Abstract
Alzheimer’s disease is a chronic and irreversible pathological process that has become the most prevalent neurodegenerative disease. Currently, it is considered a multifactorial disease where oxidative stress and chronic neuroinflammation play a crucial role in its onset and development. Its characteristic neuronal loss has been related to the formation of neurofibrillary tangles mainly composed by hyperphosphorylated tau protein. Hyperphosphorylation of tau protein is related to the over-activity of GSK-3β, a kinase that participates in several pathological mechanisms including neuroinflammation. Neuronal loss is also related to cytosolic Ca2+ homeostasis dysregulation that triggers apoptosis and free radicals production, contributing to oxidative damage and, finally, neuronal death. Under these premises, we have obtained a new family of 4,7-dihydro-2H-pyrazolo[3–b]pyridines as multitarget directed ligands showing potent antioxidant properties and able to scavenge both oxygen and nitrogen radical species, and also, with anti-inflammatory properties. Further characterization has demonstrated their capacity to inhibit GSK-3β and to block L-type voltage dependent calcium channels. Novel derivatives have also demonstrated an interesting neuroprotective profile on in vitro models of neurodegeneration. Finally, compound 4g revokes cellular death induced by tau hyperphosphorylation in hippocampal slices by blocking reactive oxygen species (ROS) production. In conclusion, the multitarget profile exhibited by these compounds is a novel therapeutic strategy of potential interest in the search of novel treatments for Alzheimer’s disease.
1. Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disease characterized by a progressive memory loss and cognitive dysfunction. Currently, 46.8 million people suffer AD and its prevalence is forecasted to increase to 152 million by 2050 [1]. The World Health Organization considers AD as the pandemic of this century, being a major health and socioeconomic problem due to its chronicity and the tremendous burden that it imposes on caregivers. Furthermore, the only available treatments are symptomatic and they are not able to halt the progression of the disease. Thus, there is an urgent medical need to find an effective drug able to delay or stop the advance of the disease.
AD pathophysiology is characterized by a progressive neuronal loss, mainly at the cholinergic system, and two major hallmarks, intracellular neurofibrillary tangles (NFTs) [2], mainly composed of hyperphosphorylated tau protein (τ), and extracellular plaques constituted by amyloid-β peptide (Aβ) [3]. During the past decades, most of the drug development programs have focused on targeting Aβ, which is consistent with the genetic mutations observed in hereditary cases of AD. However, 95% of the cases are sporadic, indicating different initial causes of the disease. The complex etiology of AD and the mechanistic study of sporadic AD cases have pointed to several pathological pathways involved in the onset of the disease, including mitochondrial failure, oxidative stress (OS), chronic neuroinflammation and proteostasis deregulation. More importantly, these pathological modifications are thought to be initiated almost twenty years earlier than the appearance of the first symptoms [4].
Oxidative stress (OS) is defined as the imbalance between pro-oxidant species (reactive oxygen and nitrogen species, ROS and RNS) and antioxidant defences. Complexes I and III of the mitochondrial electron transport chain are the main ROS generators. Under normal conditions, ROS are key signalling mediators in redox homeostasis, cell death, cell senescence, cellular proliferation, synaptic plasticity and cognitive enhancement, and they can trigger an immune response [5,6]. Under pathological conditions, free radicals can oxidize lipids, DNA and proteins leading to DNA mutations, protein loss of function and, finally, cell death [7]. During aging, the main risk factor of AD, there is a chronic OS imbalance, energy deprivation and decline of antioxidant defense systems that, under certain conditions, have been pointed as potential triggers of the disease [5]. In this line, mitochondrial dysfunction has been described in AD brains [8], together with extensive OS damage and decreased activity of mitochondrial complex IV, as demonstrated in postmortem brains of AD patients [9].
Excessive ROS/RNS production during AD development is thought to participate in the accumulation and aggregation processes of Aβ [10]. Moreover, the Aβ peptide induces OS by increasing ROS production, mitochondrial failure and energy depletion before the appearance of Aβ plaques [11], due to its ability to inhibit complex IV [12]. Recent studies have also demonstrated the capacity of Aβ peptides to impair the import of necessary proteins into the mitochondria through coaggregation, inducing mitochondrial failure and increasing ROS production [13]. Similarly, OS plays a key role in tau hyperphosphorylation through activation of p38 and inactivation of calcineurin (a Ser/Thr phosphatase involved in tau de-phosphorylation) by increasing its natural repressor RCAN1 [14]. Increased levels of RCAN1 also induce the expression of glycogen synthase kinase 3β (GSK-3β) the main kinase related to tau hyperphosphorylation in AD [15], thereby accelerating the formation of hyperphosphorylated tau. Thus, OS induces the aberrant processing of proteins that, in turn, induce mitochondrial damage and additional OS, generating a feedback loop that accelerates neuronal death [16,17]. Recently, a good correlation between the accumulation of NFTs [18] and the cognitive decline associated with AD [19] was demonstrated, highlighting the relevance of tau hyperphosphorylation in this process. GSK-3β is also related to Aβ deposition [20], OS, neuroinflammation [21] and apoptosis, participating in the neuronal death observed in AD [22]. Furthermore, GSK-3β knock-down or inhibition avoids Aβ-induced neurodegeneration [23]. GSK-3β related pathological links in AD include the negative regulation of the most important antioxidant natural defense of cells, the Nrf2-ARE pathway [24]. GSK-3β directly phosphorylates Nrf2, increasing the affinity of the Cul1 adaptor β-TrCP that induces Nrf2 proteasomal degradation [25], thus inhibiting the phase II antioxidant response and contributing to increase OS status. Therefore, renewed interest in GSK-3β as a target has emerged in the context of the multitarget approach by combining it with other emerging targets [26,27,28,29].
Increased OS at early disease stages also initiates the immune response, mainly mediated by glial cells (microglia and astrocytes), by directly acting on scavenger receptors or by the formation of oxidized products that are recognized by other receptors. Activated glia increases the expression of pro-inflammatory enzymes, such as inducible nitric oxide synthase (iNOS) and NADPH oxidase (NOX), among others, increasing the production of RNS and ROS. Nitric oxide (NO) produced by iNOS readily reacts with ROS to produce peroxynitrite, an intermediate that is able to react with Aβ at Tyr10 to increase its aggregation capacity and neurotoxicity [30]. Importantly, nitrated Aβ is the main component found in the core of senile plaques found in the brain of AD patients [31]. Furthermore, Aβ activates the cell surface receptor CD36, inducing an acute immune response that elicits the toll-like receptor 4–6 (TLR4–TLR6) heterodimer that activates the nuclear factor-κB (NF-κB) [32]. In addition, CD36 stimulates Aβ aggregation multiplying its activating capacity [33]. Uncontrolled glial activation induces the excessive expression of cytokines, such as tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β) or IL-18, and chemokines and adhesion molecules to recruit peripheral immune cells [34]. In addition, IL-1β induces neurite destruction and increases P38-MAPK (P38 mitogen-activated protein kinase) kinases [35] and GSK-3β activity, contributing to tau hyperphosphorylation and the appearance of NFTs [36]. Therefore, there is an intense crosstalk between OS and neuroinflammation, starting at the very initial phases of the disease where neither Aβ nor NFT pathologies are initiated [37].
Among the multiple pathological pathways that operate in AD, Ca2+ dysregulation has been widely related to the neuronal loss [38,39]. Cytosolic Ca2+ ([Ca2+]c) overload accelerates Aβ production [40], activates GSK-3β [41], increases OS [42] and triggers cellular apoptosis [43]. In turn, Aβ induces [Ca2+]c overload leading to ROS production and neuronal death [44]. Recently, it was further demonstrated that selective Ca2+ entry through l-type voltage-dependent Ca2+ channels (VDCC) increases ROS production and neuronal death [42]. Accordingly, OS toxicity induced by the l-type VDCC Ca2+ current was successfully blocked by isradipine, a selective l-type VDCC antagonist [42,45]. In consequence, l-type VDCC antagonists, such as 1,4-dihydropyridines (1,4-DHPs), have been intensively pursued as potential agents to decrease [Ca2+]c overload, Aβ production and NFT formation, a combination of targets that may stop the progression of the disease [46].
