Combination of PDT and NOPDT with a Tailored BODIPY Derivative

 ,
,  , , ,
, , ,  ,
, 
 and
and
Abstract
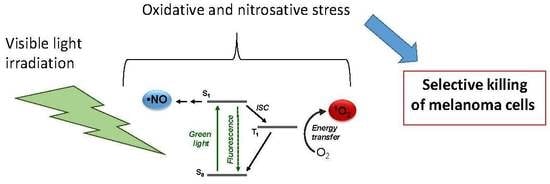
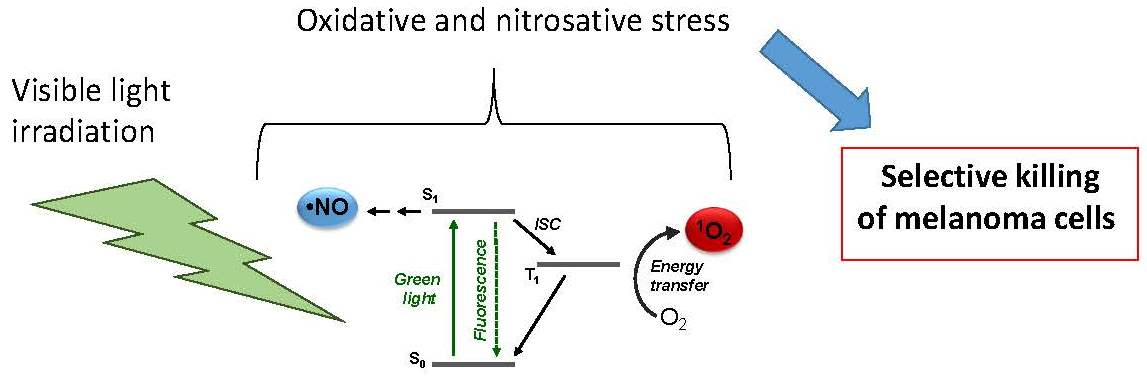
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthetic Procedures
2.2.1. Synthesis of 2-((4,4-difluoro-2,6-diiodo-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacen-8-yl) methoxy)-1-phenyldiazene oxide 2
2.2.2. Synthesis of 4,4-difluoro-2,6-diiodo-8-methoxymethyl-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene 4
2.3. Instrumentation
Laser Flash Photolysis
2.4. Biological Experiments
2.4.1. Cell Cultures
2.4.2. NO Photorelease in Cells
2.4.3. Cytotoxicity
2.4.4. Statistical Analysis
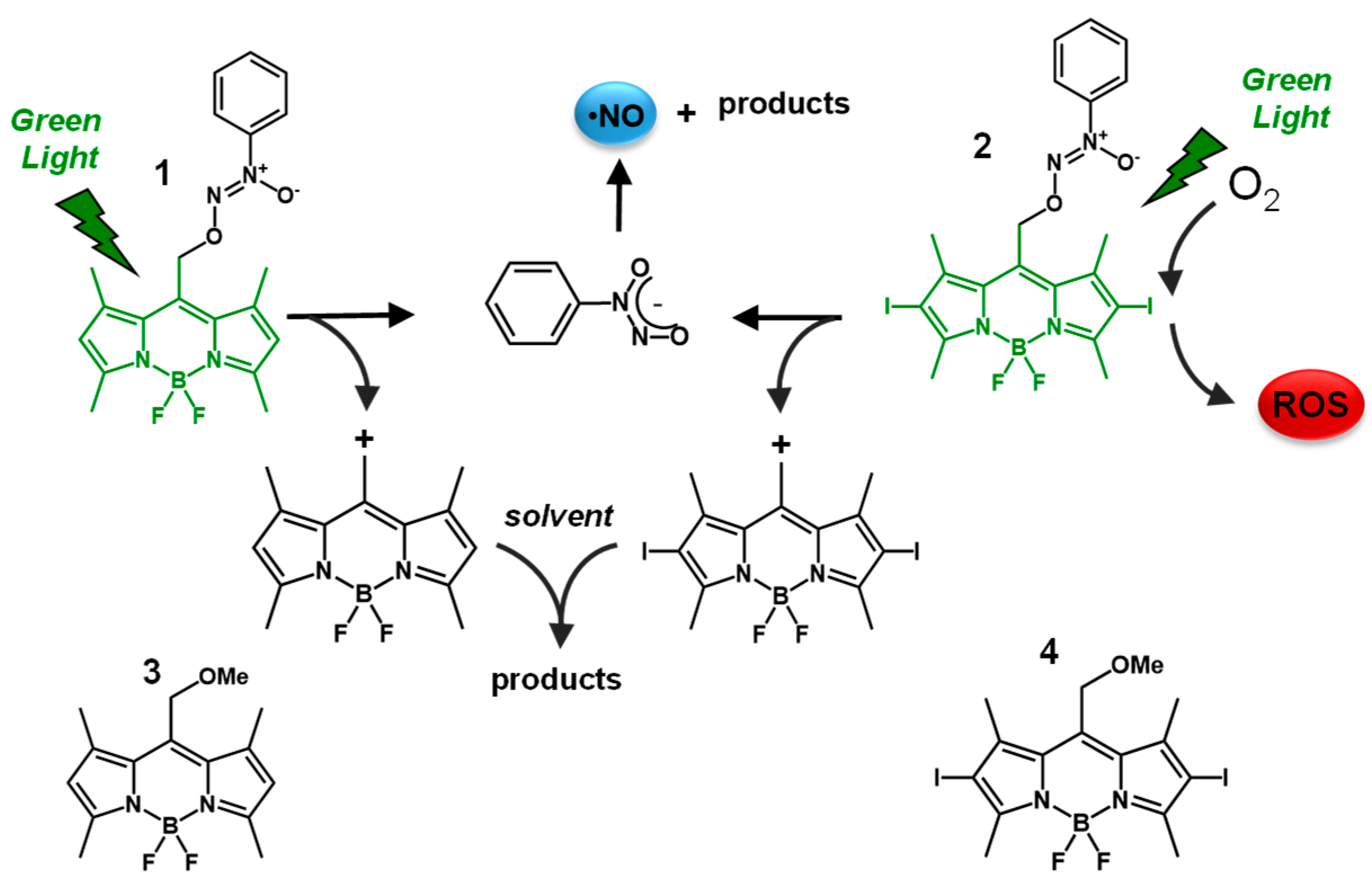
3. Results and Discussion
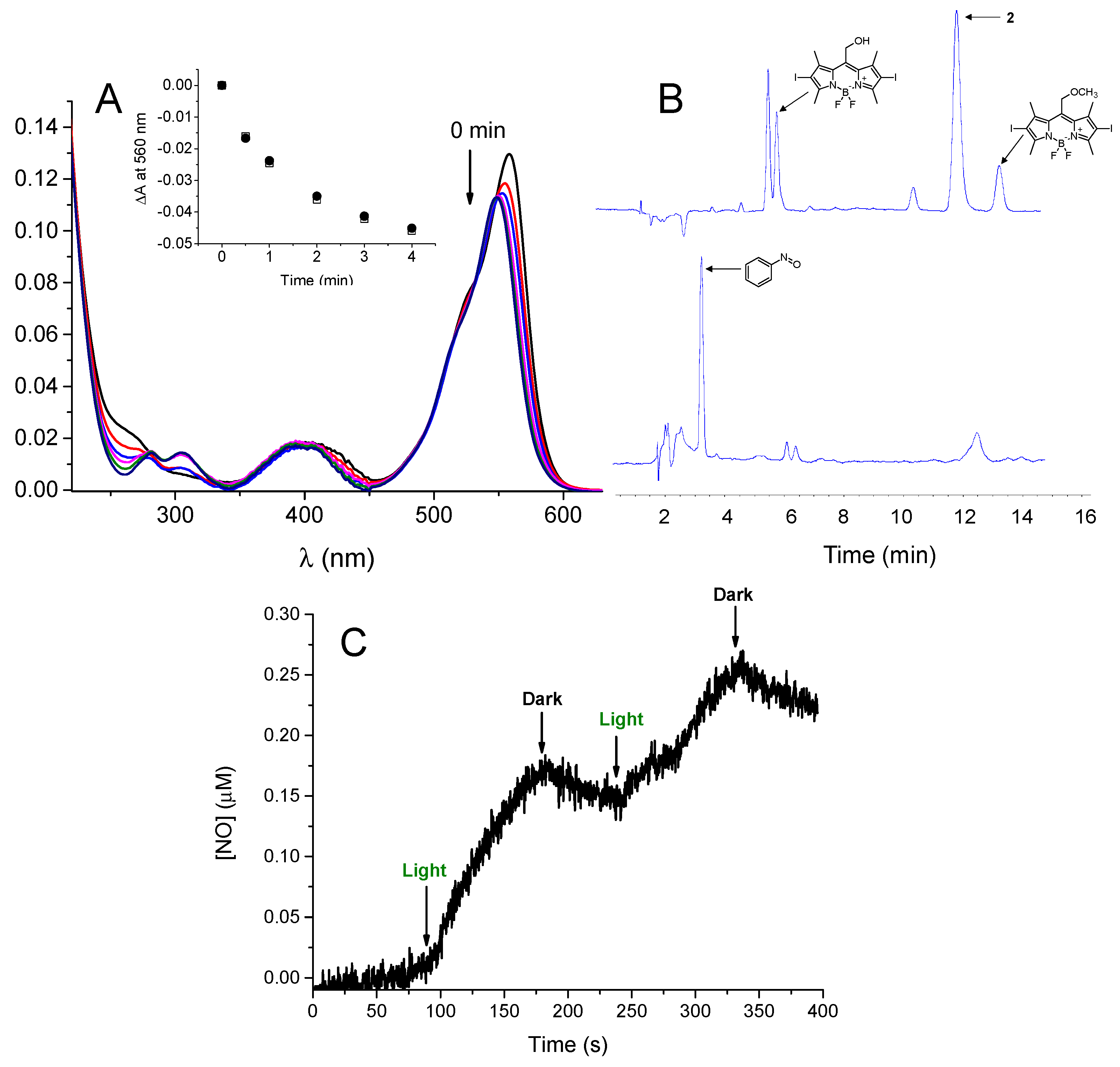
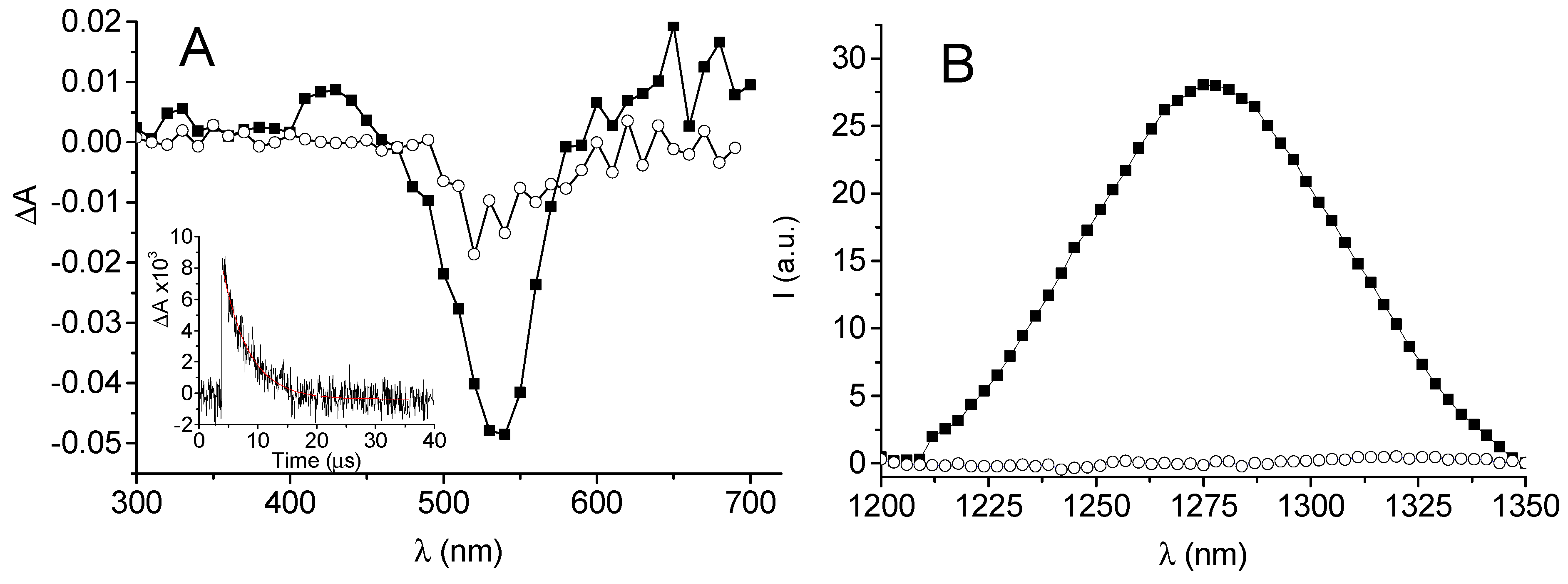
3.1. Photochemical and Spectroscopic Properties
3.2. Biological Experiments
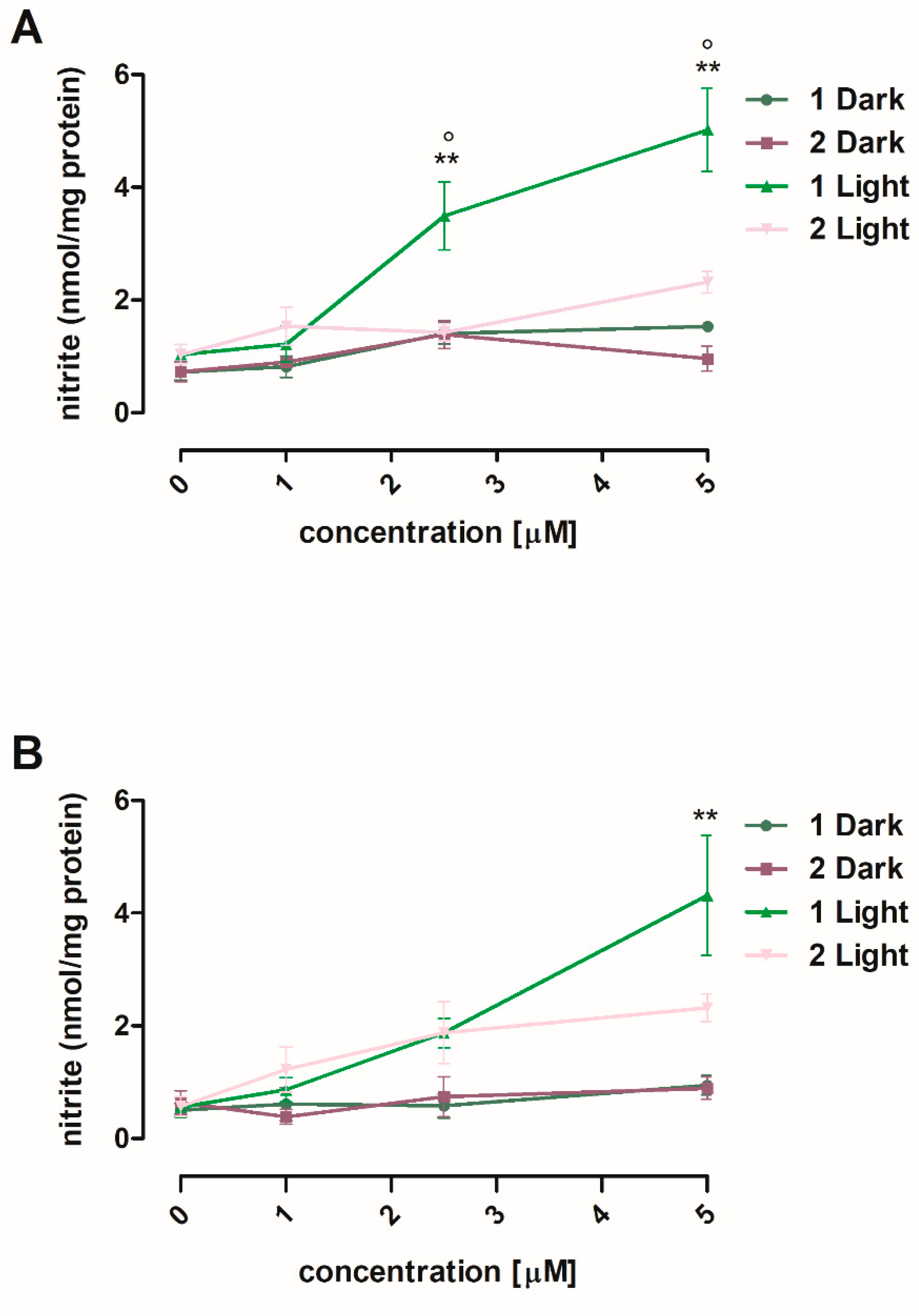
3.2.1. NO Photorelease in Cells
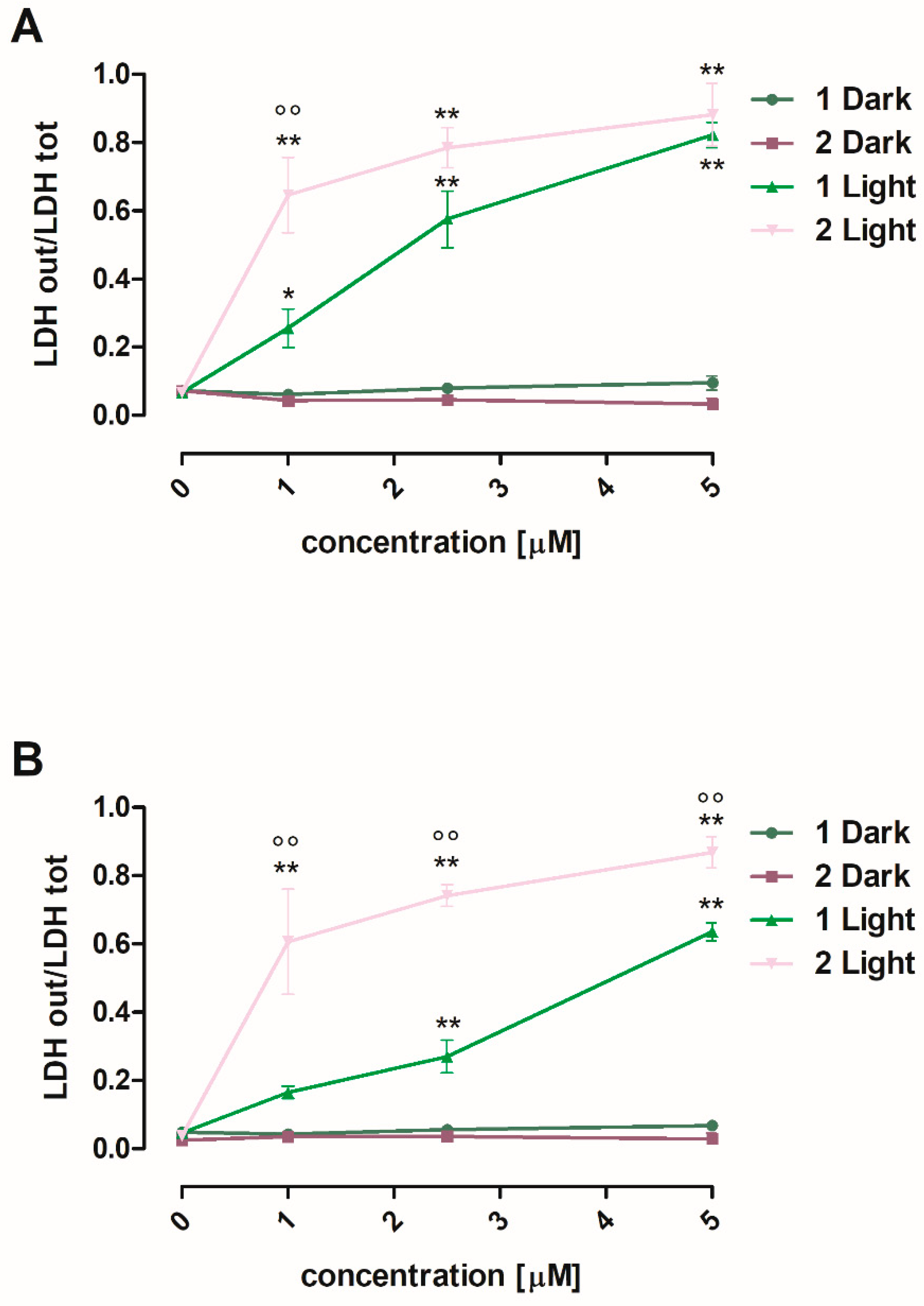
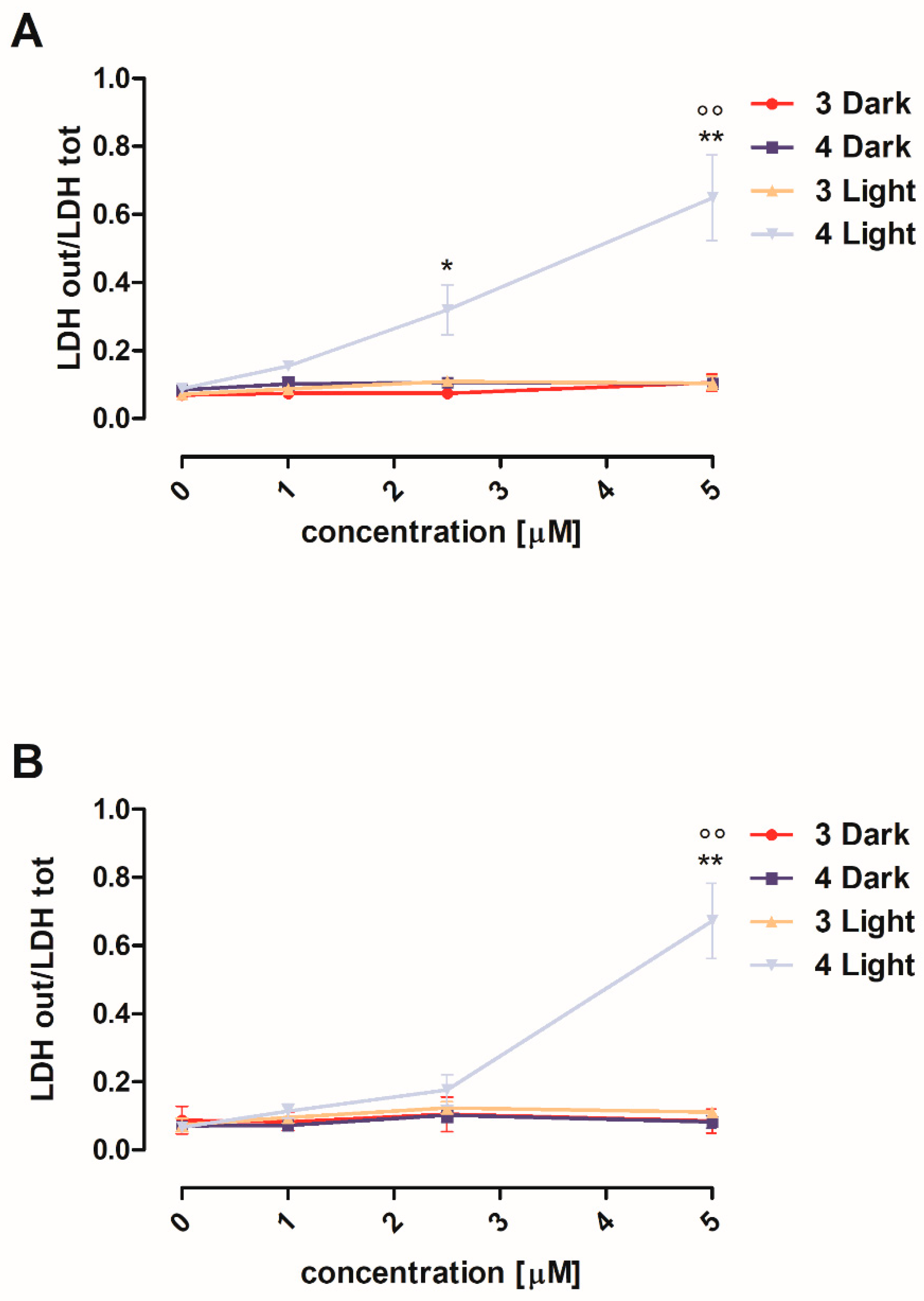
3.2.2. Cytotoxicity against Human Melanoma A375 and SKMEL28 Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wainwright, M. Photosensitizers in Biomedicine; John Wiley & Sons Ltd.: West Sussex, UK, 2009. [Google Scholar]
