszewski, M.; Maciejewska, N.; Brankiewicz, W.; Baginski, M. Cancer immune escape: The role of antigen presentation machinery. J. Cancer Res. Clin. Oncol. 2023, 149, 8131–8141. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Ginter, T.; Bier, C.; Knauer, S.K.; Sughra, K.; Hildebrand, D.; Münz, T.; Liebe, T.; Heller, R.; Henke, A.; Stauber, R.H.; et al. Histone deacetylase inhibitors block IFNγ-induced STAT1 phosphorylation. Cell. Signal. 2012, 24, 1453–1460. [Google Scholar] [CrossRef]
- Xu, W.; Liu, H.; Liu, Z.G.; Wang, H.S.; Zhang, F.; Wang, H.; Zhang, J.; Chen, J.J.; Huang, H.J.; Tan, Y.; et al. Histone deacetylase inhibitors upregulate Snail via Smad2/3 phosphorylation and stabilization of Snail to promote metastasis of hepatoma cells. Cancer Lett. 2018, 420, 1–13. [Google Scholar] [CrossRef]
- Chen, X.; Pan, X.; Zhang, W.; Guo, H.; Cheng, S.; He, Q.; Yang, B.; Ding, L. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm. Sin. B 2020, 10, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Idso, J.M.; Lao, S.; Schloemer, N.J.; Knipstein, J.; Burns, R.; Thakar, M.S.; Malarkannan, S. Entinostat augments NK cell functions via epigenetic upregulation of IFIT1-STING-STAT4 pathway. Oncotarget 2020, 11, 1799–1815. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Wang, D.; Xiong, W.; Wang, X.; Huang, W.H.; Wu, G.H.; Liu, W.Z.; Wang, Q.; Chen, J.S.; Kuai, Y.Y.; et al. Correction: Unveiling the role of HP1alpha-HDAC1-STAT1 axis as a therapeutic target for HP1alpha-positive intrahepatic cholangiocarcinoma. J. Exp. Clin. Cancer Res. 2024, 43, 186. [Google Scholar] [CrossRef]
- Bouchecareilh, M.; Hutt, D.M.; Szajner, P.; Flotte, T.R.; Balch, W.E. Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)-mediated correction of alpha1-antitrypsin deficiency. J. Biol. Chem. 2012, 287, 38265–38278. [Google Scholar] [CrossRef]
- Raghavan, M.; Del Cid, N.; Rizvi, S.M.; Peters, L.R. MHC class I assembly: Out and about. Trends. Immunol. 2008, 29, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Halilovic, M.; Marx-Blumel, L.; Marx, C.; Buder, K.; Beck, J.F.; Sonnemann, J. Assessment of HDAC Inhibitor-Induced Endoplasmic Reticulum (ER) Stress. Methods Mol. Biol. 2023, 2589, 253–268. [Google Scholar] [PubMed]
- Muller, I.K.; Winter, C.; Thomas, C.; Spaapen, R.M.; Trowitzsch, S.; Tampe, R. Structure of an MHC I-tapasin-ERp57 editing complex defines chaperone promiscuity. Nat. Commun. 2022, 13, 5383. [Google Scholar] [CrossRef]
- Xiao, W.; Dong, W.; Zhang, C.; Saren, G.; Geng, P.; Zhao, H.; Li, Q.; Zhu, J.; Li, G.; Zhang, S.; et al. Effects of the epigenetic drug MS-275 on the release and function of exosome-related immune molecules in hepatocellular carcinoma cells. Eur. J. Med. Res. 2013, 18, 61. [Google Scholar] [CrossRef]
- Ploegh, H.L.; Orr, H.T.; Strominger, J.L. Major histocompatibility antigens: The human (HLA-A, -B, -C) and murine (H-2K, H-2D) class I molecules. Cell 1981, 24, 287–299. [Google Scholar] [CrossRef]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Torngren, T.; Kvist, A.; et al. Author Correction: Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2020, 11, 1714. [Google Scholar] [CrossRef]
- Tout, I.; Bougarn, S.; Toufiq, M.; Gopinath, N.; Hussein, O.; Sathappan, A.; Chin-Smith, E.; Rehaman, F.; Mathew, R.; Mathew, L.; et al. The integrative genomic and functional immunological analyses of colorectal cancer initiating cells to modulate stemness properties and the susceptibility to immune responses. J. Transl. Med. 2025, 23, 193. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, A.; Chybicka, K.; Ziolo, E.; Strzadala, L.; Kalas, W. The Contrasting Delayed Effects of Transient Exposure of Colorectal Cancer Cells to Decitabine or Azacitidine. Cancers 2022, 14, 1530. [Google Scholar] [CrossRef]