Based on the complexity and connectivity of the different pathological pathways involved in AD onset and development, it is described as a multifactorial disease [47] whose treatment should be addressed by acting on several targets simultaneously. These reports have boosted the development of novel small molecules designed to act on several targets at the same time, the so-called “multitarget strategy”, to find a neuroprotective drug with disease modifying capacity. Considering the implication of exacerbated OS and neuroinflammation in the onset and development of AD, we report here the synthesis and pharmacological evaluation of a new family of 4,7-dihydro-2H-pyrazolo[3,4-b]pyridine derivatives, based on the 1,4-DHP core. As mentioned above, 1,4-DHPs are well-characterized selective l-type VDCC blockers with antioxidant and neuroprotective properties [48]. Based on previous results, we have included a pyrazole ring moiety in the structure of new compounds in order to implement GSK-3β inhibitory capacity to decrease tau hyperphosphorylation and, given the relationship of GSK-3β and OS with neuroinflammation, the potential inclusion of anti-inflammatory properties. The obtained derivatives showed potent free radical scavenger capacity, exerted an interesting anti-inflammatory effect, GSK-3β inhibitory capacity at low micromolar range, and moderate VDCC antagonist capacity. Additionally, these compounds showed potent neuroprotective properties against toxicity induced by OS, tau hyperphosphorylation and [Ca2+]c overload. Finally, a selected compound reduced cell death in a model of hippocampal slices treated with okadaic acid (OA) by reducing OS.
2. Materials and Methods
2.1. Synthesis
4,7-Dihydro-2H-pyrazolo[3,4-b]pyridine derivatives were prepared and purified as described in the Supplementary Material.
2.2. Oxygen Radical Absorbance Capacity (ORAC) Assay
ORAC test developed by Cao et al. [49] and modified by Ou et al. [50] was carried out to evaluate the oxygen free radical scavenger capacity of the compounds. (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) (1, 2, 4, 6 and 8 µM) was used as reference compound and melatonin as positive control. Compounds 4a–l were diluted (0.03, 0.1, 0.3, 1, 3 and 10 µM) in phosphate buffered saline (PBS) buffer (10 mM, pH 7.4) at 37 °C and placed a 96-well black microplate (COSTAR 3904). 150 µL of fluorescein (70 nM, final concentration), 25 µL of PBS buffer for blank, 25 µL of Trolox solution for standard and 25 µL of melatonin or compound were added to each well. A fluorescence measurement was done first to determine the basal signal. Then, 25 µL of 2,2′-azobis-amidinopropane dihydrochloride (AAPH) (12 mM, final concentration) were quickly added, since the reaction starts immediately after addition. All samples were carried out in duplicate in three different experiments. Fluorescence was recorded every min for 90 min at 37 °C to obtain the area under the fluorescence decay curve (fluorescence vs time) (AUC) in a FLUOstar Optima plate reader (BMG Labtech, Offenburg, Germany) with 485 nm excitation and 520 nm emission filters. After blank correction and plotting the net AUC vs antioxidant concentration, linear regressions were calculated for all the samples. Final results were expressed in Trolox equivalents (T.eq.), where the ORAC value of Trolox was taken as 1.0.
2.3. DPPH Reduction Assay
Experimental conditions were modified from a previously described procedure [51]. Briefly, compounds 4a–l (150 µL) at the desired concentration (1, 10, 60 and 100 µM) in methanol/water (80/20) were added to a solution of 2,2-diphynyl-1-picrylhydrazyl (DPPH) in methanol/water (80/20) (150 µL, 100 µM), in a clear bottom 96-well plate. The final solution was incubated during 1 h in the dark. Then, DPPH absorbance of blank (methanol/water), control (DPPH 100 µM) and compounds plus DPPH were measured at 540 nm in a SPECTROstar Nano plate-reader (BMG Labtech, Ortenberg, Germany) in duplicate. Ascorbic acid and melatonin were used as positive controls. Results are expressed as a percentage of DPPH reduction by subtracting blank absorbance (Absblank) to sample absorbance (Abssample) and corrected by control absorbance, (Abscontrol, DPPH alone, as expressed below:
% DPPH reduction = [100 − ((Abssample − Absblank) × 100/Abscontrol)]
2.4. Mixed Primary Glial Culture
Experimental procedures performed following the Guide for the Care and Use of Laboratory Animals and were previously approved by the Ethics Committee of the Autonomous University of Madrid, Spain (PROEX: 252/16). These procedures were in agreement with the European Guidelines for the use and care of animals for research in accordance with the European Union Directive of 22 September 2010 (2010/63/UE) and with Spanish Royal Decree of 1 February 2013 (53/2013). Mixed glial cultures were obtained from cerebral cortex of 2–5-days-old Sprague-Dawley rats. After the removal of blood vessels and meninges, the forebrains were dissociated mechanically in DMEM/F12 medium. Then, cells were plated (3 × 105 cells/well) in DMEM/F12 medium with 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin (10,000 units), at 37 °C and in a 5% CO2-supplemented air atmosphere. After 5 days, medium was substituted by DMEM/F12 medium with 10% FBS. Cells were cultured for 7–10 days before treatment.
2.5. Measurement of the Anti-inflammatory Capacity Measured as Nitrite Production Reduction in Mixed Primary Glial Cultures
Cells were pre-incubated with increasing concentrations (0.1, 1, 3, 10 and 30 μM) of compounds for 24 h. Then, treatments were removed and cells were incubated with Lipopolysaccharide (LPS, O26:B6 serotype, 1 μg/mL) in presence of each compound at the desired concentration. Nitrite production was assessed 18 h later by modified Griess assay. Briefly, samples (150 µL) were mixed with 4,4′-diamino-diphenylsulfone (75 µL) and n-(1-naphthyl)ethylenediamine (75 µL), and the mixture was incubated at room temperature for 5 min. Light absorption was measured at 550 nm in a microplate reader (Labtech, Offenburg, Germany). All data were normalized to basal nitrite production, considering this value as 100% of nitrite production. EC50 values were calculated from dose-response curves represented as percentage of nitrite production reduction induced by the different concentrations of each compound.
2.6. GSK-3β Inhibitory Capacity
The method of Baki et al. [52], with some modifications, was followed to analyze the ability of the compounds to inhibit GSK-3β. Tested compounds were dissolved in assay buffer, containing 40 mM Tris (pH 7.5), 20 mM MgCl2, 0.1 mg/mL bovine serum albumin (BSA) and 50 µM dithiothreitol (DTT), to achieve final reaction concentrations of 0.1, 1, 10 and 30 µM. Compound SB216763 was used as reference compound. The GSK-3β inhibition assay was performed in white 96-well plates, in duplicates. Firstly, 10 µL of the compounds at the desired concentration were added to each well, followed by 10 µL of enzyme (10 ng). After 30 min of incubation, 20 µL of a mixture of adenosine triphosphate (ATP) and GSK-3β-peptide substrate were added to each well. After a 60 min incubation at 30 °C, the enzymatic reaction was stopped, and the remaining ATP concentration was measured by adding 40 µL/well of Kinase-Glo reagent. IC50 values were calculated by non-linear regression analysis of individual concentration-response curves using GraphPad Prism software (San Diego, CA, USA). Ki values were calculated using the Chen-Prusoff equation simplified:
Ki = IC50/(1 + (S/Km))
Being S the substrate concentration (ATP) in the experiment, and Km the Michaelis-Menten constant of the substrate for the enzyme (Km = 2.345 μM for ATP).
2.7. SH-SY5Y Neuroblastoma Cell Line Culture
SH-SY5Y human neuroblastoma cells (obtained from ATCC, Manassas, VA, United States, CRL-2266) were cultured according to supplier directions in a 1:1 mixture of F12 (Ham 12) and Eagle’s Minimum Essential Medium (MEM), supplemented with 15% non-essential amino acids, 10% heat-inactivated FBS, 0.5 mM sodium pyruvate, 24 mM NaHCO3, 100 μg/mL streptomycin and 100 units/mL penicillin. Cells were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2. For experimental procedures, cells were cultured in 96-well plates (6 × 104 cells per well). Treatments were carried out in 1% FBS medium unless other concentration was specified. Cells were used from 4 up to 13 passages.
2.8. Voltage Dependent Calcium Channels Blockade Assay
Free cytosolic Ca2+ was measured using the fluorescence Ca2+ indicator Fluo-4/AM. SH-SY5Y cells were seeded in 96-well black bottom transparent plates at a density of 60,000 cells per well and cultured until they reach confluence. Cells were incubated with the Ca2+ sensitive dye Fluo-4/AM at 5 µM in Krebs-HEPES buffer (KH) (145 mM NaCl, 4.9 mM KCl, 1.2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 11 mM glucose, pH 7.4), and 0.05% pluronic acid, for 1 h at 37 °C in the dark. Then, cells were washed twice with KH solution to remove the excess of probe and compounds in solution were added. Test compounds at 10 µM were incubated 10 min before K+ 70 mM was applied to trigger cytosolic Ca2+ increase. Fluorescence was measured in a fluorescence microplate reader (FLUOstar Optima, BMG, Ortenberg, Baden-Wuerttemberg, Germany) at excitation and emission wavelengths 485 and 520 nm, respectively. To normalize Fluo-4 signals, Triton X-100 (5%) and subsequently MnCl2 1 M were applied to the cells to register maximal and minimal fluorescence, respectively. Data were calculated as a percentage of Fluorescence increment with respect to Fmax − Fmin. The percentage of response was calculated considering basal conditions as 100% response.
