Abstract
Amyotrophic Lateral Sclerosis (ALS) is a progressive motor neurodegenerative disease. Cell damage in ALS is the result of many different, largely unknown, pathogenetic mechanisms. Astrocytes and microglial cells play a critical role also for their ability to enhance a deranged inflammatory response. Excitotoxicity, due to excessive glutamate levels and increased intracellular Ca2+ concentration, has also been proposed to play a key role in ALS pathogenesis/progression. Reactive Oxygen Species (ROS) behave as key second messengers for multiple receptor/ligand interactions. ROS-dependent regulatory networks are usually mediated by peroxides. Superoxide Dismutase 1 (SOD1) physiologically mediates intracellular peroxide generation. About 10% of ALS subjects show a familial disease associated with different gain-of-function SOD1 mutations. The occurrence of sporadic ALS, not clearly associated with SOD1 defects, has been also described. SOD1-dependent pathways have been involved in neuron functional network as well as in immune-response regulation. Both, neuron depolarization and antigen-dependent T-cell activation mediate SOD1 exocytosis, inducing increased interaction of the enzyme with a complex molecular network involved in the regulation of neuron functional activity and immune response. Here, alteration of SOD1-dependent pathways mediating increased intracellular Ca2+ levels, altered mitochondria functions and defective inflammatory process regulation have been proposed to be relevant for ALS pathogenesis/progression.
1. Introduction
Amyotrophic Lateral Sclerosis (ALS) is a progressive motor neuron degeneration associated with death of motoneurons of the spinal cord, brain stem and cortical pyramidal neurons network. This fatal neurodegenerative disease is not limited to α-motoneuron death since in many patients a cortical neuron degeneration and frontal dementia is present [1,2].
ALS is associated with muscle atrophy, weakness, fasciculations and spasticity [3] and sometimes accompanied by speech and swallowing dysfunctions leading to progressive paralysis and death due to respiratory failure.
Despite intense investigation efforts, little is known about the primum movens that generates ALS development.
More than 50 different genes have been linked to ALS [2,4,5,6,7]. Indeed, this neurodegenerative disease probably has a multifactorial genesis involving multiple mechanisms and transduction pathways, including oxidative distress [8,9].
As reported by Halliwell and Gutteridge, Reactive Oxygen Species (ROS) include both free radicals, with a very short half-life that ranges from 1 × 10−6 s to 1 × 10−10 s, and hydrogen peroxide (H2O2) [10], a relatively stable, non-radical derivative of oxygen molecule that is able to freely circulate inside cells and cross cell membranes. The ability of ROS to participate in the complex molecular network involved in the regulation of cell functional activity has been largely demonstrated in multiple cell/tissue contexts. Such regulatory functions have been observed to be essentially mediated by hydrogen peroxide, stable molecule that is able to freely diffuse inside cell membranes. SOD1, discovered by Mc Cord and Fridovich [11], is an ubiquitous CuZn binding dimeric enzyme, mainly localized in cytoplasm and also present in mitochondrial intermembrane space, involved in the physiological scavenging of O2•− radicals, that are converted to molecular oxygen and hydrogen peroxide (H2O2) through the alternate reduction and re-oxidation of Cu2+.
SOD1 secretion has been observed in multiple cell models [12,13,14,15,16]; moreover, the ability of SOD1 to act as a signaling molecule, independent of its enzymatic activity, has been described [17,18].
Both redox stress and inflammation are associated with ALS pathogenesis/progression. Neuroinflammation represents a complex phenomenon, not only associated with the dysregulation of the glial cells surrounding neurons in the Central Nervous System (CNS), but also dependent on multiple regulatory molecular networks involving both innate and adaptive immune effectors [19,20,21]; furthermore, the role of T lymphocytes in inflammatory process regulation has been also demonstrated [22].
Hydrogen peroxide and superoxide anion have been described to participate in fine tuning of antigen-dependent immune response [23]. Indeed, activation of human T lymphocytes has been described to increase their SOD1 intracellular mRNA production and microvesicle secretion [23]. Moreover, in vitro administration of recombinant human SOD1 to activated human T cells has been observed to increase their production of the pro-inflammatory cytokine IL17 [24], playing a key role in the regulation of chronic inflammatory conditions [25,26].
Although ALS mostly affects patients without disease family histories, about 10% of ALS subjects show a familial form of this disease (fALS), mainly due to different gain-of-function mutations of SOD1 enzyme; indeed, the presence of a sporadic ALS (sALS), not clearly associated with SOD1 defects [27], has been described. However, in both conditions, the key events involved in ALS initiation and progression are still largely unclear.
Here, we propose that derangement of SOD1-dependent pathways, in neurons as well as in immune effectors, might play a key pathogenetic role in ALS and also in the absence of a mutated form of the enzyme.
2. Pathogenic Mechanisms of Mutant SOD1 in ALS
J.D. Atkin [28] demonstrated that mutated SOD1 (mSOD1) can inhibit secretory protein transport from the endoplasmic reticulum (ER) to Golgi apparatus, due to ER stress, Golgi fragmentation and altered axonal transport. The toxic effect carried out by SOD mutations in fALS has been attributed to its gain-of-function activity, in some cases associated with the occurrence of copper-dependent oxidative stress, largely demonstrated in patients affected by this condition. This “copper-dependent” oxidative hypothesis is challenged by the observation that the removal of copper chaperon from both wild-type SOD1 and mSOD1 is not effective in preventing motoneuron degeneration in transgenic mouse models of fALS [29]. In addition, it should be highlighted that SOD1 could itself be strongly oxidized, which may not only compromise the antioxidation function, but also acquire new binding and toxic properties [29,30].
Toxic effects of mSOD1 have also been related to its ability to produce insoluble intracellular aggregates, likely caused by the altered folding of the mutated protein, able to generate amyloid-like fibrillary structures, as proposed by the ‘aggregation hypothesis’ [31,32,33,34,35]. This hypothesis is particularly interesting because protein aggregation is frequently observed in ALS [36,37,38]. In addition, a zinc-deficient SOD1 has also been described to increase its aggregation ability upon oxidation [31], thus promoting the peroxynitrite-dependent protein tyrosine nitration, inducing motoneuron apoptosis [39]. Moreover, other studies, using transgenic rat/mouse models [40] as well as bone marrow transplants or chimeric animals, demonstrated that mSOD1 expression, in microglia cells, can also induce direct neuronal toxicity [41].
Between the several SOD1 mutations, the SOD1G93A represents the mSOD1 molecule mostly used for in vitro and in vivo studies regarding human fALS [42,43]. In the transgenic SOD1G93A ALS rodent model, unfolded protein response and ER stress-induced apoptosis have been observed [44].