- Roemer, M.G.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced beta2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, H.; Dai, J.; Lin, J.; Zhao, K.; Hu, H.; Zhong, C. Targeting macrophages in cancer immunotherapy: Frontiers and challenges. J. Adv. Res. 2025. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Zielinska, M.K.; Ciazynska, M.; Sulejczak, D.; Rutkowski, P.; Czarnecka, A.M. Mechanisms of Resistance to Anti-PD-1 Immunotherapy in Melanoma and Strategies to Overcome It. Biomolecules 2025, 15, 269. [Google Scholar] [CrossRef] [PubMed]
- Serra, P.; Garabatos, N.; Singha, S.; Fandos, C.; Garnica, J.; Sole, P.; Parras, D.; Yamanouchi, J.; Blanco, J.; Tort, M.; et al. Increased yields and biological potency of knob-into-hole-based soluble MHC class II molecules. Nat. Commun. 2019, 10, 4917. [Google Scholar] [CrossRef]
- Turner, T.B.; Meza-Perez, S.; Londono, A.; Katre, A.; Peabody, J.E.; Smith, H.J.; Forero, A.; Norian, L.A.; Straughn, J.M., Jr.; Buchsbaum, D.J.; et al. Epigenetic modifiers upregulate MHC II and impede ovarian cancer tumor growth. Oncotarget 2017, 8, 44159–44170. [Google Scholar] [CrossRef]
- Hake, S.B.; Masternak, K.; Kammerbauer, C.; Janzen, C.; Reith, W.; Steimle, V. CIITA leucine-rich repeats control nuclear localization, In Vivo recruitment to the major histocompatibility complex (MHC) class II enhanceosome, and MHC class II gene transactivation. Mol. Cell. Biol. 2000, 20, 7716–7725. [Google Scholar] [CrossRef]
- Ting, J.P.; Trowsdale, J. Genetic control of MHC class II expression. Cell 2002, 109, S21–S33. [Google Scholar] [CrossRef]
- Ducellier, S.; Demeules, M.; Letribot, B.; Gaetani, M.; Michaudel, C.; Sokol, H.; Hamze, A.; Alami, M.; Nascimento, M.; Apcher, S. Dual molecule targeting HDAC6 leads to intratumoral CD4+ cytotoxic lymphocytes recruitment through MHC-II upregulation on lung cancer cells. J. Immunother. Cancer 2024, 12, e007588. [Google Scholar] [CrossRef] [PubMed]
- Pitter, M.R.; Kryczek, I.; Zhang, H.; Nagarsheth, N.; Xia, H.; Wu, Z.; Tian, Y.; Okla, K.; Liao, P.; Wang, W.; et al. PAD4 controls tumor immunity via restraining the MHC class II machinery in macrophages. Cell Rep. 2024, 43, 113942. [Google Scholar] [CrossRef]
- Holling, T.M.; Schooten, E.; van Den Elsen, P.J. Function and regulation of MHC class II molecules in T-lymphocytes: Of mice and men. Hum. Immunol. 2004, 65, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; De Palma, R.; Altucci, L. HDAC inhibitors as epigenetic regulators for cancer immunotherapy. Int. J. Biochem. Cell Biol. 2018, 98, 65–74. [Google Scholar] [CrossRef]
- Alfaro, C.; Sanmamed, M.F.; Rodriguez-Ruiz, M.E.; Teijeira, A.; Onate, C.; Gonzalez, A.; Ponz, M.; Schalper, K.A.; Perez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef]
- Sun, Y.; Ai, J.Z.; Jin, X.; Liu, L.R.; Lin, T.H.; Xu, H.; Wei, Q.; Yang, L. IL-8 protects prostate cancer cells from GSK-3beta-induced oxidative stress by activating the mTOR signaling pathway. Prostate 2019, 79, 1180–1190. [Google Scholar] [CrossRef]