2.9. Blood-Brain Barrier Permeation Assay (PAMPA)
Prediction of the capacity of compounds to cross the blood–brain barrier (BBB) by passive diffusion was evaluated using a Parallel Artificial Membrane Permeation Assay (PAMPA), in a similar manner as described previously [53]. The effective permeability of the compounds was measured in duplicate at an initial concentration of 100 µM, including reference positive and negative compounds (see Supporting Information, Table SI1). The compounds of interest were dissolved in 10 mM phosphate-buffered saline (PBS) buffer (pH 7.4) to the desired concentration in the donor well (Multiscreen IP sterile clear plate PDVF membrane, pore size 0.45 μM). The acceptor 96-well plate (Multiscreen, Millipore Corp. Burlington, MA, United States) was filled with 180 µL of PBS (VA). The filter membrane of the donor 96-well plate was impregnated with 4 µL of porcine brain lipid (PBL) (Avanti Polar Lipids, Inc., Alabaster, AL, United States) in dodecane (20 mg/mL) (Sigma-Aldrich, Madrid, Spain), and after 5 min, 180 µL of each compound solution was added to determine their ability to pass the brain barrier (VD). Then, the donor filter plate was carefully put on the acceptor plate to form a “sandwich”, which was left undisturbed for 4 h at 25 °C. After incubation, the donor plate was carefully removed. An ultraviolet (UV) plate reader determined the concentration of compounds and commercial drugs in both the acceptor and the donor wells (150 µL/well) as the maximum absorption wavelength of each compound. Concentration of the compounds in the donor and acceptor well and equilibrium concentration were calculated from the standard curve and expressed as the permeability (Pe), according the equation [1] (see supporting information). Results are expressed as mean ± SEM of three different experiments in duplicate.
2.10. Molecular Docking Calculations
GSK-3β structure was prepared and minimized using MacroModel (v2018-1, Schrödinger, Inc., New York, NY, United States) with Optimized Potentials for Liquid Simulations 3 (OPLS3) force field. Next, molecular docking was performed using AutoDock Vina (v1.1.2, Molecular Graphics Lab, Scripps Research Institute, San Diego, CA, United State [54] with center of the box located on the ATP binding site, and the best nine poses were considered. The selected 4,7-dihydro-2H-pyrazolo[3,4-b]pyridine 4f (R)- and (S)- enantiomers were submitted to molecular docking with the previous specifications [28]. The same docking strategy was used for predictions with CaV1.2 L-type channel and selected 4,7-dihydro-2H-pyrazolo[3,4-b]pyridine compound 4f in the 1,4-DHP binding site. As a crystal structure of this subtype is not available, we used the CaV1.2 l-subtype VDCC model developed by Denis Tikhonov and Boris S. Zhorov and kindly shared with us by Prof. Zhorov [55].
2.11. Neuroprotection Studies in the SH-SY5Y Human Neuroblastoma Cell Line
Cells were pre-incubated with the corresponding compound at 1 μM in neuroblastoma culture medium. After 24 h, the medium was removed and replaced with 1% FBS neuroblastoma culture media containing the corresponding compound at 1 µM and the toxic stimuli, namely a mixture of rotenone and oligomycin A (R/O, 30 μM/10 μM, respectively), okadaic acid (OA) at 20 nM or a high concentration of KCl (70 mM). Cells were co-incubated for further 24 h with the R/O mixture or KCl solution, or 18 h with the OA solution. Melatonin (1 µM) or nimodipine (1 µM) were used as positive control and reference compounds in the R/O, OA or high concentration of potassium models, respectively. Control cells were incubated with the same amount of dimethyl sulfoxide (DMSO) without any drug. After the co-incubation period, cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction method [56]; basal condition was considered as 100% survival.
2.12. Acute Treatment of Rat Hippocampal Slices
The 3–4 month-old Sprague-Dawley rats were decapitated and both hippocampi were dissected and placed in a previously oxygenated (95% O2 and 5% CO2) ice-cold Krebs’s dissection buffer (120 mM NaCl; 2 mM KCl; 26 mM NaHCO3; 1.18 mM KH2PO4; 10 mM MgSO4; 0.5 mM CaCl2; 11 mM glucose and 200 mM sucrose at pH 7.4). Hippocampi were cut into 250 µm thick slices using a McIlwain Tissue Chopper (Cavey Laboratory Engineering, Surrey, United Kingdom) and separated using a Leica SE6 microscope (Leica, Madrid, Spain). To allow slice recovery from the previous slicing trauma, they were transferred to a vial containing dissection buffer without sucrose, bubbled with 95% O2 and 5% CO2 for 45 min at 34 °C (stabilization). Thereafter, the slices were placed in a 48-well plate containing new medium composed of 1:1 control buffer (120 mM NaCl; 2 mM KCl; 26 mM NaHCO3; 1.18 mM KH2PO4; 10 mM MgSO4; 2 mM CaCl2 and 11 mM glucose) and DMEM/F12 medium (Invitrogen, Madrid, Spain). Slices were treated with saline, OA (1 µM) or OA co-incubated with the corresponding compounds (10 µM), and the plate was maintained under culture conditions (at 37 °C, 5 % CO2 and 95 % O2) for 6 h. Finally, cell viability and ROS production were measured.
2.13. Measurement of ROS Production in Rat Hippocampal Slices
ROS quantification was performed using the fluorescent probe 2´,7´-dichlorodihydrofluorescein diacetate (H2DCFDA) (λEx/Em = 495/529 nm) (Thermo Fisher, Waltham, MA, USA). Slices were incubated with the probe for 45 min at a concentration of 10 µL/mL, in the presence of 1 µL/mL of Hoechst at 37 °C before finalizing the experiment. Fluorescence from the cornu Ammonis 1 (CA1) was measured as mean intensity at an excitation wavelength of 480 nm and an emission of 520 nm for DCFDA in an inverted Nikon Eclipse T2000-U microscope (Nikon, Tokyo, Japan), with a 2X objective. Results were normalized to basal condition, which was considered as 100 %. NIS-Elements BR 4.10.04 64-bit software (NIS BR 4.10.04, Nikon, Tokyo, Japan) was used to analyze the images.
2.14. Statistical Analysis
All values are expressed as mean ± S.E.M. IC50 and LD50 values were calculated by non-linear regression analysis of individual concentration-response curves using GraphPad Prism software (v6.0, GraphPad, San Diego, CA, USA). Analysis of the results was performed by comparison between experimental and control data using one-way ANOVA followed by Newman-Keuls post-hoc test when three groups were implicated. Differences were considered to be statistically significant when p ≤ 0.05; “n” represents the number of different cultures used or enzyme inhibition assays performed.
3. Results and Discussion
3.1. Synthesis of 4,7-Dihydro-2H-Pyrazolo[3,4-b]Pyridine Derivatives
The synthesis of the 4,7-dihydro-2H-pyrazolo[3,4-b]pyridine derivatives 4 was performed by using a Hantzsch-like multicomponent-reaction approach from 3-methyl-1H-pyrazol-5-amine 1, aromatic aldehydes 2a–l and malononitrile 3 [28]. This mixture was refluxed in dry ethanol in the presence of dry ammonium acetate (Scheme 1) to obtain the desired compounds 4a–l.
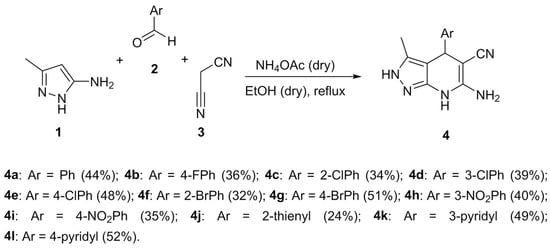
Scheme 1.
Multicomponent synthetic approach to the 4,7-dihydro-2H-pyrazolo[3,4-b]pyridine derivatives 4a–l.
Under these conditions, we were able to obtain a library of substituted Ph derivatives bearing electron-withdrawing substituents at the aromatic ring (compounds 4a–i), as well as three compounds bearing heterocyclic substituents (4j–l). Regarding reactions involving aromatic aldehydes with electron-donating groups, we obtained only traces of the corresponding pyrazolo[3,4-b]pyridine derivatives from highly complex reaction mixtures, due to full oxidation to the completely aromatic pyridine system.