Autophagy and mitophagy are conserved degradation mechanisms finalized to maintain cellular homeostasis and to respond to cellular stress conditions. These processes have been described to be specifically tuned in highly differentiated cells, like neurons [45]. Several genes implicated in control of autophagy and mitophagy have been observed to be associated with ALS. Thus, their involvement in the impairment of the protein clearance and in the formation of aggregates, severely damaging motor neurons of ALS patients, may be hypothesized [46]. Data on SOD1G93A expressing neurons reveal in vitro a limited ability of such cells to respond to proteotoxic stress [47]. Moreover, in transgenic SOD1G93A mouse models, the presence of a protein complex positive for p62, a product of the ALS-associated gene SQSTM1 that is linked with protein aggregates formation and to the presence of damaged mitochondria, has been observed [48].
3. ALS and Mitochondrial Damage
Cellular motoneuron damage in ALS has been described to be also associated with the mitochondrial alterations frequently observed in motor axon terminals of muscle biopsies of patients with early diagnosis of sporadic ALS. In fact, misfolded, metal-free SOD1 mutants form insoluble aggregates in motoneurons; such a condition represents an early event in the pathogenesis of ALS [49]. These mitochondrial alterations might generate an impairment in the respiratory chain metabolic processes, thus increasing oxidative stress induced damage [50,51].
Many SOD1 mutants show altered metal-binding ability; therefore, it has been suggested that metal-deficient SOD1, which is more readily taken up by mitochondria, may acquire copper in the intermembrane space causing protein aggregation and extensive mitochondria and cellular damages [52,53].
Physiological mitochondrial activity associates with outer and inner mitochondrial membrane separation, ensured by the integrity of the intermembrane space. Loss of such integrity generates a permeability alteration associated with the formation of the Mitochondrial Permeability Transition Pore (mPTP) [54,55]. Wild-type and mutant SOD1 have been observed to associate with mitochondria [47,56]. Post-translational oxidized wild-type SOD1 as well as its mutants have been observed to link Bcl-2, physiologically expressed in mitochondria, perturbing its structure and function with consequent release of cytochrome C and neuron death induction [30,57]. Moreover, this alteration generates an impairment of electron chain transport with further increased ROS production. This event might induce a pathological loop able to perturbate the redox mitochondrial homeostasis in motoneurons. SOD1G93A has been observed, in mouse models, to access mitochondria matrix, thus inducing strong perturbation of oxidative phosphorylation [58,59,60,61].
4. Microglia Activation, NADPH Oxidase and SOD1 in ALS
Activated microglia represents a key element in Alzheimer’s disease as well as in the pathogenesis of ALS and other neurodegenerative conditions [62,63,64]. Microglia activation can be observed in transgenic mice expressing human SOD1 mutants, before neuron loss [65]; thus, dysregulated microglia functions, together with astrocyte activation, carry out an important role in ALS pathogenesis/progression [62,63,64,66].
ROS generated from NADPH oxidases play a role in signaling events leading to microglia activation [67]. Seven structural homologues of the phagocyte NOX enzyme (NOX2) have been identified, such as NOX1-5, DUOX1 and DUOX2 [68]. On the other hand, activated microglia produces ROS, primarily by NADPH oxidase 2 (NOX2), that result in enhanced microglia activation. It has also been shown that redox distress, caused by NOX1 and NOX2, significantly influences the progression of motor neuron disease, in mutant SOD1G93A ALS mice [69]. Generation of ROS represents a general phenomenon in human cells. However, the excess of ROS production or an imbalance between ROS production and antioxidant defense are thought to represent important factors of disease progression in ALS [70,71].
Recent studies have shown that SOD1G93A ALS transgenic mice generate high levels of gp91PHOX (NOX2) and superoxide in spinal cord microglia [72]. Moreover, it has been proposed that SOD1 interacts directly with Rac1, a cytosolic regulator of NOX2 [73], resulting in overproduction of ROS. In addition, ROS generated by Rac-dependent NADPH oxidases have been observed to be involved in cell signaling as well as in microbial killing.
Harraz [74] hypothesized that Rac-GTP-mediated activation of the NADPH oxidase complex might lead to production of O2•− and H2O2, which were able to mediate Nox complex autoregulation by reducing Rac-GTP levels. Interestingly, Marden [69] showed that female ALS mice, lacking a copy of the X-chromosomal Nox1 or Nox2 genes, exhibited significantly increased survival rates, thus suggesting that a 50% reduction in NOX1/NOX2 expression levels might be associated with a substantial improvement of ALS outcome in mice. Moreover, the observation that multiple Nox genes might contribute to ALS progression clearly expands the potential therapeutic targets for this disease.
5. SOD1, Immunity and Neuroinflammation Processes in ALS
Chronic inflammation has been considered an important element in the pathogenesis/progression of different diseases, as well as in neurodegenerative processes. [75]. In this context, multiple immune dysfunctions, represented by extensive, dysregulated inflammatory processes, auto immunity phenomena, and deranged immune responses have been described in ALS. In addition, mutations in several genes, directly involved in immune response, have recently been reported in ALS patients [76,77,78,79].
Neuroinflammation, in particular, behaves as a key modulator of ALS progression potentially representing a prospective therapeutic target for this disease. In ALS, inflammatory responses are not restricted to the proximity of motoneurons but have been detected in muscles, peripheral axons, skin, liver and blood [80,81,82,83,84,85].
Multiple immune cell subsets have been described to participate in ALS pathogenetic mechanisms. Indeed, T cells, monocytes and other immune effectors have been observed to directly or indirectly access the CNS through the choroid plexus [86], thus mediating neuron damaging as well as neuroprotective processes.
E. Coque et al. [87] showed that ablation of CD8+ T cells in ALS mice increased the number of surviving motoneurons. Moreover, CD8+ T cells expressing the ALS-causing mSOD protein have been described to recognize and selectively kill motoneurons in vitro. To exert their cytotoxic function, CD8+ T cells carrying mSOD1 must be able to recognize neuron antigens inside MHC-I complex at the surface of the motoneurons. Moreover, analysis of the T cell receptor (TCR) diversity supports the evidence that self-reactive CD8+ T lymphocytes infiltrate the CNS of ALS mice and exert cytotoxic functions.