- Ormond, A.B.; Freeman, H.S. Dye sensitizers for photodynamic therapy. Materials 2013, 6, 817–840. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—Photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef]
- Li, X.; Kwon, N.; Guo, T.; Liu, Z.; Yoon, J. Innovative strategies for hypoxic-tumor photodynamic therapy. Angew. Chem. Int. Ed. 2018, 57, 11522–11531. [Google Scholar] [CrossRef] [PubMed]
- Kervin, J.F., Jr.; Lancaster, J.R., Jr.; Feldman, P.L. Nitric oxide: A new paradigm for second messengers. J. Med. Chem. 1995, 38, 4343–4362. [Google Scholar]
- Gross, S.S.; Wolin, M.S. Nitric oxide: Pathophysiological mechanisms. Annu. Rev. Physiol. 1995, 57, 737–769. [Google Scholar] [CrossRef]
- Kroncke, K.-D.; Fehsel, K.; Kolb-Bachofen, V. Nitric oxide: Cytotoxic versus cytoprotection—How, why, and where? Nitric Oxide 1997, 1, 107–120. [Google Scholar] [CrossRef]
- Wink, D.A.; Mitchell, J.B. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanism of nitric oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Thomas, D.D.; Donzelli, S.; Espey, M.G.; Roberts, D.D.; Wink, D.A.; Isenberg, J.S. The biphasic nature of nitric oxide responses in tumor biology. Antioxid. Redox Signal. 2006, 8, 1329–1337. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 2018, 118, 1330–1408. [Google Scholar] [CrossRef]