3.2. Pharmacological Evaluation
3.2.1. Antioxidant and Anti-inflammatory Properties of Novel 4,7-Dihydro-2H-Pyrazolo[3,4-b]Pyridine Derivatives 4a–l
As described previously, OS is considered a prominent pathological factor, not only in the progression of AD, but also in the onset and initial phases of the disease. Particularly, 1,4-DHPs are considered a specific type of antioxidants due to their ability to inhibit free radical reactions by acting as hydrogen donors and their capacity to be oxidized under biological conditions [48]. Thus, we tested the potential antioxidant effect of our 4,7-dihydro-2H-pyrazolo[3,4-b]pyridine derivatives as direct ROS scavengers using the ORAC test, and melatonin, a potent natural antioxidant molecule, as positive control. As reported in Table 1, melatonin successfully scavenged oxygen free radicals being 2.14-fold more potent than trolox. Compounds 4a–l demonstrated scavenger properties showing ORAC values between 0.63 T.eq. (4d, Ar = 3-ClPh) and 2.93 T.eq. (4j, Ar = 2-thienyl), the latter being more potent than melatonin. Considering phenyl derivatives, only compound 4b showed higher antioxidant capacity than trolox (1.86 T. eq.) being the most potent of the carbocyclic derivatives. To delve into the antioxidant characterization of compounds 4a–l, we evaluated their potential capacity to scavenge RNS by using the DPPH method. Ascorbic acid and melatonin were included as positive control and reference for comparative purposes. In general, compounds 4a–l showed increased scavenger effect towards RNS compared to melatonin, being in all cases more potent. Furthermore, they showed almost 3-fold higher antioxidant capacity than ascorbic acid, a potent RNS scavenger [57]. RNS scavenger capacity was similar in all cases with IC50 values ranging from 17.1 μM (4k, Ar = 3-Py) to 25.2 μM (4a, Ar = Ph). This result indicates that the scavenger effect is directly related to the 4,7-dihydro-2H-pyrazolo[3,4-b]pyridine core, as it has been shown for different 1,4-DHPs described previously [58,59].

Table 1.
Antioxidant and anti-inflammatory properties of derivatives 4a–l and reference compounds.
Neuroinflammation is a widely described pathological pathway present also at the initial phases of AD [60,61]. Furthermore, as depicted in the introduction section, extensive OS induces glial activation by raising the production of pro-inflammatory mediators. These mediators augment ROS and RNS to generate a positive feedback loop leading to chronic neuroinflammation, a highly toxic environment for neurons described as one of the pathological hallmarks of AD. With these premises, we tested the potential anti-inflammatory capacity of compounds 4a–l in primary rat glial cultures stimulated with LPS. TLR4/2 activation by LPS induces iNOS overexpression that increases NO production. Compounds 4a–l were evaluated at increasing concentrations (0.1, 1, 3, 10 and 30 μM) and EC50 values were calculated from non-lineal regression of nitrite production reduction. Sulforaphane, a potent anti-inflammatory agent was included as positive control and for comparative purposes. Interestingly, all compounds showed anti-inflammatory capacity with EC50 values ranging from 780 nM of compound 4d (Ar = 3-ClPh) to 22.3 μM of compound 4k (Ar = 3-Py) (Table 1). With respect to the heterocyclic derivatives, only the 2-thienyl derivative showed an EC50 lower than 10 μM.
3.2.2. GSK-3β Inhibition, Voltage-Dependent Ca2+ Channels Blockade and BBB Permeation Properties
Among the pro-inflammatory pathways triggered before and after disease onset, GSK-3β is extensively described as an important kinase related to tau hyperphosphorylation and pro-inflammatory signal transduction [62,63]. Thus, we envisaged studying the GSK-3β inhibitory capacity of derivatives 4a–l. To this end, we used the luminescence Kinase-Glo assay at four different concentrations (0.1, 1, 10 and 30 μM). IC50 values and their related apparent equilibrium dissociation constants (Ki) are summarized in Table 2, including compound SB216763, a potent GSK-3β inhibitor, as a positive control. Compound 4a showed an improved inhibitory capacity compared to its pyrano[2,3-c]pyrazole isoster [28], indicating an important role of the 1,4-DHP ring in the interaction with GSK-3β. Interestingly, the inclusion of substituents at the aromatic moiety at C-4 position increased their capacity to inhibit GSK-3β, showing IC50 values from 820 nM of derivative 4h (Ar = 3-NO2Ph) to 10.2 μM of compound 4l (Ar = 4-Py). Considering substituent patterns, the tendency indicated that GSK-3β inhibitory potency increased when the substituent was inserted in ortho and meta positions compared to para position. Focusing on heterocyclic derivatives, the addition of an electron-rich heterocycle (thiophene) increased the inhibitory potency, compared to an electron-deficient ring (pyridine), the thienyl derivative being 4j two-fold more potent than 4k (Ar = 3-Py) and 5-fold more potent than 4l (Ar = 4-Py).
Table 2.
GSK-3β inhibition (IC50 and Ki values), voltage-dependent Ca2+ channels (VDCC) antagonism properties of derivatives 4a–l and reference compounds.
As discussed, inflammatory pathway activation is partially mediated by GSK-3β; thus, the inhibitory properties of this kinase by compounds 4a–l might be related to their anti-inflammatory capacity. Indeed, the anti-inflammatory properties of ClPh and NO2Ph derivatives correlated with GSK-3β inhibitory capacity, being meta derivatives most potent in both assays. Similar conclusions can be extracted considering the heterocyclic substituents. In this case, the most potent GSK-3β inhibitor, compound 4j (Ar = 2-thienyl, IC50 = 1.74 μM) showed the most potent anti-inflammatory properties (EC50 = 5.4 μM). However, bromine-substituted derivatives did not show the same tendency, since o-BrPh derivative 4f, with the best IC50 towards GSK-3β (IC50 = 0.83 μM) is a poor anti-inflammatory compound (EC50 = 10.1 μM), while p-BrPh derivative 4g (GSK-3β IC50 = 2.35 μM) was 2.2-fold more potent as anti-inflammatory (nitrite reduction EC50 = 4.5 μM) than 4f. These results indicate that the anti-inflammatory properties of compounds 4a–l could be partially dependent on the GSK-3β inhibitory capacity, and that their antioxidant properties participate in its anti-inflammatory mechanism of action.
As previously described, specific Ca2+ overload through L-type VDCC has been directly related to OS generation [42,45] and neuronal death. In this regard, the 1,4-DHP ring has specific activity towards L-type VDCCs. Thus, compounds 4a–l were tested as potential VDCC antagonist at 10 μM and the results are shown as percentage of response after membrane depolarization induced by 70 mM K+ stimuli in the SH-SY5Y neuronal cell line. Nimodipine was included as positive control at the same concentration. In general, compounds 4a–l showed interesting antagonistic activity reducing Ca2+ entrance to 85.6 % (4k) up to only 6.46 % response (4e). Compounds 4e and 4f (Ar = 4-ClPh and Ar = 2-BrPh derivatives, respectively) showed a highly effective blockade of [Ca2+]c elevation, being more effective than nimodipine at the same concentration. Interestingly, they contain a halogen substitution, as other 1,4-DHPs used in the clinic as VDCC antagonists that are being tested for mild cognitive impairment [66].
Finally, considering the potential use of our compounds as AD treatment, we explored the BBB penetration using the parallel artificial membrane permeation assay (PAMPA) prediction method. The in vitro permeabilities (Pe) of compounds 4a–l were calculated using the lipid extract of porcine brain (Table 2). We have used a range of positive and negative controls (see Supporting Information, Table SI1). The results depicted in Table 2 indicate that, excluding 4h (Ar = 3-NO2Ph), 4i (Ar = 4-NO2Ph) and 4k (Ar = 3-Py), all compounds are predicted to be able to cross the BBB by passive diffusion with Pe values higher than 4, a widely accepted value that marks the limit for passive diffusion [53].