Fine-tuning of immune response is usually obtained by multiple regulatory processes, all belonging to the immune tolerance network, that are in place to prevent potentially deleterious immune responses against self-tissues. Regulatory T cells (Treg) are CD4 T lymphocytes characterized by the expression of the Foxp3 transcription factor. This T cell subset controls the immune effector response in terms of clonal expansion, differentiation, cytokine profile and tissue migration and is indispensable for the maintenance of immune self-tolerance [88]. Clinical studies in humans and in transgenic mouse models pointed out the role of Tregs in ALS pathophysiology. In humans and in mouse ALS models, Tregs infiltrating the Central Nervous System have been observed to suppress neuroinflammatory processes and promote the activation of neuroprotective microglia. Thus, immune-modulation strategies aimed at increasing Treg number and enhancing its functional effectiveness might be considered relevant to promote neuroprotective activity in ALS [89].
mTOR is an evolutionarily conserved serine/threonine protein kinase that directly influences T cell differentiation and proliferation by integrating environmental cues (nutrients, energy stores and growth factors) with immunity functions [90]. mTOR activity exerts opposite effects on effector T lymphocytes and on Treg. Indeed, mTOR inhibition strongly favors Treg differentiation and expansion while modulating T cell effector functions [91]. Compelling evidence indicate that progressive metabolic conversions, usually mediated by mTOR-dependent pathways, underlie the generation of proper effector functions during T cell response. Recent evidence indicates that SOD1 represents one of the major targets of the mTOR enzyme. Indeed, reversible mTOR-dependent SOD1 phosphorylation has been described to mediate SOD1 inhibition [91,92]. These observations propose a complex scenario in which SOD1/mTOR intracellular interplay may finely tune T cell activity [93,94]; instead, no data are available on the role of SOD1 in regulating mTOR dependent pathways. In this context, the recent correlation of increasing SOD1 intracellular levels in T cells with the presence of circulating Tregs in a cohort of subjects affected by Multiple Sclerosis undergoing effective immune-modulating treatment [24], strongly supports the idea that a SOD1-mTOR regulatory network [93,94] may participate in the complex mechanisms regulating immune-modulation processes.
Hydrogen peroxide and superoxide anion are involved in TCR-dependent signaling and adaptive immune response activation [23]. Indeed, antigen-dependent stimulation of human T lymphocytes can modify SOD1 intracellular localization in T cells, mediating a clear SOD1 recruitment by TCR clusters. In addition, increased SOD1 mRNA production and microvesicle secretion of the enzyme, by activated human T lymphocytes, has been observed [23].
We found [24] that in vitro administration of recombinant human SOD1 to activated human T cells increases their IL17 production, a key cytokine in induction/maintenance of chronic inflammatory processes [25,26]. This effect is mediated by SOD1-dependent enzymatic activity, since SOD1 molecule lacking dismutase activity (Apo SOD1), it is unable to affect T cell cytokine production. Furthermore, hydrogen peroxide addition to activated T-cells mimics the SOD1 effect [24]. These data indicate, as summarized in Figure 1, that SOD1 effects on inflammatory process may be, at least partially, linked to its hydrogen peroxide production; this molecule is more stable than other ROS and can freely migrate outside cells and between different cell compartments [24].
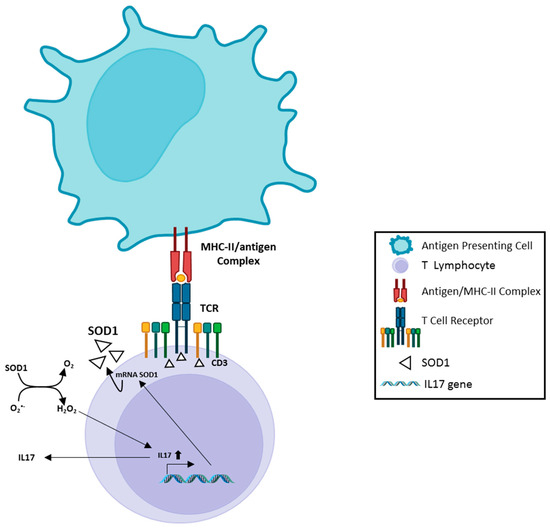
Figure 1.
A scheme of SOD1 involvement in antigen-dependent T cell response. Recognition of antigen by TCR, inside MHC complex expressed on the surface of the Antigen Presenting Cell, induces rapid recruitment of SOD1 molecules near the TCR clusters; this phenomenon is accompanied by increased SOD1-mRNA, followed by SOD1 export by a microvesicle pathway. Extracellular SOD1 has been observed to mediate enhanced IL17 production by activated T lymphocytes. This effect depends on enzymatic activity of SOD1; the involvement of IL17 in induction/maintenance of chronic proinflammatory processes has been extensively studied by others. The image has been partially created by using BioRender.com.
Thus, a very complex scenario involving multiple molecular networks, including SOD1-dependent pathways, might be involved in the regulation of T cell activation and differentiation in the neuroinflammatory context.
6. Constitutive and Inducible SOD1 Secretion
The first demonstration of constitutive SOD1 secretion in many eukaryotic cells date back to many years ago when we, for the first time, showed the export of this protein in hepatocytes and fibroblasts [95], in neuroblastoma SK-N-BE cells [96] and in thymus-derived epithelial cells [12]. Many others researchers confirmed our observations [13,14,15,16]. It is of relevance that although more than 33% of all proteins are exported through the ER and Golgi compartments [97], wild-type SOD1 is secreted by an alternative pathway, bypassing the canonical ER–Golgi apparatus [13].
SOD1 constitutively produced and released by microglia cells, by a lysosomal secretory pathway, has been also described to play a neuroprotective role [15]. Moreover, an association between impaired constitutive extracellular secretion of mutant SOD1, the presence of cytoplasmic insoluble protein inclusions and toxicity in NSC-34 cell line [98] has also been reported.
In the ALS transgenic rat model, the chronic intraspinal infusion of exogenous unmutated human SOD1 significantly delayed disease progression, suggesting a novel extracellular role for SOD1 in ALS pathogenesis and therapy [16]. In this context, a chromogranin-mediated secretion of mutant SOD1, but not wild-type SOD1 proteins, linked to ALS progression, has been observed [16], while a clear association of microglia activation with the occurrence of motor neurons toxicity has been revealed in transgenic mice carrying SOD1 mutations [16,99].
The constitutive SOD1 secretion suggests a paracrine antioxidant and protective role of this protein against oxygen radical in extracellular milieu. In addition, the mechanisms underlying the observed proinflammatory effects of SOD1 secretion [24] and the ability of the enzyme to potentiate the kinase activity in certain cell types by directly or indirectly modulating phosphatase functional effectiveness [100,101,102] need to be further investigated.
In vitro experiments performed by our group demonstrated that SOD1 is able to directly interact with cell surface of human neuroblastoma SK-N-BE cells activating a phospholipase C-dependent (PLC-PKC) pathway, with consequent massive intracellular Ca2+ increase; interestingly, this effect is independent from the dismutase activity of SOD1 molecule, since apo SOD1 (free metal SOD1) or mimetic SOD1 (MnTMPyP) were observed to mediate the same effects. Moreover, U73122, a powerful PLC inhibitor, was observed to strongly reduce SOD1-induced intracellular Ca2+ increase [17].