- Dunbar, A.J.; Kim, D.; Lu, M.; Farina, M.; Bowman, R.L.; Yang, J.L.; Park, Y.; Karzai, A.; Xiao, W.; Zaroogian, Z.; et al. CXCL8/CXCR2 signaling mediates bone marrow fibrosis and is a therapeutic target in myelofibrosis. Blood 2023, 141, 2508–2519. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.J.; Li, Y.B.; Yang, L.X.; Cheng, H.J.; Liu, X.; Chen, H. Roles of the CXCL8-CXCR1/2 Axis in the Tumor Microenvironment and Immunotherapy. Molecules 2021, 27, 137. [Google Scholar] [CrossRef]
- Yuen, K.C.; Liu, L.F.; Gupta, V.; Madireddi, S.; Keerthivasan, S.; Li, C.; Rishipathak, D.; Williams, P.; Kadel, E.E., 3rd; Koeppen, H.; et al. Author Correction: High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 2021, 27, 560. [Google Scholar] [CrossRef]
- Schalper, K.A.; Carleton, M.; Zhou, M.; Chen, T.; Feng, Y.; Huang, S.P.; Walsh, A.M.; Baxi, V.; Pandya, D.; Baradet, T.; et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 2020, 26, 688–692. [Google Scholar] [CrossRef]
- Wang, H.C.; Haung, L.Y.; Wang, C.J.; Chao, Y.J.; Hou, Y.C.; Yen, C.J.; Shan, Y.S. Tumor-associated macrophages promote resistance of hepatocellular carcinoma cells against sorafenib by activating CXCR2 signaling. J. Biomed. Sci. 2022, 29, 99. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.W.; Zhang, L.; Cai, X.R.; Wang, X.; She, F.F.; Chen, Y.L. Author Correction: IL-8 is a novel prometastatic chemokine in intrahepatic cholangiocarcinoma that induces CXCR2-PI3K/AKT signaling upon CD97 activation. Sci. Rep. 2024, 14, 8478. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Zhou, Z.J.; Xia, G.K.; Zhang, X.H.; Wei, Z.W.; Zhu, J.T.; Yu, J.; Chen, W.; He, Y.; Schwarz, R.E.; et al. A positive crosstalk between CXCR4 and CXCR2 promotes gastric cancer metastasis. Oncogene 2017, 36, 5122–5133. [Google Scholar] [CrossRef]
- Leslie, J.; Mackey, J.B.G.; Jamieson, T.; Ramon-Gil, E.; Drake, T.M.; Fercoq, F.; Clark, W.; Gilroy, K.; Hedley, A.; Nixon, C.; et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut 2022, 71, 2093–2106. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hao, S.; Gao, M.; Liu, J.; Xu, X.; Huang, J.; Cheng, G.; Yang, H. HDAC3 Inhibition Promotes Antitumor Immunity by Enhancing CXCL10-Mediated Chemotaxis and Recruiting of Immune Cells. Cancer Immunol. Res. 2023, 11, 657–673. [Google Scholar] [CrossRef]
- Guo, C.; Li, J.; Steinauer, N.; Wong, M.; Wu, B.; Dickson, A.; Kalkum, M.; Zhang, J. Histone deacetylase 3 preferentially binds and collaborates with the transcription factor RUNX1 to repress AML1-ETO-dependent transcription in t(8;21) AML. J. Biol. Chem. 2020, 295, 4212–4223. [Google Scholar] [CrossRef]
- Yang, W.; Feng, Y.; Zhou, J.; Cheung, O.K.; Cao, J.; Wang, J.; Tang, W.; Tu, Y.; Xu, L.; Wu, F.; et al. A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci. Transl. Med. 2021, 13, eaaz6804. [Google Scholar] [CrossRef]
- van der Oost, J.; Patinios, C. The genome editing revolution. Trends. Biotechnol. 2023, 41, 396–409. [Google Scholar] [CrossRef]
- Li, J.; Zhao, D.; Zhang, T.; Xiong, H.; Hu, M.; Liu, H.; Zhao, F.; Sun, X.; Fan, P.; Qian, Y.; et al. Precise large-fragment deletions in mammalian cells and mice generated by dCas9-controlled CRISPR/Cas3. Sci. Adv. 2024, 10, eadk8052. [Google Scholar] [CrossRef]
- Wang, S.W.; Gao, C.; Zheng, Y.M.; Yi, L.; Lu, J.C.; Huang, X.Y.; Cai, J.B.; Zhang, P.F.; Cui, Y.H.; Ke, A.W. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol. Cancer 2022, 21, 57. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.; Huang, X. Genome modification by CRISPR/Cas9. Febs. J. 2014, 281, 5186–5193. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, L.; Lin, G.; Hu, Y.; Jiao, Y.; Wang, Y.; Liu, J.; Yang, S.; Yao, S. HDAC inhibitors improve CRISPR-Cas9 mediated prime editing and base editing. Mol. Ther. Nucleic. Acids 2022, 29, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.Y.; Zhao, Y.T.; Lamonica, J.M.; Zhou, Z. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun. 2017, 8, 15315. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed]