3.3. Molecular Docking: Proposed Binding Mode of Compound 4f at the ATP Binding Site of GSK-3β and the 1,4-DHP Binding Site of L-Type VDCC
In order to understand the GSK-3β inhibition ability of compounds 4a–l, we studied the binding mode of the most potent GSK-3β inhibitor, derivative 4f (Ar = 2-BrPh), to GSK-3β (Figure 1) by means of molecular docking experiments. To this end, we evaluated both enantiomers at the ATP binding site of the enzyme. Results afforded similar poses for the (R)- and (S)- enantiomers of 4f, overlapping the pyridine ring with a 180° flip, leaving the pyrazole core on opposite sides. Both enantiomers form stable hydrogen bonds with Q185 and N186 residues (Figure 1A,B). Remarkably, the hydrogen bond between the NH group of the 1,4-DHP ring of both enantiomers and N186 emphasizes the importance of this group for its inhibitory activity. Additionally, both enantiomers showed hydrogen bonding with Q185. The (R)-enantiomer forms this interaction through the amino group at the 1,4-DHP ring (Figure 1A), while the (S)-enantiomer uses the NH moiety at the pyrazole ring (Figure 1B), in line with the predicted 180° flip binding mode shown in Figure 1. Compound 4f predicted poses were located in the surroundings of D200, key residue for ATP stabilization in the substrate cavity (see Figure 1 and Supporting Information, Figure SI1).
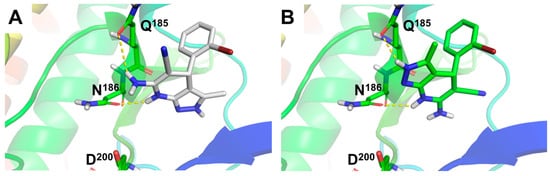
Figure 1.
Molecular docking of compound 4f (Ar = 2-BrPh), (R)- and (S)- enantiomers, at the adenosine triphosphate (ATP) binding site of GSK-3β. (A) Predicted binding mode for 4f (R)-enantiomer and (B) (S)-enantiomer represented as white and green sticks, respectively. Hydrogen bond interactions showed as yellow dotted lines. GSK-3β protein (PDB-ID: 3I4B) represented as colored cartoon and key residues as colored sticks.
Considering that compound 4f showed a potent L-type VDCC blockade capacity (% response = 7.39%), we also carried out molecular docking studies to determine the binding mode and potential interactions with the CaV1.2 L-type VDCC. The L-type VDCC crystal structure is not available; thus, we used a structural homology model of the channel developed by Zhorov et al. [55]. As previously done, we performed docking studies of both enantiomers of 4f at the 1,4-DHP L-type VDCC binding site located at the interface of domains III and IV of the α1 pore-constituent subunit (Figure 2). Docking results predicted a binding mode similar to that previously described for nimodipine (see Supporting Information, Figure SI2) [55]. The results obtained indicate that compound 4f places the 1,4-DHP ring of both enantiomers in a similar position to (S)-nimodipine, although they are slightly displaced towards the I3i14 and Y3i10 residues (Figure 2 and Supporting Information, Figure SI2). In this sense, the stern and bowsprit positions of 4f are close to those of (S)-nimodipine (see Supporting Information, Figure SI2). Both enantiomers showed a similar pose with a 180° flip generating similar interactions with key residues known to be involved in 1,4-DHPs stabilization from experimental data [67], such as (i) halogen bond between bromine present in the bowsprit and Y4i11; (ii) hydrogen bond between N atom of pyrazole core from starboard side of (S)-enantiomer and Q3o18. Interestingly, compound 4f also shows a π-π stacking interaction with F3i22, defined as one of the major contributors to the ligand-receptor energy stabilization of 1,4-DHPs at the CaV1.2 l-subtype VDCC model [55]. In summary, the predicted binding mode for compound 4f is analogous to other 1,4-DHPs such as (S)-nimodipine, showing interactions with key DHP-sensing residues of CaV1.2 l-subtype VDCCs.
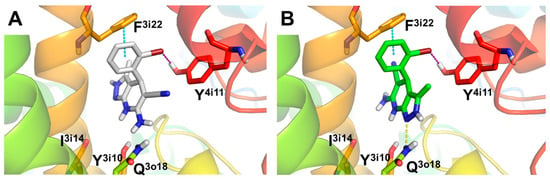
Figure 2.
Molecular docking of compound 4f (Ar = 2-BrPh), (R)- and (S)- enantiomers, at the 1,4-DHP binding site of CaV1.2 l-type VDCC. (A) Predicted binding mode for 4f (R)-enantiomer and (B) (S)-enantiomer represented as white and green sticks, respectively. Hydrogen bond, halogen bond and π-π stacking interactions showed as yellow, purple and blue dotted lines, respectively. CaV1.2 L-type channel structure represented as colored cartoon with key residues as sticks.
3.4. Neuroprotective Properties of Derivatives 4a–l towards Tau-Hyperphosphorylation, [Ca2+]c Overload and Oxidative Stress
As mentioned earlier, OS strongly contributes to AD onset and development, being a key pathological factor in the neurodegeneration observed in this disease [68]. Compounds 4a–l exert significant antioxidant properties (Table 1); thus, we tested the potential neuroprotection towards OS toxicity induced by rotenone (30 μM) and oligomycin A (10 μM) (R/O). Compounds 4a–l were tested at 1 μM concentration including melatonin as positive control and for comparative purposes. Results are expressed as percentage of survival considering non-treated cells as 100% survival. As shown in Figure 3A, all compounds significantly increased SH-SY5Y cells survival compared to the toxic challenge, eliciting protection values ranging from to 32.0% for compound 4d (Ar = 3-ClPh) to 71.6% of compound 4l (Ar = 4-Py). Interestingly, compounds 4b (Ar = 4-FPh), 4g (Ar = 4-BrPh) and 4l (Ar = 4-Py), with protective percentages of 62.7%, 59.7% and 71.6%, respectively, showed a better neuroprotective effect than melatonin (52.5%). In this case, considering that the R/O combination induces high amounts of ROS, we observed an interesting correlation between the neuroprotective ability of compounds 4a–l and their capacity to scavenge ROS. In general, the neuroprotective effect increased with higher scavenger capacity, except in the case of compound 4j. Compounds with ORAC values lower than 0.9 T.eq. showed the lowest percentages of survival (% survival < 75.7%), while compounds showing higher ORAC values presented percentages of survival higher than 78% in all cases. For example, chlorine derivatives 4c, 4d and 4e (o-, m- and p-ClPh, respectively) showed scavenger capacities of 0.94, 0.63 and 0.66 T.eq., respectively, directly correlated with percentages of survival of 81.3%, 72.3% and 73.6%. In this line, derivative 4b (Ar = 4-FPh) with ROS scavenger ability of 1.86 showed an improvement on cell viability to 84.3%. The same trend can be extracted for bromophenyl, nitrophenyl and pyridyl derivatives.
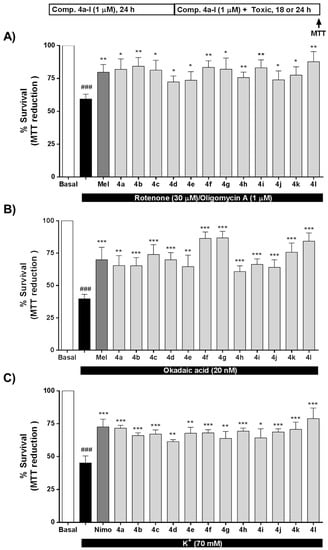
Figure 3.
Neuroprotective activity of compounds 4a–l against the toxicity exerted by (A) oxidative stress, (B) tau hyperphosphorylation and (C) [Ca2+]c overload. SH-SY5Y neuroblastoma cells were treated with compounds 4a-l (1 μM) or reference compounds (melatonin or nimodipine, 1 μM) during 24 h. Thereafter, cells were co-incubated with compounds 4a-l, melatonin or nimodipine (1 μM) and the corresponding toxic stimuli, R/O combination (30/10 μM, oxidative stress, 24 h), okadaic acid (OA) (20 nM, tau hyperphosphorylation, 18 h) or 70 mM K+ ([Ca2+]c overload, 24 h). Cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. Values are expressed as mean ± S.E.M of five independent experiments in triplicate and results were normalized to basal conditions (100%). ### p < 0.001 compared to basal condition; * p < 0.05, ** p < 0.01, *** p < 0.001 compared to toxic stimuli treated cells.
