PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation

 ,
,  ,
, 
 , , , and
, , , and 
Abstract
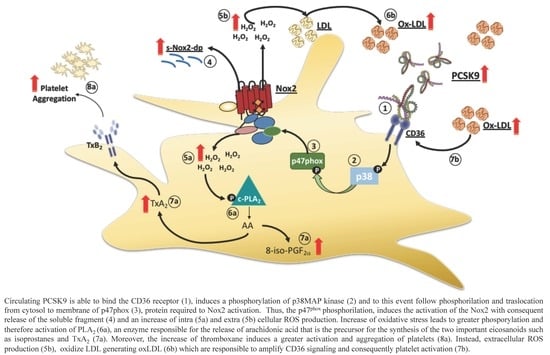
1. Introduction
2. Materials and Methods
2.1. Platelet Preparation
2.2. Platelet Aggregation and Recruitment
2.3. Serum and Platelet sNox2-dp
2.4. Serum and Platelet H2O2 Production
2.5. Platelet and Urinary 8-Iso-PGF2α Assay
2.6. Serum Detection of Oxidization of Low-Density Lipoprotein (ox-LDL)
2.7. Serum and Platelet TxB2 Assay
2.8. Serum and Platelet sP-selectin Assay
2.9. LDL Isolation and Determination of Conjugated Dienes
2.10. Co-Immunoprecipitation Assays and Western Blot Analysis
2.11. Statistical Analysis
3. Results
3.1. Ex-Vivo Study
3.2. In Vitro Study
3.2.1. PCSK9 and Platelet Activation
3.2.2. PCSK9 and Oxidative Stress
3.2.3. Intra-Signalling Pathway of Platelet Activation PCSK9-Mediated p38MAP Kinase, p47phox and cPLA2 Phosphorylation
3.2.4. PCSK9 and LDL-Mediated Platelet Activation
3.2.5. PCSK9 Activates Platelets by CD36 Signalling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- El Khoury, P.; Elbitar, S.; Ghaleb, Y.; Khalil, Y.A.; Varret, M.; Boileau, C.; Abifadel, M. PCSK9 mutations in familial hypercholesterolemia: From a groundbreaking discovery to anti-PCSK9 therapies. Curr. Atheroscler. Rep. 2017, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Shahanawaz, J.; Shmookler Reis, R.J.; Varughese, K.I.; Sawamura, T.; Mehta, J.L. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 2015, 107, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Theus, S.; Deng, X.; Fan, Y.; Zhou, S.; Mehta, J.L. PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc. Res. 2018, 114, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Nocella, C.; Farcomeni, A.; Bartimoccia, S.; Santulli, M.; Vasaturo, F.; Carnevale, R.; Menichelli, D.; Violi, F.; Pignatelli, P.; et al. Relationship of PCSK9 and urinary thromboxane excretion to cardiovascular events in patients with atrial fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Navarese, E.P.; Kolodziejczak, M.; Winter, M.-P.; Alimohammadi, A.; Lang, I.M.; Buffon, A.; Lip, G.Y.; Siller-Matula, J.M. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: The PCSK9-REACT study. Int. J. Cardiol. 2017, 227, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; Russo, I. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Kassassir, H.; Siewiera, K.; Sychowski, R.; Watała, C. Can the antiplatelet effects of cangrelor be reliably studied in mice under in vivo and in vitro conditions using flow cytometry? Pharmacol. Rep. 2013, 65, 870–883. [Google Scholar] [CrossRef]
- Camera, M.; Rossetti, L.; Barbieri, S.S.; Zanotti, I.; Canciani, B.; Trabattoni, D.; Ruscica, M.; Tremoli, E.; Ferri, N. PCSK9 as a positive modulator of platelet activation. J. Am. Coll. Cardiol. 2018, 71, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Navarese, E.P.; Tantry, U.S. Exploration of PCSK9 as a cardiovascular risk factor. J. Am. Coll. Cardiol. 2017, 70, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Carnevale, R.; Di Santo, S.; Bartimoccia, S.; Sanguigni, V.; Lenti, L.; Finocchi, A.; Mendolicchio, L.; Soresina, A.R.; Plebani, A.; et al. Inherited human gp91 phox deficiency is associated with impaired isoprostane formation and platelet dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 423–434. [Google Scholar] [CrossRef] [PubMed]