- Mandegar, M.A.; Huebsch, N.; Frolov, E.B.; Shin, E.; Truong, A.; Olvera, M.P.; Chan, A.H.; Miyaoka, Y.; Holmes, K.; Spencer, C.I.; et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 2016, 18, 541–553. [Google Scholar] [CrossRef]
- Li, C.; Brant, E.; Budak, H.; Zhang, B. CRISPR/Cas: A Nobel Prize award-winning precise genome editing technology for gene therapy and crop improvement. J. Zhejiang Univ. Sci. B 2021, 22, 253–284. [Google Scholar] [CrossRef]
- Chen, P.J.; Liu, D.R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 2023, 24, 161–177. [Google Scholar] [CrossRef]
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A.V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A.; et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022, 40, 402–410. [Google Scholar] [CrossRef]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 2021, 184, 5635–5652.e5629. [Google Scholar] [CrossRef]
- Zhang, H.X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Shukla, S.; Yang, C.; Zhang, M.; Fatma, Z.; Lingamaneni, M.; Abesteh, S.; Lane, S.T.; Xiong, X.; Wang, Y.; et al. TALEN outperforms Cas9 in editing heterochromatin target sites. Nat. Commun. 2021, 12, 606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yao, J.; Ajmal, I.; Farooq, M.A.; Jiang, W. shRNA-mediated gene silencing of HDAC11 empowers CAR-T cells against prostate cancer. Front. Immunol. 2024, 15, 1369406. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Yang, H.; Patel, D.J. Structures, mechanisms and applications of RNA-centric CRISPR-Cas13. Nat. Chem. Biol. 2024, 20, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, A.; Kawamoto, N.; Chow, S.Y.A.; Tsuiji, H.; Ikeuchi, Y.; Shichino, Y.; Iwasaki, S. dCas13-mediated translational repression for accurate gene silencing in mammalian cells. Nat. Commun. 2024, 15, 2205. [Google Scholar] [CrossRef]
- Ma, E.; Chen, K.; Shi, H.; Stahl, E.C.; Adler, B.; Trinidad, M.; Liu, J.; Zhou, K.; Ye, J.; Doudna, J.A. Improved genome editing by an engineered CRISPR-Cas12a. Nucleic Acids Res. 2022, 50, 12689–12701. [Google Scholar] [CrossRef]
- Kang, W.; Liu, L.; Yu, P.; Zhang, T.; Lei, C.; Nie, Z. A switchable Cas12a enabling CRISPR-based direct histone deacetylase activity detection. Biosens. Bioelectron. 2022, 213, 114468. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Kumar, S.; Lonial, S.; Mateos, M.V. Smoldering multiple myeloma current treatment algorithms. Blood Cancer J. 2022, 12, 129. [Google Scholar] [CrossRef]
- Nooka, A.K.; Wang, M.; Yee, A.J.; Kaufman, J.L.; Bae, J.; Peterkin, D.; Richardson, P.G.; Raje, N.S. Assessment of Safety and Immunogenicity of PVX-410 Vaccine With or Without Lenalidomide in Patients With Smoldering Multiple Myeloma. JAMA Oncol. 2018, 4, e183267. [Google Scholar] [CrossRef]
- Laino, A.S.; Betts, B.C.; Veerapathran, A.; Dolgalev, I.; Sarnaik, A.; Quayle, S.N.; Jones, S.S.; Weber, J.S.; Woods, D.M. HDAC6 selective inhibition of melanoma patient T-cells augments anti-tumor characteristics. J. Immunother. Cancer 2019, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hong, J.A.; Kunst, T.F.; Bond, C.D.; Kenney, C.M.; Warga, C.L.; Yeray, J.; Lee, M.J.; Yuno, A.; Lee, S.; et al. Randomized phase II trial of a first-in-human cancer cell lysate vaccine in patients with thoracic malignancies. Transl. Lung Cancer Res. 2021, 10, 3079–3092. [Google Scholar] [CrossRef] [PubMed]