In addition to the constitutive SOD1 secretion, we showed that SOD1 is present in large dense core vesicles actively released from rat brain synaptosomes, as well as from rat pituitary GH3 cells. This rapid SOD1 export, mediated by depolarization and induced by high extracellular K+ concentration, is strictly associated with an increase in intracellular Ca2+-mediated SOD1 exocytosis; this SOD1 export has been observed to rely on the SNARE protein-dependent synaptic exocytosis machinery [103].
7. Role of SOD1 Interaction with the Muscarinic M1 Receptor
Accumulation of Ca2+ in neurons, with consequent activation of Ca-dependent pathways, has been largely associated with cell death [104]. Intracellular Ca2+ increase might be mediated by different mechanisms, as represented by several voltage-gate-dependent calcium channels (VGCCS), metabotropic (M1 muscarinic) receptors and ionotropic glutamate-dependent receptors (N-Methyl-D-aspartate (NMDA) and non-NMDA molecules). In addition, the intracellular Ca2+ increase can be mediated by the involvement of metabotropic glutamate receptors (GRM) [105]. Reduced glutamate re-uptake, mediated by altered transporter availability, has been also associated with increased Ca2+ concentration at neuron post-synaptic level. Moreover, Ca2+ overload inside mitochondria has been observed to induce deranged generation of ROS with subsequent availability of pro-apoptotic factors. Such altered ROS production may be involved in damaging processes along with ‘free radical buffering depletion’, mediated by glutamate competing with cysteine at the glutamate–cysteine pump [106].
Excitotoxicity has been defined as a phenomenon dependent on massive glutamate release related to depolarization, as well as on impaired glutamate re-uptake. Both mechanisms lead to over-activation of NMDA receptors thus inducing heavy intracellular Ca2+ influx [107]. Still largely unknown are the potential downstream effects due to interactions between mSOD1, M1 receptors and glutamate excitotoxicity.
Increased levels of glutamate in the cerebrospinal fluid, likely dependent on the selective decrease in the glial glutamate transporter EAAT2, associated with reduced glutamate transport into astrocytes, have been characterized in subjects affected by sporadic ALS [108]. In this context, excitotoxicity, generated by excessive glutamate levels, has been proposed as a relevant mechanism involved in ALS pathogenesis.
Our group recently found that SOD1 interaction with muscarinic M1 receptor, in human neuroblastoma SK-N-BE cells, can activate ERK1/2 and AKT kinases in a dose/time-dependent manner. This effect was strongly reduced by M1 receptor silencing, as well as by using the M1 antagonist pirenzepine. Moreover, the incubation of SK-N-BE and the neuroblastoma–spinal motoneuron fusion NSC-34 cell line with mutant SOD1G93A significantly increased their intracellular Ca2+ concentration, as compared with wild-type SOD1 treatment [109].
Primary myocytes and skeletal muscle fibers derived from SOD1G93A transgenic mice have been observed to show perturbed expression of Ca2+ transporters, likely responsible for their altered mitochondrial Ca2+ fluxes [110]. These defects occur in young SODG93A mice prior to the disease onset. Thus, mSOD1 mutants might be involved in the alterations in the skeletal myocytes’ functions largely reported in ALS.
The elevation of mitochondrial Ca2+ concentration, induced by muscle contraction or muscle inactivity, has been hypothesized to strongly interfere with ROS production. Moreover, temporal profile of mitochondrial Ca2+ intracellular levels has been suggested to behave as a physiological switch flipping between the beneficiary versus destructive outcomes [111,112].
The potential ability of SOD1 mutants to affect Ca2+ mitochondrial levels, consequently interfering with mitochondrial ROS production, needs to be investigated.
These observations, as a whole, are conceivable with the hypothesis, summarized in Figure 2, that the Ca2+-dependent excitotoxicity, associated with depolarization-dependent increase in SOD1 extracellular levels with consequent M1 receptor/SOD1 interaction, may be involved in the derangement of key neuro-modulatory networks during ALS pathogenesis.
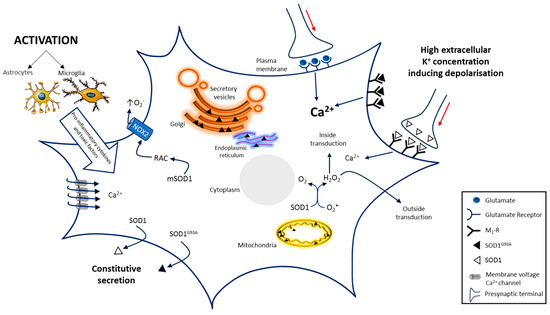
Figure 2.
A scheme of the SOD1-dependent molecular mechanisms involved in motoneuron death in ALS. SOD1-dependent pathways may damage motoneurons increasing ROS production through RAC activation, that impairs protein export, by protein aggregation in the ER–Golgi apparatus and by inducing deranged mitochondrial activity generating further ROS production Excitotoxicity induced by glutamatergic synapses hyper-stimulation or by impaired glutamate transporter into glial cells and presynaptic membrane, also contributes to motoneuron death. Similar effects could be due to SOD1 and even more due to SOD1G93A interaction with metabotropic muscarinic M1 receptor. Both mechanisms generate excitotoxicity by increasing intracellular Ca2+ concentration that alters multiple Ca2+-dependent signaling pathways. Activation of glial cells, generating pro-inflammatory cytokines and toxic factors, also contributes to the progression of ALS. Others factors involved in the pathogenesis of ALS progression are not represented. The red arrows indicate signal propagation inside axon; the increasing black indicate higher concentration. Image has been partially created by using BioRender.com.
Five muscarinic receptor subtypes (m1–m5) have been identified in T lymphocytes [113,114]; m1, m3 and m5 muscarinic receptor subtypes are coupled to Gq/11, which, upon stimulation, mediate activation of phospholipase C activity, resulting in increases in Ca2+ availability inside cells [115]. Moreover, acetylcholine is synthesized and released by T-lymphocytes, acting as an autocrine/paracrine factor, likely involved in immune function regulation [115].
These observations add further complexity to the biological scenario involved in the regulation of the neuroinflammatory context.
8. Discussion
ALS is a progressive motor neurodegenerative disease whose pathogenetic mechanisms are still largely unknown. Gain-of-function mutations of SOD1 enzyme, physiologically involved in the intracellular generation of hydrogen peroxide, have been associated with the familial form of ALS, usually accounting for 10% of all the cases, involving multiple gene mutations.