As summarized in the introduction, GSK-3β is the main kinase related to the formation of neurotoxic hyperphosphorylated tau, the core component of NFTs in AD. Considering the antioxidant and GSK-3β inhibitory properties of compounds 4a–l, we envisaged their evaluation as potential neuroprotective agents in an in vitro model of tau hyperphosphorylation induced by OA. OA inhibits protein phosphatases 1 and 2A [69], increasing the phosphorylation status of tau [70], being an accepted in vitro model of AD. The toxic cascade activated by OA includes increased levels of OS that also contributes to cellular death [71]. All compounds were tested at 1 μM concentration and melatonin was included as positive control [28] and for comparative purposes (Figure 3B). Successfully, compounds 4a–l showed neuroprotective capacity ranging from 34.6% protection of compound 4h (Ar = 3-NO2Ph) to 77.9% protection of compound 4g (Ar = 4-BrPh). Compounds 4c (Ar = 2-ClPh), 4f (Ar = 2-BrPh), 4g (Ar = 4-BrPh), 4k (Ar = 3-Py) and 4l (Ar = 4-Py) showed neuroprotective percentages over 50%, being the compounds 4f, 4g and 4l more potent than the reference compound, melatonin. In this particular model, the neuroprotective effect appears to be dependent on both, the antioxidant effect and the GSK-3β inhibitory potency.
In this line, compound 4g with the best neuroprotective profile increased cell viability to 86.7%, and showed good GSK-3β inhibition (IC50 = 2.35 μM), although it was less potent than compound 4h, the most potent GSK-3β inhibitor (IC50 = 0.82 μM). However, compound 4h increased cell viability to only 60.8% and, interestingly, the difference between them was the antioxidant capacity, being compound 4g (ORAC = 0.96 T.eq) more potent than compound 4h (ORAC = 0.73 T.eq.). Similarly, compound 4c exhibited an increased survival percentage (74.0%) compared to compound 4e (percentage survival = 64.6%), compound 4c being a slightly more potent GSK-3β inhibitor (4c IC50 = 1.51 μM compared to 4e IC50 = 1.94) and a better antioxidant (4c = 0.94 T.eq. compared to 4e = 0.66 T.eq). These results notwithstanding, and similarly to results obtained in the OS toxicity evaluation, compound 4j did not follow the same tendency. Although it was the most potent antioxidant (ORAC = 2.93 T.eq.) and a good GSK-3β inhibitor (IC50 = 1.74 μM), it increased cell viability to just 64.0%.
Regarding the second target pursued in compounds 4a–l, VDCC antagonism, we evaluated their potential neuroprotective profile against [Ca2+]c overload induced by high K+ (70 mM). Nimodipine, a potent VDCC antagonist, was included as positive control at 1 μM. As shown in Figure 3C, compounds 4a–l exhibited an interesting neuroprotective profile with survival percentages ranging from 61.4 % of compound 4d (Ar = 3-ClPh) to 78.9 % for compound 4l (Ar = 4-Pyridyl). In this case, neuroprotection capacity was similar for all compounds with percentages of survival in the range of 70%, except in the case of compound 4l. Compounds 4e and 4f showed highly potent VDCC antagonism capacity. This potency correlated with interesting neuroprotection capacity (67.6% and 68.0%, respectively); however, they were moderated neuroprotectants compared to compound 4l. Regarding their antioxidant properties, in this case, the correlation between their neuroprotective capacity against [Ca2+]c overload and their scavenger capacity was not clearly correlated. However, the most potent neuroprotectant in this model, compound 4l, showed moderate VDCC antagonist capacity and high antioxidant ability (ORAC = 1.1 T.eq.) indicating a potential implication in its neuroprotective mechanism of action. Recently, we described the potential dependence of the neuroprotective effect in this toxicity model with the antioxidant effect [72] following results previously published and demonstrating the relationship between Ca2+ entry through VDCC and increased levels of free radicals [42,45,73].
In addition to compounds 4a–l neuroprotective profile, we evaluated their safety profile by testing their neurotoxicity in the SH-SY5Y neuroblastoma cell line and their gliotoxicity in primary mixed glial cultures. Our results indicate that compounds 4a–l are neither neurotoxic nor gliotoxic with LD50 values higher than 100 μM (Table SI2, Supporting Information).
3.5. Compound 4g Reduces Cellular Death Induced by Okadaic Acid in Hippocampal Slices by Reducing Oxidative Stress
These encouraging results prompted us to test our compounds in a more complex model of AD. For this, we selected an acute treatment with OA of hippocampal slices. Recently, we have demonstrated the deleterious effect of OA (1 μM) in hippocampal slices after 6 h treatment [74]. It increases ROS production, decreases cellular viability and deregulates the autophagy system; thus, it was described as an ex vivo model of tauopathy mimicking AD pathology. For this purpose, we selected compound 4g (Ar = 4-BrPh) due to its overall pharmacological profile as a potent antioxidant able to scavenge ROS and RNS free radicals, a potent anti-inflammatory properties at low micromolar range, a GSK-3β inhibitor in the same low micromolar range and a moderate VDCC antagonist. The multitarget profile integrated into compound 4g afforded potent neuroprotective effect against OS and [Ca2+]c overload, and it was the most potent compound against tau hyperphosphorylation. Compound 4g was tested at 10 μM, a concentration established for control and reference compounds used in these ex vivo models. For these experiments, we included melatonin, known to be highly effective in this model, as a reference and positive control.
As shown in Figure 4A, OA reduced cell viability by 30% (p < 0.001) measured by the MTT reduction assay. Successfully, compound 4g recovered cell viability to 92.1%, being a slightly more potent neuroprotector than melatonin, although the differences were not significant (Figure 4B). As demonstrated previously, treatment with OA induces a significant increase in free radical production [74], measured as H2DCFDA fluorescence intensity increase (p < 0.01), and this ROS increase is related to the toxicity elicited by OA. Under these experimental conditions, compound 4g effectively reduced free radical production to basal conditions (p < 0.01) with an equal potency to melatonin, a well-known antioxidant molecule (Figure 4A,C). As summarized above, we have demonstrated the scavenger capacity of this compound, and thus this antioxidant capacity should be part of its protective mechanism of action.
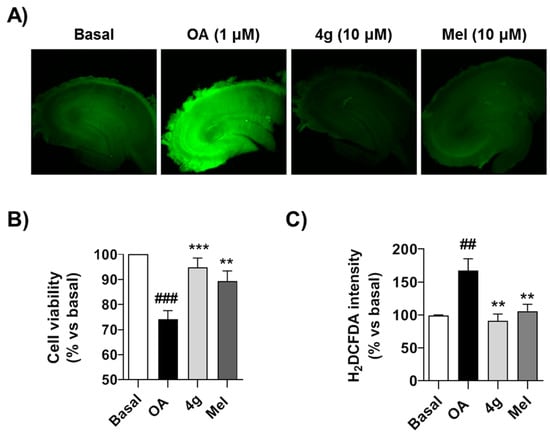
Figure 4.
Compound 4g protects against acute toxicity induced by OA in hippocampal slices: (A) Representative H2DCFDA fluorescence images of hippocampal slices treated with OA in presence or absence of 4g (10 μM) or melatonin (10 μM) during 6 h. (B) Cell viability of hippocampal slices treated with saline (Basal), OA (1 μM), OA (1 μM) + 4g (10 μM) or OA (1 μM) + melatonin (Mel, 10 μM) during 6 h. Cell viability was then evaluated by MTT reduction assay and normalized to basal conditions (100%). (C) Quantitation of H2DCFDA fluorescence intensity as free radical production measurement after the experimental conditions described in (B). Data are expressed as mean ± S.E.M of four different experiments. ## p < 0.01, ### p < 0.001 compared to basal condition; ** p < 0.01, *** p < 0.001, compared to OA treated hippocampal slices.