- Truong, A.S.; Zhou, M.; Krishnan, B.; Utsumi, T.; Manocha, U.; Stewart, K.G.; Beck, W.; Rose, T.L.; Milowsky, M.I.; He, X.; et al. Entinostat induces antitumor immune responses through immune editing of tumor neoantigens. J. Clin. Invest. 2021, 131, e138560. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, P.J.; Lee-Wisdom, K.; Donahue, R.N.; Madan, R.A.; Karzai, F.; Schwab, A.; Palena, C.; Jochems, C.; Floudas, C.; Strauss, J.; et al. Phase 1 open-label trial of intravenous administration of MVA-BN-brachyury-TRICOM vaccine in patients with advanced cancer. J. Immunother. Cancer 2021, 9, e003238. [Google Scholar] [CrossRef]
- Sidiropoulos, D.N.; Rafie, C.I.; Jang, J.K.; Castanon, S.; Baugh, A.G.; Gonzalez, E.; Christmas, B.J.; Narumi, V.H.; Davis-Marcisak, E.F.; Sharma, G.; et al. Entinostat Decreases Immune Suppression to Promote Antitumor Responses in a HER2+ Breast Tumor Microenvironment. Cancer Immunol. Res. 2022, 10, 656–669. [Google Scholar] [CrossRef]
- Yi, M.; Zheng, X.; Niu, M.; Zhu, S.; Ge, H.; Wu, K. Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Mol. Cancer 2022, 21, 28. [Google Scholar] [CrossRef]
- Gao, Y.; Nihira, N.T.; Bu, X.; Chu, C.; Zhang, J.; Kolodziejczyk, A.; Fan, Y.; Chan, N.T.; Ma, L.; Liu, J.; et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol. 2020, 22, 1064–1075. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Jaccard, A.; Ho, P.C. The hidden side of PD-L1. Nat. Cell Biol. 2020, 22, 1031–1032. [Google Scholar] [CrossRef]
- Xu, P.; Xiong, W.; Lin, Y.; Fan, L.; Pan, H.; Li, Y. Histone deacetylase 2 knockout suppresses immune escape of triple-negative breast cancer cells via downregulating PD-L1 expression. Cell Death Dis. 2021, 12, 779. [Google Scholar] [CrossRef]
- Du, W.; Zhu, J.; Zeng, Y.; Liu, T.; Zhang, Y.; Cai, T.; Fu, Y.; Zhang, W.; Zhang, R.; Liu, Z.; et al. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Differ. 2021, 28, 1284–1300. [Google Scholar] [CrossRef]
- Yu, J.; Zhuang, A.; Gu, X.; Hua, Y.; Yang, L.; Ge, S.; Ruan, J.; Chai, P.; Jia, R.; Fan, X. Nuclear PD-L1 promotes EGR1-mediated angiogenesis and accelerates tumorigenesis. Cell Discov. 2023, 9, 33. [Google Scholar] [CrossRef]
- Ji, Z.; Cheng, Y.; Yuan, P.; Dang, X.; Guo, X.; Wang, W. Panax notoginseng stimulates alkaline phosphatase activity, collagen synthesis, and mineralization in osteoblastic MC3T3-E1 cells. Vitr. Cell. Dev. Biol. Anim. 2015, 51, 950–957. [Google Scholar] [CrossRef]
- Li, T.; Xing, R.; Xiang, L.; Liu, H.; Wei, J.; Lu, J.; Li, T.; Yang, J. MIIP downregulates PD-L1 expression through HDAC6 in cutaneous melanoma. Holist. Integr. Oncol. 2024, 3, 25. [Google Scholar] [CrossRef]
- Que, Y.; Zhang, X.L.; Liu, Z.X.; Zhao, J.J.; Pan, Q.Z.; Wen, X.Z.; Xiao, W.; Xu, B.S.; Hong, D.C.; Guo, T.H.; et al. Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J. Immunother. Cancer 2021, 9, e001696. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]
- Peng, H.; Li, L.; Zuo, C.; Chen, M.Y.; Zhang, X.; Myers, N.B.; Hogg, G.D.; DeNardo, D.G.; Goedegebuure, S.P.; Hawkins, W.G.; et al. Combination TIGIT/PD-1 blockade enhances the efficacy of neoantigen vaccines in a model of pancreatic cancer. Front. Immunol. 2022, 13, 1039226. [Google Scholar] [CrossRef]