Neuron functional effectiveness and survival have been largely described to depend on a complex molecular network involving intracellular neuronal and extra-neuronal circuits. The function of neuron-surrounding microglia cells as well as the presence of neuroprotective immune-dependent pathways, usually regulated by multiple cell subsets, have been extensively studied. In this context, a deranged regulation of each component of this complex scenario may be hypothesized to be relevant for ALS pathogenesis/progression.
Compelling evidence indicate that in familial and sporadic ALS, SOD1 misfolded proteins are able to mediate endoplasmic reticulum (ER) stress and fragmentation of the Golgi apparatus, resulting in impaired protein secretion and cellular apoptosis. Moreover, extensive mitochondrial alterations, with severe impairment in the respiratory chain metabolic processes, have been described to increase oxidative stress-induced damage [49,50,51].
SOD1 mutants have been largely demonstrated to maintain their enzymatic activity, suggesting that the role of mSOD1 in ALS is characterized by a gain-of-toxic function, rather than by a loss of function feature [116,117].
The secretion of SOD1 by normal and transformed cells has been previously reported [12,13,95,96]; in addition, our in vitro experiments demonstrated that SOD1, in SK-NB-E cells, activates the PLC-PKC pathway increasing intracellular Ca2+ concentration; these data are confirmed by the ability of the PLC inhibitor U73122 to revert SOD1-dependent Ca2+ increase in SK-NB-E cells [17].
The mechanistic difference between the pathogenesis of familiar and sporadic forms of ALS has not yet been clarified, since in the latter, misfolded wild-type SOD1 protein seems to activate the same neurotoxic pathway, that is mediated by SOD1 mutants in familiar ALS [118,119]. The mechanism by which mutated SOD1G93A induces ER stress, Golgi apparatus fragmentation and accumulation of misfolded or unfolded proteins at the endoplasmic reticulum lumen has been extensively studied, but is still not quite clear [98,120,121].
In this review, we highlight the observations that SOD1 enzyme acts as a signaling molecule modulating different subcellular processes to adjust molecular homeostasis of the cell. SOD1 deficiency is associated with oxidative distress in Multiple Sclerosis, an autoimmune demyelinating disease, evidencing the importance of this molecule in maintaining normal function of the central nerve system [122].
Recent data obtained in SODG93A mouse ALS model [123] reveal that the treatment outcome may significantly vary with the disease progression. Indeed, targeting of oxidative stress in pre-onset, of excitability in near onset, of inflammation in post-onset, and of apoptosis in the end-stage condition have been shown to be associated with disease improvement. In this context, SOD1 molecule can be considered as a potential unifying molecular pathway, whose proper targeting might improve ALS treatment at different disease stages.
Many effects of SOD1 could be ascribed to its peroxide generation obtained by radical oxygen dismutation; in this context, hydrogen peroxide concentration shift, from physiological (in eustress condition) to pathological (higher H2O2) levels, may participate in SOD1-mediated pathological effects. In fact, in eustress condition, H2O2 can be considered as a key signaling molecule that is able to regulate many physiological functions in relation to its concentration, like innate and adaptive immunity activities [124], biosynthesis of thyroid hormones, multiple cells signaling pathways, gene expression and cellular growth [125,126].
Besides the role of H2O2 as a physiological signal transduction molecule, our in vitro experiments indicate that the availability of SOD1-containing extracellular microvesicles, dependent on the depolarization of excitable cells, induces a markedly higher extracellular SOD1 concentration, as compared with the SOD1 concentration released in the constitutive secretion. This effect generates an increase in intracellular Ca2+ concentration that is strongly enhanced by incubating excitable cells with SOD1G93A [109].
Therefore, it can be conceivable that an enhanced export of SOD1G93A from excitable cells, with consequent increased interaction with muscarinic M1 membrane neuron receptor, results in a significant increase in intracellular Ca2+ level associated with neuron cells apoptotic death, as suggested by our in vitro observations.
Thus, besides the Ca2+ cytotoxic effects induced by ROS and NMDA glutamate receptors stimulations, a further marked increase in intracellular Ca2+ concentration may be likely carried out by the deranged SOD1G93A muscarinic M1 receptor interaction. Further experiments by depolarization of human neuroblastoma NS-K-BE cells transfected with SOD1G93A are necessary to strengthen such hypothesis.
Neuron depolarization and antigen-dependent T cell activation both contribute to the exocytosis of SOD1, leading to an increased interaction of the enzyme with a complex receptor network. This interaction could play a crucial role in neuroinflammation during ALS pathogenesis.
The involvement of deranged inflammatory processes in motoneuron degeneration as well as the key role of immune-mediated neuroprotective pathways, closely related to T cell activity and differentiation, has been largely recognized. Indeed, T cell response in CNS has been described as able to mediate both neuroprotection as well as damaging effects on neuronal tissues [127,128,129]. In this context, Treg subset, physiologically involved in the tolerance control, has been observed to play a relevant role for neuroprotection against neuroinflammatory processes and for neurological recovery [128,130]. Our recent observations [24] have also been proposing the relevant role of SOD1 intracellular levels in Treg differentiation. Consistently, the ability of mTOR enzyme, a key regulator of Treg subset, to control SOD1 intracellular activity has been revealed [93,94].
9. Conclusions
The collective findings here support the notion that SOD1, including its gain-of-function mutant SOD 1 isoforms, plays a pivotal role in multiple cellular pathways, whose derangement might represent a key element in ALS pathogenesis/progression.
Specifically, derangement of SOD1-dependent pathways has been proposed to perturbate the complex immune regulatory mechanisms, likely crucial for the control of neuroinflammatory processes and neurological recovery. Moreover, excitotoxicity, largely described in ALS, may be enhanced by the ability of SOD1 and even more by its mSOD1G93A mutant to generate, also by interaction with muscarinic M1 receptor, a pathological increase in intracellular Ca2+ levels, with consequent neuron apoptosis.
These observations bring new insights on the complex interplay between SOD1, neurotoxicity and neuroinflammation, with the potential to inspire novel therapeutic strategies to control the detrimental effects of SOD1 dysregulation in neurodegenerative disorders.