- Huerta, S.; Chilka, S.; Bonavida, B. Nitric oxide donors: Novel cancer therapeutics. Int. J. Oncol. 2008, 33, 909–927. [Google Scholar] [CrossRef]
- Huang, Z.; Fu, J.; Zhang, Y. Nitric-oxide donor-based cancer therapy: Advances and prospects. J. Med. Chem. 2017, 60, 7617–7635. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Nagae, O.; Kato, Y.; Nakagawa, H.; Fukuhara, K.; Miyata, N. Photoinduced nitric oxide release from nitrobenzene derivatives. J. Am. Chem. Soc. 2005, 127, 11720–11726. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Kawaguchi, M.; Ieda, N.; Miyata, N.; Nakagawa, H. Visible light-controlled nitric oxide release from hindered nitrobenzene derivatives for specific modulation of mitochondrial dynamics. ACS Chem. Biol. 2016, 11, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Caruso, E.B.; Petralia, S.; Conoci, S.; Giuffrida, S.; Sortino, S. Photodelivery of nitric oxide from water soluble platinum nanoparticles. J. Am. Chem. Soc. 2007, 129, 480–481. [Google Scholar] [CrossRef] [PubMed]
- Parisi, C.; Failla, M.; Fraix, A.; Rolando, B.; Gianquinto, E.; Spyrakis, F.; Gazzano, E.; Riganti, C.; Lazzarato, L.; Fruttero, R.; et al. Fluorescent nitric oxide photodonors based on BODIPY and rhodamine antennae. Chem. Eur. J. 2019, 47, 11080–11084. [Google Scholar] [CrossRef]