- BORN, G. V Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Silvestri, R.; Loffredo, L.; Novo, M.; Cammisotto, V.; Castellani, V.; Bartimoccia, S.; Nocella, C.; Violi, F. Oleuropein, a component of extra virgin olive oil, lowers postprandial glycaemia in healthy subjects. Br. J. Clin. Pharmacol. 2018, 84, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Emelyanov, A.; Fedoseev, G.; Abulimity, A.; Rudinski, K.; Fedoulov, A.; Karabanov, A.; Barnes, P.J. Elevated concentrations of exhaled hydrogen peroxide in asthmatic patients. Chest 2001, 120, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, R.; Pignatelli, P.; Lenti, L.; Buchetti, B.; Sanguigni, V.; Di Santo, S.; Violi, F. LDL are oxidatively modified by platelets via GP91 phox and accumulate in human monocytes. FASEB J. 2007, 21, 927–934. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Dang, P.M.-C.; Gougerot-Pocidalo, M.-A.; Marie, J.-C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Murugesan, G.; Chen, K.; Zhang, L.; Wang, Q.; Febbraio, M.; Anselmo, R.M.; Marchant, K.; Barnard, J.; Silverstein, R.L. Platelet CD36 surface expression levels affect functional responses to oxidized LDL and are associated with inheritance of specific genetic polymorphisms. Blood 2011, 117, 6355–6366. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Kakutani, M.; Naruko, T.; Ueda, M.; Narumiya, S.; Masaki, T.; Sawamura, T. Activation-dependent surface expression of LOX-1 in human platelets. Biochem. Biophys. Res. Commun. 2001, 282, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Knowles, R.B.; Kirkby, N.S.; Reed, D.M.; Edin, M.L.; White, W.E.; Chan, M.V.; Longhurst, H.; Yaqoob, M.M.; Milne, G.L.; et al. Kidney transplantation in a patient lacking cytosolic phospholipase A2 proves renal origins of urinary PGI-M and TX-M. Circ. Res. 2018, 122, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Demers, A.; Samami, S.; Lauzier, B.; Des Rosiers, C.; Sock, E.T.N.; Ong, H.; Mayer, G. PCSK9 induces CD36 degradation and affects long-chain fatty acid uptake and triglyceride metabolism in adipocytes and in mouse liver. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2517–2525. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 88) | Below Median PCSK9 (n = 44) | Above Median PCSK9 (n = 44) | p-Value | |
|---|---|---|---|---|
| Age (years) | 73.6 ± 7.7 | 74.1 ± 8.9 | 72.8 ± 6.9 | 0.424 |
| Female sex (%) | 48.9 | 43.2 | 54.5 | 0.394 |
| Arterial hypertension (%) | 88.6 | 93.2 | 84.1 | 0.314 |
| Diabetes mellitus (%) | 18.2 | 15.9 | 20.5 | 0.783 |
| Heart failure (%) | 14.8 | 15.9 | 13.6 | 0.764 |
| Prior cerebrovascular events (%) | 12.6 | 15.9 | 9.3 | 0.521 |
| Prior cardiac events (%) | 22.7 | 25.0 | 20.5 | 0.800 |
| Antiplatelet therapy (%) | 00.0 | |||
| Statins (%) | 42.0 | 47.7 | 36.4 | 0.388 |
| Proton pomp inhibitors (%) | 43.0 | 38.6 | 47.6 | 0.514 |
| CHA2DS2-VASc score | 3.46 ± 1.42 | 3.64 ± 1.50 | 3.34 ± 1.40 | 0.341 |
| PCSK9 | ||
|---|---|---|
| Rs | p-value | |
| Platelet Aggregation | 0.309 | <0.001 |
| Platelet Recruitment | 0.485 | <0.001 |
| sP-selectin | 0.330 | <0.001 |
| Serum TxB2 production | 0.538 | <0.001 |
| Serum sNox2-dp | 0.314 | <0.001 |
| Hydrogen peroxide | 0.185 | <0.001 |
| Urinary 8-iso-PGE2α | 0.336 | <0.001 |
| Ox-LDL production | 0.550 | <0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cammisotto, V.; Pastori, D.; Nocella, C.; Bartimoccia, S.; Castellani, V.; Marchese, C.; Sili Scavalli, A.; Ettorre, E.; Viceconte, N.; Violi, F.; et al. PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation. Antioxidants 2020, 9, 296. https://doi.org/10.3390/antiox9040296
Cammisotto V, Pastori D, Nocella C, Bartimoccia S, Castellani V, Marchese C, Sili Scavalli A, Ettorre E, Viceconte N, Violi F, et al. PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation. Antioxidants. 2020; 9(4):296. https://doi.org/10.3390/antiox9040296
Chicago/Turabian StyleCammisotto, Vittoria, Daniele Pastori, Cristina Nocella, Simona Bartimoccia, Valentina Castellani, Cinzia Marchese, Antonio Sili Scavalli, Evaristo Ettorre, Nicola Viceconte, Francesco Violi, and et al. 2020. "PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation" Antioxidants 9, no. 4: 296. https://doi.org/10.3390/antiox9040296
APA StyleCammisotto, V., Pastori, D., Nocella, C., Bartimoccia, S., Castellani, V., Marchese, C., Sili Scavalli, A., Ettorre, E., Viceconte, N., Violi, F., Pignatelli, P., & Carnevale, R. (2020). PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation. Antioxidants, 9(4), 296. https://doi.org/10.3390/antiox9040296