Author Contributions
V.R. and G.L.R. searched and selected the literature, participated in designing the structure of the paper and in writing the manuscript; L.P., G.L.R. and V.R. created the pictures; F.C. performed the literature search and participated in writing the paper; S.D., M.S. and G.T. reviewed and revised the manuscript and contributed toward writing the paper; G.R. and P.M. selected and evaluated the literature, designed the structure of the paper and drafted, reviewed and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Phukan, J.; Pender, N.P.; Hardiman, O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007, 6, 994–1003. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Cozzolino, M.; Pesaresi, M.G.; Gerbino, V.; Grosskreutz, J.; Carrì, M.T. Amyotrophic lateral sclerosis: New insights into underlying molecular mechanisms and opportunities for therapeutic intervention. Antioxid. Redox Signal 2012, 17, 1277–1330. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Clarendon Press: Oxford, UK, 1989. [Google Scholar]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Cimini, V.; Ruggiero, G.; Buonomo, T.; Seru, R.; Sciorio, S.; Zanzi, C.; Santangelo, F.; Mondola, P. CuZn-superoxide dismutase in human thymus: Immunocytochemical localisation and secretion in thymus-derived epithelial and fibroblast cell lines. Histochem. Cell Biol. 2002, 118, 163–169. [Google Scholar] [CrossRef]
- Mondola, P.; Ruggiero, G.; Serù, R.; Damiano, S.; Grimaldi, S.; Garbi, C.; Monda, M.; Greco, D.; Santillo, M. The Cu,Zn superoxide dismutase in neuroblastoma SK-N-BE cells is exported by a microvesicles dependent pathway. Brain Res. Mol. Brain Res. 2003, 110, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef]
- Polazzi, E.; Mengoni, I.; Caprini, M.; Peña-Altamira, E.; Kurtys, E.; Monti, B. Copper-zinc superoxide dismutase (SOD1) is released by microglial cells and confers neuroprotection against 6-OHDA neurotoxicity. NeuroSignals 2013, 21, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Sik, A.; Sakurai, T.; Nukina, N.; Takahashi, R.; Julien, J.P. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci. 2006, 9, 108–118. [Google Scholar] [CrossRef]
- Mondola, P.; Santillo, M.; Serù, R.; Damiano, S.; Alvino, C.; Ruggiero, G.; Formisano, P.; Terrazzano, G.; Secondo, A.; Annunziato, L. Cu,Zn superoxide dismutase increases intracellular calcium levels via a phospholipase C-protein kinase C pathway in SK-N-BE neuroblastoma cells. Biochem. Biophys. Res. Commun. 2004, 324, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Viggiano, A.; Serù, R.; Damiano, S.; De Luca, B.; Santillo, M.; Mondola, P. Inhibition of long-term potentiation by CuZn superoxide dismutase injection in rat dentate gyrus: Involvement of muscarinic M1 receptor. J. Cell Physiol. 2012, 227, 3111–3115. [Google Scholar] [CrossRef]
- Becher, B.; Prat, A.; Antel, J.P. Brain-immune connection: Immuno-regulatory properties of CNS-resident cells. Glia 2000, 29, 293–304. [Google Scholar] [CrossRef]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Castellani, G.; Croese, T.; Peralta Ramos, J.M.; Schwartz, M. Transforming the understanding of brain immunity. Science 2023, 380, eabo7649. [Google Scholar] [CrossRef]
- Schnell, A.; Littman, D.R.; Kuchroo, V.K. T(H)17 cell heterogeneity and its role in tissue inflammation. Nat. Immunol. 2023, 24, 19–29. [Google Scholar] [CrossRef]
- Terrazzano, G.; Rubino, V.; Damiano, S.; Sasso, A.; Petrozziello, T.; Ucci, V.; Palatucci, A.T.; Giovazzino, A.; Santillo, M.; De Felice, B.; et al. T cell activation induces CuZn superoxide dismutase (SOD)-1 intracellular re-localization, production and secretion. Biochim. Biophys. Acta 2014, 1843, 265–274. [Google Scholar] [CrossRef]
- Rubino, V.; Palatucci, A.T.; La Rosa, G.; Giovazzino, A.; Aruta, F.; Damiano, S.; Carriero, F.; Santillo, M.; Iodice, R.; Mondola, P.; et al. Superoxide Dismutase-1 Intracellular Content in T Lymphocytes Associates with Increased Regulatory T Cell Level in Multiple Sclerosis Subjects Undergoing Immune-Modulating Treatment. Antioxidants 2021, 10, 1940. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Jin, M.; Akgün, K.; Ziemssen, T.; Kipp, M.; Günther, R.; Hermann, A. Interleukin-17 and Th17 Lymphocytes Directly Impair Motoneuron Survival of Wildtype and FUS-ALS Mutant Human iPSCs. Int. J. Mol. Sci. 2021, 22, 8042. [Google Scholar] [CrossRef] [PubMed]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Soo, K.Y.; Walker, A.K.; Halloran, M.; Turner, B.J.; Nagley, P.; Horne, M.K. Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J. Neurochem. 2014, 129, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Ezzi, S.A.; Urushitani, M.; Julien, J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef]
- Elam, J.S.; Taylor, A.B.; Strange, R.; Antonyuk, S.; Doucette, P.A.; Rodriguez, J.A.; Hasnain, S.S.; Hayward, L.J.; Valentine, J.S.; Yeates, T.O.; et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat. Struct. Biol. 2003, 10, 461–467. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Rumfeldt, J.A.O.; Scholz, G.A.; Irani, R.A.; Frey, H.E.; Hallewell, R.A.; Lepock, J.R.; Meiering, E.M. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc. Natl. Acad. Sci. USA 2003, 100, 7021–7026. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Valentine, J.S.; Eggers, D.K.; Roe, J.A.; Tiwari, A.; Brown, R.H., Jr.; Hayward, L.J. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J. Biol. Chem. 2002, 277, 15932–15937. [Google Scholar] [CrossRef]
- Bendotti, C.; Carrì, M.T. Lessons from models of SOD1-linked familial ALS. Trends Mol. Med. 2004, 10, 393–400. [Google Scholar] [CrossRef]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef]
- Alexander, G.M.; Erwin, K.L.; Byers, N.; Deitch, J.S.; Augelli, B.J.; Blankenhorn, E.P.; Heiman-Patterson, T.D. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res. Mol. Brain Res. 2004, 130, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Nilsson, P.; Ala-Hurula, V.; Keränen, M.L.; Tarvainen, I.; Haltia, T.; Nilsson, L.; Binzer, M.; Forsgren, L.; Marklund, S.L. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat. Genet. 1995, 10, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Alexianu, M.E.; Ho, B.K.; Mohamed, A.H.; La Bella, V.; Smith, R.G.; Appel, S.H. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 1994, 36, 846–858. [Google Scholar] [CrossRef]