- Ford, P.C. Polychromophoric metal metal complexes for generating the bioregulatory agent nitric oxide by single- and two-photon excitation. Acc. Chem. Res. 2008, 41, 190–200. [Google Scholar] [CrossRef]
- Ford, P.C. Photochemical delivery of nitric oxide. Nitric Oxide 2013, 34, 56–64. [Google Scholar] [CrossRef]
- Ieda, N.; Hotta, Y.; Miyata, N.; Kimura, K.; Nakagawa, H. Photomanipulation of vasodilation with a blue-light controllable nitric oxide releaser. J. Am. Chem. Soc. 2014, 136, 7085–7091. [Google Scholar] [CrossRef]
- Sortino, S. Light-controlled nitric oxide delivering molecular assemblies. Chem. Soc. Rev. 2010, 39, 2903–2913. [Google Scholar] [CrossRef]
- Makings, L.R.; Tsien, R.Y. Caged nitric oxide. J. Biol. Chem. 1994, 269, 6282–6285. [Google Scholar]
- Ruane, P.H.; Bushan, K.M.; Pavlos, C.M.; D’Sa, R.A.; Toscano, J.P. Controlled photochemical release of nitric oxide from O2-benzyl-substituted diazeniumdiolates. J. Am. Chem. Soc. 2002, 124, 9806–9811. [Google Scholar] [CrossRef] [PubMed]