- Jin, C.; Zhang, Y.; Li, B.; Gao, T.; Wang, B.; Hua, P. Robust anti-tumor immunity through the integration of targeted lipid nanoparticle-based mRNA nanovaccines with PD-1/PD-L1 blockade. Mater Today Bio. 2024, 27, 101136. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar]
- Li, C.; Lee, A.; Grigoryan, L.; Arunachalam, P.S.; Scott, M.K.D.; Trisal, M.; Wimmers, F.; Sanyal, M.; Weidenbacher, P.A.; Feng, Y.; et al. Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine. Nat. Immunol. 2022, 23, 543–555. [Google Scholar] [CrossRef]
- Shah, D.; Soper, B.; Shopland, L. Cytokine release syndrome and cancer immunotherapies - historical challenges and promising futures. Front. Immunol. 2023, 14, 1190379. [Google Scholar] [CrossRef]
- Cobb, D.A.; Lee, D.W. Cytokine Release Syndrome Biology and Management. Cancer J. 2021, 27, 119–125. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef]
- Vink, P.; Delgado Mingorance, I.; Maximiano Alonso, C.; Rubio-Viqueira, B.; Jung, K.H.; Rodriguez Moreno, J.F.; Grande, E.; Marrupe Gonzalez, D.; Lowndes, S.; Puente, J.; et al. Immunogenicity and safety of the adjuvanted recombinant zoster vaccine in patients with solid tumors, vaccinated before or during chemotherapy: A randomized trial. Cancer 2019, 125, 1301–1312. [Google Scholar] [CrossRef]
- Increasing the dose intensity of chemotherapy by more frequent administration or sequential scheduling: A patient-level meta-analysis of 37 298 women with early breast cancer in 26 randomised trials. Lancet 2019, 393, 1440–1452. [CrossRef]
- Kim, K.; Chie, E.K.; Han, W.; Noh, D.Y.; Oh, D.Y.; Im, S.A.; Kim, T.Y.; Bang, Y.J.; Ha, S.W. Concurrent versus sequential administration of CMF chemotherapy and radiotherapy. Tumori J. 2018, 97, 280–285. [Google Scholar] [CrossRef]
- Maruyama, M.; Torii, R.; Matsui, H.; Hayashi, H.; Ogawara, K.I.; Higaki, K. Repeated sequential administration of pegylated emulsion of SU5416 and liposomal paclitaxel enhances anti-tumor effect in 4T1 breast cancer-bearing mice. Eur. J. Pharm. Biopharm. 2025, 209, 114663. [Google Scholar] [CrossRef]
- Poggio, F.; Ceppi, M.; Lambertini, M.; Bruzzi, P.; Ugolini, D.; Bighin, C.; Levaggi, A.; Giraudi, S.; D’Alonzo, A.; Vaglica, M.; et al. Concurrent versus sequential adjuvant chemo-endocrine therapy in hormone-receptor positive early stage breast cancer patients: A systematic review and meta-analysis. Breast 2017, 33, 104–108. [Google Scholar] [CrossRef]
- Wang, H.; Fu, B.B.; Gale, R.P.; Liang, Y. NK-/T-cell lymphomas. Leukemia 2021, 35, 2460–2468. [Google Scholar] [CrossRef]
- Angelika Ihle, M.; Merkelbach-Bruse, S.; Hartmann, W.; Bauer, S.; Ratner, N.; Sonobe, H.; Nishio, J.; Larsson, O.; Åman, P.; Pedeutour, F.; et al. HR23b expression is a potential predictive biomarker for HDAC inhibitor treatment in mesenchymal tumours and is associated with response to vorinostat. J. Pathol. Clin. Res. 2016, 2, 59–71. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, G.; Tang, T.; Liang, T. Identification of tumor antigens and immune subtypes of pancreatic adenocarcinoma for mRNA vaccine development. Mol. Cancer 2021, 20, 44. [Google Scholar] [CrossRef]
- Masopust, D.; Schenkel, J.M. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 2013, 13, 309–320. [Google Scholar] [CrossRef]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]
- Movassaghian, S.; Merkel, O.M.; Torchilin, V.P. Applications of polymer micelles for imaging and drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 691–707. [Google Scholar] [CrossRef]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef]