- Beckman, J.S.; Estévez, A.G.; Crow, J.P.; Barbeito, L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001, 24, S15–S20. [Google Scholar] [CrossRef] [PubMed]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Sun, H.; Bénardais, K.; Stanslowsky, N.; Thau-Habermann, N.; Hensel, N.; Huang, D.; Claus, P.; Dengler, R.; Stangel, M.; Petri, S. Therapeutic potential of mesenchymal stromal cells and MSC conditioned medium in Amyotrophic Lateral Sclerosis (ALS)--in vitro evidence from primary motor neuron cultures, NSC-34 cells, astrocytes and microglia. PLoS ONE 2013, 8, e72926. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Turner, B.J.; Tomas, D.; Lysaght, J.A.; Nunan, J.; Rembach, A.; Nagley, P.; Beart, P.M.; Cheema, S.S.; et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 2006, 281, 30152–30165. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown Jr, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef]
- Johnston, J.A.; Dalton, M.J.; Gurney, M.E.; Kopito, R.R. Formation of high molecular weight complexes of mutant Cu,Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 12571–12576. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondria and the pathogenesis of ALS. Brain 2000, 123, 1291–1292. [Google Scholar] [CrossRef]
- Siklós, L.; Engelhardt, J.; Harati, Y.; Smith, R.G.; Joó, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophic lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar] [CrossRef]
- Okado-Matsumoto, A.; Fridovich, I. Amyotrophic lateral sclerosis: A proposed mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 9010–9014. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, V.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Martin, L.J. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Biochim. Biophys. Acta 2010, 1802, 186–197. [Google Scholar] [CrossRef]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef] [PubMed]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, E.; Van Damme, P.; Van Den Bosch, L.; Robberecht, W. Vascular endothelial growth factor in amyotrophic lateral sclerosis and other neurodegenerative diseases. Muscle Nerve 2006, 34, 391–405. [Google Scholar] [CrossRef]
- Bomsztyk, K.; Denisenko, O.; Ostrowski, J. hnRNP K: One protein multiple processes. Bioessays 2004, 26, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Lee, M.K.; Slunt, H.S.; Guarnieri, M.; Xu, Z.S.; Wong, P.C.; Brown, R.H., Jr.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8292–8296. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Bonvicini, C.; Alberici, A.; Buratti, E.; Agosti, C.; Archetti, S.; Papetti, A.; Stuani, C.; Di Luca, M.; Gennarelli, M.; et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 2009, 30, E974–E983. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; McGeer, E.G. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. 1988, 76, 550–557. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. History of innate immunity in neurodegenerative disorders. Front. Pharmacol. 2011, 2, 77. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Pramatarova, A.; Laganière, J.; Roussel, J.; Brisebois, K.; Rouleau, G.A. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J. Neurosci. 2001, 21, 3369–3374. [Google Scholar] [CrossRef]
- Qin, L.; Liu, Y.; Wang, T.; Wei, S.J.; Block, M.L.; Wilson, B.; Liu, B.; Hong, J.S. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J. Biol. Chem. 2004, 279, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Investig. 2007, 117, 2913–2919. [Google Scholar] [CrossRef]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef]
- Wu, D.C.; Ré, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef]
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schöneich, C.; et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Investig. 2008, 118, 659–670. [Google Scholar] [CrossRef]
- Harraz, M.M.; Park, A.; Abbott, D.; Zhou, W.; Zhang, Y.; Engelhardt, J.F. MKK6 phosphorylation regulates production of superoxide by enhancing Rac GTPase activity. Antioxid. Redox Signal 2007, 9, 1803–1813. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Dobson-Stone, C.; Hallupp, M.; Bartley, L.; Shepherd, C.E.; Halliday, G.M.; Schofield, P.R.; Hodges, J.R.; Kwok, J.B. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology 2012, 79, 995–1001. [Google Scholar] [CrossRef]
- Ono, S.; Hu, J.; Shimizu, N.; Imai, T.; Nakagawa, H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 187, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef]
- Chiu, I.M.; Phatnani, H.; Kuligowski, M.; Tapia, J.C.; Carrasco, M.A.; Zhang, M.; Maniatis, T.; Carroll, M.C. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20960–20965. [Google Scholar] [CrossRef] [PubMed]
- Gasco, S.; Zaragoza, P.; García-Redondo, A.; Calvo, A.C.; Osta, R. Inflammatory and non-inflammatory monocytes as novel prognostic biomarkers of survival in SOD1G93A mouse model of Amyotrophic Lateral Sclerosis. PLoS ONE 2017, 12, e0184626. [Google Scholar] [CrossRef] [PubMed]
- Paré, B.; Gros-Louis, F. Potential skin involvement in ALS: Revisiting Charcot’s observation—A review of skin abnormalities in ALS. Rev. Neurosci. 2017, 28, 551–572. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Trolese, M.C.; de Vito, G.; Cecchi, R.; Riva, N.; Dina, G.; Heath, P.R.; Quattrini, A.; Shaw, P.J.; Piazza, V.; et al. Immune response in peripheral axons delays disease progression in SOD1(G93A) mice. J. Neuroinflammation 2016, 13, 261. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8(+) T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 2006, 212, 8–27. [Google Scholar] [CrossRef]
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion With Progression of Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model. JAMA Neurol. 2018, 75, 681–689. [Google Scholar] [CrossRef]
- de Candia, P.; Procaccini, C.; Russo, C.; Lepore, M.T.; Matarese, G. Regulatory T cells as metabolic sensors. Immunity 2022, 55, 1981–1992. [Google Scholar] [CrossRef]
- Procaccini, C.; Matarese, G. Regulatory T cells, mTOR kinase, and metabolic activity. Cell Mol. Life Sci. 2012, 69, 3975–3987. [Google Scholar] [CrossRef]
- Tsang, C.K.; Zheng, X. F S. A balancing act: mTOR integrates nutrient signals to regulate redox-dependent growth and survival through SOD1. Mol. Cell Oncol. 2018, 5, e1488372. [Google Scholar] [CrossRef]
- Tsang, C.K.; Chen, M.; Cheng, X.; Qi, Y.; Chen, Y.; Das, I.; Li, X.; Vallat, B.; Fu, L.W.; Qian, C.N.; et al. SOD1 Phosphorylation by mTORC1 Couples Nutrient Sensing and Redox Regulation. Mol. Cell 2018, 70, 502–515.e508. [Google Scholar] [CrossRef]