- Behara, K.K.; Rajesh, Y.; Venkatesh, Y.; Pinninti, B.R.; Mandal, M.; Singh, N.D.P. Cascade photocaging of diazeniumdiolates: A novel strategy for one and two photon triggered uncaging with real time reporting. Chem. Commun. 2017, 53, 9470–9473. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wu, J.; Lv, T.; Lai, Y.; Zhang, H.; Lu, J.-J.; Zhang, Y.; Huang, Z. Synthesis and evaluation of novel O2-derived diazeniumdiolates as photochemical and real-time monitoring nitric oxide delivery agents. Org. Chem. Front. 2017, 4, 2445–2449. [Google Scholar] [CrossRef]
- Stroppel, A.S.; Paolillo, M.; Ziegler, T.; Feil, R.; Stafforst, T. Npom-protected NONOate enables light-triggered NO/cGMP signalling in primary vascular smooth muscle cells. ChemBioChem 2018, 19, 1312–1318. [Google Scholar] [CrossRef]
- Blangetti, M.; Fraix, A.; Lazzarato, L.; Marini, E.; Rolando, B.; Sodano, F.; Fruttero, R.; Gasco, A.; Sortino, S. A nonmetal-containing nitric oxide donor activated with single-photon green light. Chem. Eur. J. 2017, 23, 9026–9029. [Google Scholar] [CrossRef] [PubMed]
- Fraix, A.; Sortino, S. Combination of PDT photosensitizers with NO-photodonors. Photochem. Photobiol. Sci. 2018, 17, 1709–1727. [Google Scholar] [CrossRef] [PubMed]