4. Conclusions
AD is currently the most common type of dementia affecting an increasing number of patients and is one of the most important unmet clinical needs for our society. The development of an effective therapy with real disease modifying capacity is an urgent goal for medicinal chemists and biomedical research. The complexity of AD has led to the proposal of the “multitarget hypothesis” for drug development in which a unique chemical entity combines different activities to tackle the pathological pathways involved in AD onset and development. Following this hypothesis, we have obtained a new set of multitarget compounds based on the chemical core of the 1,4-DHPs due to their antioxidant properties and well-known VDCC antagonism capacity. In addition, the fusion of a pyrazole ring generated a suitable scaffold able to inhibit GSK-3β, a kinase related to tau hyperphosphorylation and pro-inflammatory pathways. The combination of pyrazole and 4-aryl-1,4-dihydropyridine moieties into a single molecular entity was achieved through a Hantzsch-like three-component reaction from 3-methyl-1H-pyrazol-5-amine, aromatic aldehydes and malononitrile. Compounds 4a–l, thus generated, have demonstrated ROS and RNS antioxidant capacity, anti-inflammatory properties, GSK-3β inhibitory capacity, at low micromolar range and l-type VDCC blockade with micromolar potency. Moreover, the multi-target directed ligands developed were found to lack neurotoxicity or gliotoxicity. They also showed good neuroprotective activity towards OS, tau-hyperphosphorylation and [Ca2+]c overload induced toxicity in cell cultures, with interesting correlations between their primary targets and this neuroprotective effect indicating their participation in the neuroprotective mechanism of action. From our set of compounds we have identified 6-amino-4-(4-bromophenyl)-3-methyl-4,7-dihydro-2H-pyrazolo[3,4-b]pyridine-5-carbonitrile (4g) as an interesting multitarget directed ligand for further optimization. Compound 4g showed good antioxidant activity (ORAC = 0.96 T.eq.; DPPH IC50 = 24.1 μM), potent anti-inflammatory capacity (EC50 = 4.50 μM), GSK-3β inhibitory capacity (IC50 = 2.35 μM) and VDCC antagonist activity (percentage response reduced to 62%) and it is predicted to cross the BBB. Moreover, it showed potent neuroprotective properties against OS, tau hyperphosphorylation and [Ca2+]c overload. When tested in a more complex ex vivo model of AD, the acute treatment of hippocampal slices with OA, it demonstrated an extreme reduction of ROS production and also showed potent cytoprotecting capacity. Thus, this compound can be regarded as a good lead for future efforts to increase its potency towards primary targets in a hit to lead project that is currently under development in our laboratories following the promising and novel multitarget approach described here.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-3921/9/8/650/s1, Table SI1 containing PAMPA model control compounds, Table SI2 showing toxicological evaluation, full methodological description including chemical synthetic protocols, pharmacological experiments and images of the 1H and 13C NMR spectra images, Figures SI1 and SI2 showing molecular docking experiments of reference compounds.
Author Contributions
P.M. (Patrycja Michalska), P.M. (Paloma Mayo), P.D. and I.B. performed experimental in vitro pharmacology evaluation; C.F.-M. performed the ex vivo experiments. G.T. carried out the chemistry. M.T.R. and J.C.M. supervised the chemical synthesis of compounds and critically revised the manuscript. M.G.L. supervised the ex vivo experiments and critically revised the manuscript. R.L. designed the project, supervised experiments and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from IS Carlos III co-financed by the European Regional Development’s funds (FEDER), Programa Miguel Servet II (CP16/00014) and research project (grant PI17/01700) to R.L.; Fundación La Caixa, CaixaImpulse program (grant CI17-00048) to R.L.; Comunidad Autónoma de Madrid (grants B2017/BMD-3827 and B2017/BMD-3813) and Spanish Ministry of Science, Innovation and Education (grants RTI2018-095793-B-I00 to M.G.L. and CTQ2015-68380-R and RTI2018-097662-B-I00 to J.C.M.).
Acknowledgments
P.M. (Patrycja Michalska), P.D. and C.F.-M. thank MECD for FPU fellowships (13/03737, 16/03977 and 15/03269 respectively). P.M. (Paloma Mayo) thanks Universidad Autónoma de Madrid for a FPI fellowship. G.T. thanks Universidad Complutense for a predoctoral fellowship. I.B. thanks MECD for Juan de la Cierva fellowship (FJCI-2016/28282). We would also like to thank “Fundación Teófilo Hernando” for its continued support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Alzheimer Report 2019 Attitudes to Dementia. 2019. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2019.pdf (accessed on 16 June 2020).
- Lee, V.M.Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Molinuevo, J.L.; Sanchez-Valle, R.; Lladó, A.; Fortea, J.; Bartres-Faz, D.; Rami, L. Identifying Earlier Alzheimer’s Disease: Insights from the Preclinical and Prodromal Phases. Neurodegener. Dis. 2012, 10, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef]
- Mecocci, P.; Boccardi, V.; Cecchetti, R.; Bastiani, P.; Scamosci, M.; Ruggiero, C.; Baroni, M. A Long Journey into Aging, Brain Aging, and Alzheimer’s Disease Following the Oxidative Stress Tracks1. J. Alzheimer’s Dis. 2018, 62, 1319–1335. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Sheehan, J.P.; Swerdlow, R.H.; Miller, S.W.; Davis, R.E.; Parks, J.K.; Parker, W.D.; Tuttle, J.B. Calcium Homeostasis and Reactive Oxygen Species Production in Cells Transformed by Mitochondria from Individuals with Sporadic Alzheimer’s Disease. J. Neurosci. 1997, 17, 4612–4622. [Google Scholar] [CrossRef]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Beringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Du Yan, S. Mitochondrial Aβ: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef]
- Canevari, L.; Clark, J.B.; Bates, T.E. β-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999, 457, 131–134. [Google Scholar] [CrossRef]
- Cenini, G.; Rüb, C.; Bruderek, M.; Voos, W. Amyloid β-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 2016, 27, 3257–3272. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badia, M.-C.; Giraldo, E.; Ermak, G.; Alonso, M.-D.; Pallardó, F.V.; Davies, K.E.; Viña, J. Amyloid-β Toxicity and Tau Hyperphosphorylation are Linked Via RCAN1 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 701–709. [Google Scholar] [CrossRef]
- Leroy, K.; Yılmaz, Z.; Brion, J.-P.; Yilmaz, Z. Increased level of active GSK-3? In Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, T.; Kamimura, N.; Yokota, T.; Iuchi, K.; Nishimaki, K.; Takami, S.; Akashiba, H.; Shitaka, Y.; Katsura, K.-I.; Kimura, K.; et al. Oxidative stress accelerates amyloid deposition and memory impairment in a double-transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2015, 587, 126–131. [Google Scholar] [CrossRef]
- Coma, M.; Guix, F.X.; Ill-Raga, G.; Uribesalgo, I.; Alameda, F.; Valverde, M.A.; Muñoz, F.J. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol. Aging 2008, 29, 969–980. [Google Scholar] [CrossRef]
- Mondragón-Rodríguez, S.; Perry, G.; Luna-Muñoz, J.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein at sites Ser396–404 is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Himmelstein, D.S.; Ward, S.M.; Lancia, J.K.; Patterson, K.R.; Binder, L.I. Tau as a therapeutic target in neurodegenerative disease. Pharmacol. Ther. 2012, 136, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Hohman, T.J.; Koran, M.E.I.; Thornton-Wells, T.A.; Initiative, F.T.A.N. Interactions between GSK3β and amyloid genes explain variance in amyloid burden. Neurobiol. Aging 2014, 35, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2007, 104, 1433–1439. [Google Scholar] [CrossRef]
- Mines, M.A.; Beurel, E.; Jope, R.S. Regulation of Cell Survival Mechanisms in Alzheimer’s Disease by Glycogen Synthase Kinase-3. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef]
- Alvarez, G.; Muñoz-Montaño, J.R.; Satrústegui, J.; Avila, J.; Bogonez, E.; Díaz-Nido, J. Lithium protects cultured neurons against β-amyloid-induced neurodegeneration. FEBS Lett. 1999, 453, 260–264. [Google Scholar] [CrossRef]
- Salazar-Roa, M.; Rojo, A.I.; Velasco, D.; De Sagarra, R.M.; Cuadrado, A. Glycogen Synthase Kinase-3β Inhibits the Xenobiotic and Antioxidant Cell Response by Direct Phosphorylation and Nuclear Exclusion of the Transcription Factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.J.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Rampa, A.; Gobbi, S.; Di Martino, R.M.C.; Belluti, F.; Bisi, A. Dual BACE-1/GSK-3β Inhibitors to Combat Alzheimer’s Disease: A Focused Review. Curr. Top. Med. Chem. 2017, 17, 3361–3369. [Google Scholar] [CrossRef]