- Liang, Y.; Duan, L.; Lu, J.; Xia, J. Engineering exosomes for targeted drug delivery. Theranostics 2021, 11, 3183–3195. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Dehshahri, A.; Sagar Madamsetty, V.; Zahmatkeshan, M.; Tavakol, S.; Makvandi, P.; Khorsandi, D.; Pardakhty, A.; Ashrafizadeh, M.; Ghasemipour Afshar, E.; et al. In Vivo gene delivery mediated by non-viral vectors for cancer therapy. J. Control. Release 2020, 325, 249–275. [Google Scholar] [CrossRef]
- Yang, A.; Bai, Y.; Dong, X.; Ma, T.; Zhu, D.; Mei, L.; Lv, F. Hydrogel/nanoadjuvant-mediated combined cell vaccines for cancer immunotherapy. Acta Biomater. 2021, 133, 257–267. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Emerging HDACi Targets | Full Name | Potential Mechanism | Cancer Type |
|---|---|---|---|
| LTR12 | Long terminal repeat 12 | Activate multiple cryptic transcription start sites of LTR12 elements | Prostate cancer, Liver cancer |
| HERV | Human endogenous retroviruses | Directly influence HERV sites in T cells, and the upregulated ERVs can be recognized by Pattern Recognition Receptors (PRRs) | Colorectal cancer, Triple-negative breast cancer |
| TAP2 | Transporter 2 | Upregulate the expression of TAP2 | Melanoma |
| LMP2 | Latent Membrane Protein 2 | Upregulate the expression of LMP2 | Melanoma |
| LMP7 | Latent Membrane Protein 7 | Upregulate the expression of LMP7 | Melanoma |
| MHC class I | Major Histocompatibility Complex Class I | Induce STAT1 and Smad2/3 phosphorylation in NSCLC cells, leading to increased MHC class I expression | Melanoma |
| Trial Number | Launch | Phase | Study Status | HDACi (Targets) | INT | Other Combined Agents | Cancer Type | Patient Numbers | Endpoints | Preliminary Results |
|---|---|---|---|---|---|---|---|---|---|---|
| NCT02886065 | 2017 | Ib | Active | Citarinostat | PVX-410 | - | Smoldering Multiple Myeloma | 19 | Safety and Tolerability of the Vaccine | No Results Posted |
| NCT05898828 | 2024 | I/II | Withdrawn | Entinostat | H1299 cell lysate vaccine | Nivolumab, Montanide(R) ISA-51 VG | Advanced esophageal Cancer | 0 | Safe Dose/Frequency of Immunologic Responses | No Results Posted |
| NCT04296942 | 2021 | I | Terminated | Entinostat | BN-Brachyury vaccine | M7824, T-DM1 | Advanced Stage Breast Cancer | 1 | Overall Response | Progression/Recurrence Time is 5 Months and 17 Days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, R.; Zhou, H.; Peng, B.; Yu, S.; Zhu, J.; Chen, J. Synergistic Integration of HDAC Inhibitors and Individualized Neoantigen Therapy (INT): A Next-Generation Combinatorial Approach for Cancer Immunotherapy. Vaccines 2025, 13, 550. https://doi.org/10.3390/vaccines13060550
Han R, Zhou H, Peng B, Yu S, Zhu J, Chen J. Synergistic Integration of HDAC Inhibitors and Individualized Neoantigen Therapy (INT): A Next-Generation Combinatorial Approach for Cancer Immunotherapy. Vaccines. 2025; 13(6):550. https://doi.org/10.3390/vaccines13060550
Chicago/Turabian StyleHan, Rui, Huiling Zhou, Baoqing Peng, Shasha Yu, Jiajie Zhu, and Jiaojiao Chen. 2025. "Synergistic Integration of HDAC Inhibitors and Individualized Neoantigen Therapy (INT): A Next-Generation Combinatorial Approach for Cancer Immunotherapy" Vaccines 13, no. 6: 550. https://doi.org/10.3390/vaccines13060550
APA StyleHan, R., Zhou, H., Peng, B., Yu, S., Zhu, J., & Chen, J. (2025). Synergistic Integration of HDAC Inhibitors and Individualized Neoantigen Therapy (INT): A Next-Generation Combinatorial Approach for Cancer Immunotherapy. Vaccines, 13(6), 550. https://doi.org/10.3390/vaccines13060550