- Parisi, C.; Failla, M.; Fraix, A.; Rescifina, A.; Rolando, B.; Lazzarato, L.; Cardile, V.; Graziano, A.C.E.; Fruttero, R.; Gasco, A.; et al. A molecular hybrid producing simultaneously singlet oxygen and nitric oxide by single photon excitation with green light. Bioorg. Chem. 2019, 85, 18–22. [Google Scholar] [CrossRef]
- Krumova, K.; Cosa, G. Bodipy dyes with tunable redox potentials and functional groups for further tethering: Preparation, electrochemical, and spectroscopic characterization. J. Am. Chem. Soc. 2010, 132, 17560–17569. [Google Scholar] [CrossRef]
- Chegaev, K.; Fraix, A.; Gazzano, E.; Abd-Ellatef, G.E.; Blangetti, M.; Rolando, B.; Conoci, S.; Riganti, C.; Fruttero, R.; Gasco, A.; et al. Light-regulated NO release as a novel strategy to overcome doxorubicin multidrug resistance. ACS Med. Chem. Lett. 2017, 8, 361–365. [Google Scholar] [CrossRef]
- Riganti, C.; Costamagna, C.; Bosia, A.; Ghigo, D. The NADPH oxidase inhibitor apocynin (acetovanillone) induces oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 179–187. [Google Scholar] [CrossRef]
- Rachford, A.A.; Ziessel, R.; Bura, T.; Retailleau, P.; Castellano, F.N. Boron dipyrromethene (Bodipy) phosphorescence revealed in [Ir(ppy)2(bpy-CtC-Bodipy)]. Inorg. Chem. 2010, 49, 3730–3736. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Costamagna, C.; Doublier, S.; Miraglia, E.; Polimeni, M.; Bosia, A.; Ghigo, D. The NADPH oxidase inhibitor apocynin induces nitric oxide synthesis via oxidative stress. Toxicol. Appl. Pharmacol. 