- Damiano, S.; Sozio, C.; La Rosa, G.; Guida, B.; Faraonio, R.; Santillo, M.; Mondola, P. Metabolism Regulation and Redox State: Insight into the Role of Superoxide Dismutase 1. Int. J. Mol. Sci. 2020, 21, 6606. [Google Scholar] [CrossRef] [PubMed]
- Mondola, P.; Annella, T.; Santillo, M.; Santangelo, F. Evidence for secretion of cytosolic CuZn superoxide dismutase by Hep G2 cells and human fibroblasts. Int. J. Biochem. Cell Biol. 1996, 28, 677–681. [Google Scholar] [CrossRef]
- Mondola, P.; Annella, T.; Serù, R.; Santangelo, F.; Iossa, S.; Gioielli, A.; Santillo, M. Secretion and increase of intracellular CuZn superoxide dismutase content in human neuroblastoma SK-N-BE cells subjected to oxidative stress. Brain Res. Bull. 1998, 45, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.J.; Atkin, J.D.; Farg, M.A.; Zang, D.W.; Rembach, A.; Lopes, E.C.; Patch, J.D.; Hill, A.F.; Cheema, S.S. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci. 2005, 25, 108–117. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef]
- Fetrow, J.S.; Siew, N.; Skolnick, J. Structure-based functional motif identifies a potential disulfide oxidoreductase active site in the serine/threonine protein phosphatase-1 subfamily. FASEB J. 1999, 13, 1866–1874. [Google Scholar] [CrossRef]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Doñate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, H.Y.; Kim, H.; Park, S.M.; Joe, E.H.; Jou, I.; Choi, Y.H. Oxidative stress induces lipid-raft-mediated activation of Src homology 2 domain-containing protein-tyrosine phosphatase 2 in astrocytes. Free Radic. Biol. Med. 2009, 46, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Santillo, M.; Secondo, A.; Serù, R.; Damiano, S.; Garbi, C.; Taverna, E.; Rosa, P.; Giovedì, S.; Benfenati, F.; Mondola, P. Evidence of calcium- and SNARE-dependent release of CuZn superoxide dismutase from rat pituitary GH3 cells and synaptosomes in response to depolarization. J. Neurochem. 2007, 102, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Floyd, C.L.; Gorin, F.A.; Lyeth, B.G. Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia 2005, 51, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Chen, D.; Zhou, Q.; Hang, J.; Zhang, W.; Kuang, Z.; Sun, Z.; Li, L. Glutamate metabotropic receptor 4 (GRM4) inhibits cell proliferation, migration and invasion in breast cancer and is regulated by miR-328-3p and miR-370-3p. BMC Cancer 2019, 19, 891. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Luo, T.; Wu, W.H.; Chen, B.S. NMDA receptor signaling: Death or survival? Front. Biol. 2011, 6, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Damiano, S.; Sasso, A.; Accetta, R.; Monda, M.; De Luca, B.; Pavone, L.M.; Belfiore, A.; Santillo, M.; Mondola, P. Effect of Mutated Cu, Zn Superoxide Dismutase (SOD1(G93A)) on Modulation of Transductional Pathway Mediated by M1 Muscarinic Receptor in SK-N-BE and NSC-34 Cells. Front. Physiol. 2018, 9, 611. [Google Scholar] [CrossRef]
- Peggion, C.; Scalcon, V.; Massimino, M.L.; Nies, K.; Lopreiato, R.; Rigobello, M.P.; Bertoli, A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants 2022, 11, 614. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Ríos, E.; Deng, H.-X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef]
- Li, A.; Yi, J.; Li, X.; Zhou, J. Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front. Physiol. 2020, 11, 595800. [Google Scholar] [CrossRef] [PubMed]
- Bonner, T.I.; Young, A.C.; Brann, M.R.; Buckley, N.J. Cloning and expression of the human and rat m5 muscarinic acetylcholine receptor genes. Neuron 1988, 1, 403–410. [Google Scholar] [CrossRef]
- Bonner, T.I.; Buckley, N.J.; Young, A.C.; Brann, M.R. Identification of a family of muscarinic acetylcholine receptor genes. Science 1987, 237, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Fujii, T. Extraneuronal cholinergic system in lymphocytes. Pharmacol. Ther. 2000, 86, 29–48. [Google Scholar] [CrossRef]
- Strong, M.J.; Kesavapany, S.; Pant, H.C. The pathobiology of amyotrophic lateral sclerosis: A proteinopathy? J. Neuropathol. Exp. Neurol. 2005, 64, 649–664. [Google Scholar] [CrossRef]
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet. 2009, 10, 769–782. [Google Scholar] [CrossRef]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef]
- Forsberg, K.; Jonsson, P.A.; Andersen, P.M.; Bergemalm, D.; Graffmo, K.S.; Hultdin, M.; Jacobsson, J.; Rosquist, R.; Marklund, S.L.; Brännström, T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 2010, 5, e11552. [Google Scholar] [CrossRef] [PubMed]
- Nassif, M.; Matus, S.; Castillo, K.; Hetz, C. Amyotrophic lateral sclerosis pathogenesis: A journey through the secretory pathway. Antioxid. Redox Signal 2010, 13, 1955–1989. [Google Scholar] [CrossRef]
- Turner, B.J.; Atkin, J.D. ER stress and UPR in familial amyotrophic lateral sclerosis. Curr. Mol. Med. 2006, 6, 79–86. [Google Scholar] [CrossRef]
- Damiano, S.; Sasso, A.; De Felice, B.; Terrazzano, G.; Bresciamorra, V.; Carotenuto, A.; Orefice, N.S.; Orefice, G.; Vacca, G.; Belfiore, A.; et al. The IFN-β 1b effect on Cu Zn superoxide dismutase (SOD1) in peripheral mononuclear blood cells of relapsing-remitting multiple sclerosis patients and in neuroblastoma SK-N-BE cells. Brain Res. Bull. 2015, 118, 1–6. [Google Scholar] [CrossRef]
- Lee, A.J.B.; Kittel, T.E.; Kim, R.B.; Bach, T.-N.; Zhang, T.; Mitchell, C.S. Comparing therapeutic modulators of the SOD1 G93A Amyotrophic Lateral Sclerosis mouse pathophysiology. Front. Neurosci. 2023, 16, 1111763. [Google Scholar] [CrossRef] [PubMed]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Rigutto, S.; Hoste, C.; Dumont, J.E.; Corvilain, B.; Miot, F.; De Deken, X. Duox1 is the main source of hydrogen peroxide in the rat thyroid cell line PCCl3. Exp. Cell Res. 2007, 313, 3892–3901. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Moalem, G.; Leibowitz-Amit, R.; Yoles, E.; Mor, F.; Cohen, I.R.; Schwartz, M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat. Med. 1999, 5, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Ellwardt, E.; Walsh, J.T.; Kipnis, J.; Zipp, F. Understanding the Role of T Cells in CNS Homeostasis. Trends Immunol. 2016, 37, 154–165. [Google Scholar] [CrossRef]
- Xie, L.; Choudhury, G.R.; Winters, A.; Yang, S.H.; Jin, K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur. J. Immunol. 2015, 45, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Kallies, A. T cell responses in the central nervous system. Nat. Rev. Immunol. 2017, 17, 179–194. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).