- Gandini, A.; Bartolini, M.; Tedesco, D.; Martínez-González, L.; Roca, C.; Campillo, N.E.; Zaldivar-Diez, J.; Perez, C.; Zuccheri, G.; Miti, A.; et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3β and Tau-Aggregation Inhibitors. J. Med. Chem. 2018, 61, 7640–7656. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, Á.; Buendía, I.; Rojo, A.I.; Georgakopoulos, N.D.; Guijo, J.M.H.; Ramos, M.T.; Wells, G.; et al. Discovery of the first dual GSK3β inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef]
- Jankowska, A.; Satała, G.; Bojarski, A.J.; Pawłowski, M.; Chłoń-Rzepa, G. Multifunctional Ligands with Glycogen Synthase Kinase 3 Inhibitory Activity as a New Direction in Drug Research for Alzheimer’s Disease. Curr. Med. Chem. 2020, 27. [Google Scholar] [CrossRef]
- Kummer, M.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.-C.; König, S.; Roeber, S.; et al. Nitration of Tyrosine 10 Critically Enhances Amyloid β Aggregation and Plaque Formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef]
- Hensleya, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical Analysis of Protein Nitrotyrosine and Dityrosine in the Alzheimer Brain Indicates Region-Specific Accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; Van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2009, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Grebe, A.; Rayner, K.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, U.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Benveniste, E.N. Critical role of tumor necrosis factor-alpha and NF-kappa B in interferon-gamma-induced CD40 expression in microglia/macrophages. J. Biol. Chem. 2002, 277, 13796–13803. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Praticò, D.; Clark, C.M.; Liun, F.; Lee, V.Y.-M.; Trojanowski, J.Q. Increase of Brain Oxidative Stress in Mild Cognitive Impairment. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I.B. Dysregulation of Intracellular Calcium Signaling in Alzheimer’s Disease. Antioxid. Redox Signal. 2018, 29, 1176–1188. [Google Scholar] [CrossRef]
- Tong, B.C.-K.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochimica et Biophysica Acta BBA Bioenerg. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Hayley, M.; Perspicace, S.; Schulthess, T.; Seelig, J. Calcium enhances the proteolytic activity of BACE1: An in vitro biophysical and biochemical characterization of the BACE1–calcium interaction. Biochimica et Biophysica Acta BBA Biomembr. 2009, 1788, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Yin, X.; Yu, D.; Cao, M.; Gong, C.-X.; Iqbal, K.; Ding, F.; Gu, X.; Liu, F. Truncation and activation of GSK-3β by calpain I: A molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci. Rep. 2015, 5, 8187. [Google Scholar] [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijić, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; De Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.J.; Brandon, B.; Gentleman, S.M.; Dexter, D.T. Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 2013, 136, 2077–2097. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.B.; Schmeidler, J.; Lesser, G.T.; Beeri, M.S.; Purohit, D.P.; Grossman, H.T.; Haroutunian, V. Less Alzheimer disease neuropathology in medicated hypertensive than nonhypertensive persons. Neurology 2009, 72, 1720–1726. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer disease is multifactorial and heterogeneous. Neurobiol. Aging 2000, 21, 901–902. [Google Scholar] [CrossRef]
- Velēna, A.; Zarkovic, N.; Trošelj, K.G.; Bisenieks, E.; Krauze, A.; Poikāns, J.; Duburs, G. 1,4-Dihydropyridine Derivatives: Dihydronicotinamide Analogues—Model Compounds Targeting Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 1892412. [Google Scholar] [CrossRef]
- Cao, G.; Alessio, H.M.; Cutler, R.G. Oxygen-radical absorbance capacity assay for antioxidants. Free. Radic. Biol. Med. 1993, 14, 303–311. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Dudonné, S.; Vitrac, X.; Coutière, P.; Woillez, M.; Mérillon, J.-M. Comparative Study of Antioxidant Properties and Total Phenolic Content of 30 Plant Extracts of Industrial Interest Using DPPH, ABTS, FRAP, SOD, and ORAC Assays. J. Agric. Food Chem. 2009, 57, 1768–1774. [Google Scholar] [CrossRef]
- Baki, A.; Bielik, A.; Molnár, L.; Szendrei, G.; Keserü, G.M. A High Throughput Luminescent Assay for Glycogen Synthase Kinase-3β Inhibitors. ASSAY Drug Dev. Technol. 2007, 5, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B.; Zhorov, B.S. Structural Model for Dihydropyridine Binding to L-type Calcium Channels. J. Biol. Chem. 2009, 284, 19006–19017. [Google Scholar] [CrossRef] [PubMed]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Golonka, I.; Oleksy, M.; Junka, A.; Matera-Witkiewicz, A.; Bartoszewicz, M.; Musiał, W. Selected Physicochemical and Biological Properties of Ethyl Ascorbic Acid Compared to Ascorbic Acid. Biol. Pharm. Bull. 2017, 40, 1199–1206. [Google Scholar] [CrossRef][Green Version]
- Vijesh, A.; Isloor, A.M.; Peethambar, S.; Shivananda, K.; Arulmoli, T.; Isloor, N.A. Hantzsch reaction: Synthesis and characterization of some new 1,4-dihydropyridine derivatives as potent antimicrobial and antioxidant agents. Eur. J. Med. Chem. 2011, 46, 5591–5597. [Google Scholar] [CrossRef]
- Mulder, P.; Litwinienko, G.; Lin, S.; MacLean, P.D.; Barclay, L.R.C.; Ingold, K.U. The L-Type Calcium Channel Blockers, Hantzsch 1,4-Dihydropyridines, Are Not Peroxyl Radical-Trapping, Chain-Breaking Antioxidants. Chem. Res. Toxicol. 2006, 19, 79–85. [Google Scholar] [CrossRef]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bras, J.; Hardy, J. SnapShot: Genetics of Alzheimer’s Disease. Cell 2013, 155, 968. [Google Scholar] [CrossRef]
- Ko, C.-Y.; Wang, W.-L.; Wang, S.-M.; Chu, Y.-Y.; Chang, W.-C.; Wang, J.-M. Glycogen synthase kinase-3β–mediated CCAAT/enhancer-binding protein delta phosphorylation in astrocytes promot es migration and activation of microglia/macrophages. Neurobiol. Aging 2014, 35, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martín, M.; Jurado-Arjona, J.; Fuster-Matanzo, A.; Hernandez, F.; Rabano, A.; Avila, J. Peripherally triggered and GSK-3β-driven brain inflammation differentially skew adult hippocampal neurogenesis, behavioral pattern separation and microglial activation in response to ibuprofen. Transl. Psychiatry 2014, 4, e463. [Google Scholar] [CrossRef] [PubMed]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Gaisina, I.; Yuan, H.; Petukhov, P.A.; Blond, S.Y.; Fedolak, A.; Caldarone, B.; Mcgonigle, P. Structure-Based Design Leads to the Identification of Lithium Mimetics That Block Mania-like Effects in Rodents. Possible New GSK-3β Therapies for Bipolar Disorders. J. Am. Chem. Soc. 2007, 129, 8328–8332. [Google Scholar] [CrossRef] [PubMed]
- Tiwaskar, M.; Langote, A.; Kashyap, R.; Toppo, A. Amlodipine in the Era of New Generation Calcium Channel Blockers. J. Assoc. Physicians India 2018, 66, 64–69. [Google Scholar] [PubMed]
- Wappl, E.; Mitterdorfer, J.; Glossmann, H.; Striessnig, J. Mechanism of Dihydropyridine Interaction with Critical Binding Residues of L-type Ca2+ Channel α1 Subunits. J. Biol. Chem. 2001, 276, 12730–12735. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Kamat, P.K.; Rai, S.; Nath, C. Okadaic acid induced neurotoxicity: An emerging tool to study Alzheimer’s disease pathology. Neurotoxicology 2013, 37, 163–172. [Google Scholar] [CrossRef]
- Pérez, M.; Hernandez, F.; Gómez-Ramos, A.; Smith, M.; Perry, G.; Avila, J. Formation of aberrant phosphotau fibrillar polymers in neural cultured cells. JBIC J. Biol. Inorg. Chem. 2002, 269, 1484–1489. [Google Scholar] [CrossRef]
- Kamat, P.K.; Rai, S.; Swarnkar, S.; Shukla, R.; Nath, C. Mechanism of synapse redox stress in Okadaic acid (ICV) induced memory impairment: Role of NMDA receptor. Neurochem. Int. 2014, 76, 32–41. [Google Scholar] [CrossRef]
- Tenti, G.; Parada, E.; León, R.; Egea, J.; Martínez-Revelles, S.; Briones, A.M.; Sridharan, V.; Lopez, M.G.; Ramos, M.T.; Menéndez, J.C. New 5-Unsubstituted Dihydropyridines with Improved CaV1.3 Selectivity as Potential Neuroprotective Agents against Ischemic Injury. J. Med. Chem. 2014, 57, 4313–4323. [Google Scholar] [CrossRef] [PubMed]
- Khaliq, Z.M.; Bean, B.P. Pacemaking in dopaminergic ventral tegmental area neurons: Depolarizing drive from background and voltage-dependent sodium conductances. J. Neurosci. 2010, 30, 7401–7413. [Google Scholar] [CrossRef] [PubMed]
- Luengo, E.; Buendia, I.; Fernández-Mendívil, C.; Trigo-Alonso, P.; Negredo, P.; Michalska, P.; Hernández-García, B.; Sánchez-Ramos, C.; Bernal, J.A.; Ikezu, T.; et al. Pharmacological doses of melatonin impede cognitive decline in tau-related Alzheimer models, once tauopathy is initiated, by restoring the autophagic flux. J. Pineal Res. 2019, 67, e12578. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).