2008, 228, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Aldieri, E.; Fenoglio, I.; Cesano, F.; Gazzano, E.; Gulino, C.; Scarano, D.; Attanasio, A.; Mazzucco, G.; Ghigo, D.; Fubini, B. The role of iron impurities in the toxic effects exerted by short multiwalled carbon nanotubes (MWCNT) in murine Aalveolar Macrophages. J. Toxicol. Environ. Health Part A 2013, 76, 1056–1071. [Google Scholar] [CrossRef] [PubMed]
- Corazao-Rozas, P.; Guerreschi, P.; Jendoubi, M.; André, F.; Jonneaux, A.; Scalbert, C.; Garçon, G.; Malet-Martino, M.; Balayssac, S.; Rocchi, S.; et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget 2013, 4, 1986–1998. [Google Scholar] [CrossRef] [PubMed]
- Kalal, B.S.; Upadhya, D.; Pai, V.R. Chemotherapy resistance mechanisms in advanced skin cancer. Oncol. Rev. 2017, 11, 326. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
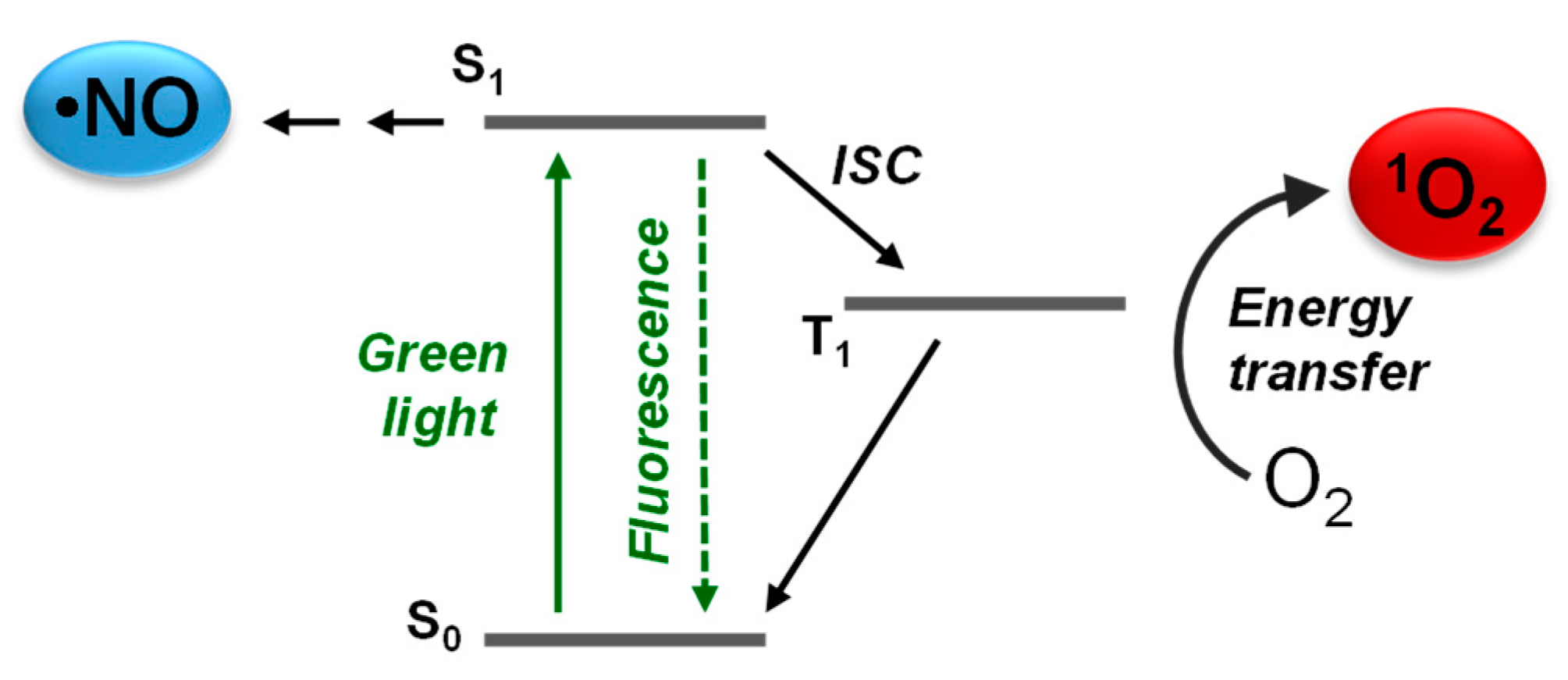{kind=link}
| Cell Lines | 1 (Dark) | 2 (Dark) | 3 (Dark) | 4 (Dark) | 1 (Light) | 2 (Light) | 3 (Light) | 4 (Light) |
|---|---|---|---|---|---|---|---|---|
| A375 | >100 | >100 | >100 | >100 | 8.6 ± 1.2 *** | 4.6 ± 0.4 ***,° | >100 | 7.3 ± 1.6 *** |
| SKMEL28 | >100 | >100 | >100 | >100 | 10.1 ± 2.5 *** | 5.9 ± 0.7 ***,° | >100 | 8.1 ± 0.7 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzarato, L.; Gazzano, E.; Blangetti, M.; Fraix, A.; Sodano, F.; Picone, G.M.; Fruttero, R.; Gasco, A.; Riganti, C.; Sortino, S. Combination of PDT and NOPDT with a Tailored BODIPY Derivative. Antioxidants 2019, 8, 531. https://doi.org/10.3390/antiox8110531
Lazzarato L, Gazzano E, Blangetti M, Fraix A, Sodano F, Picone GM, Fruttero R, Gasco A, Riganti C, Sortino S. Combination of PDT and NOPDT with a Tailored BODIPY Derivative. Antioxidants. 2019; 8(11):531. https://doi.org/10.3390/antiox8110531
Chicago/Turabian StyleLazzarato, Loretta, Elena Gazzano, Marco Blangetti, Aurore Fraix, Federica Sodano, Giulia Maria Picone, Roberta Fruttero, Alberto Gasco, Chiara Riganti, and Salvatore Sortino. 2019. "Combination of PDT and NOPDT with a Tailored BODIPY Derivative" Antioxidants 8, no. 11: 531. https://doi.org/10.3390/antiox8110531
APA StyleLazzarato, L., Gazzano, E., Blangetti, M., Fraix, A., Sodano, F., Picone, G. M., Fruttero, R., Gasco, A., Riganti, C., & Sortino, S. (2019). Combination of PDT and NOPDT with a Tailored BODIPY Derivative. Antioxidants, 8(11), 531. https://doi.org/10.3390/antiox8110531