
Hallmarks and Biomarkers of Skin Senescence: An Updated Review of Skin Senotherapeutics

 ,
,  ,
,  ,
,  and
and 
Abstract
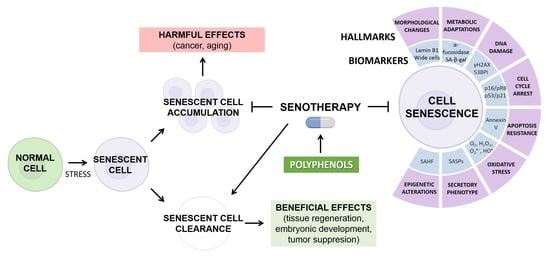
1. Introduction
2. Search Strategy
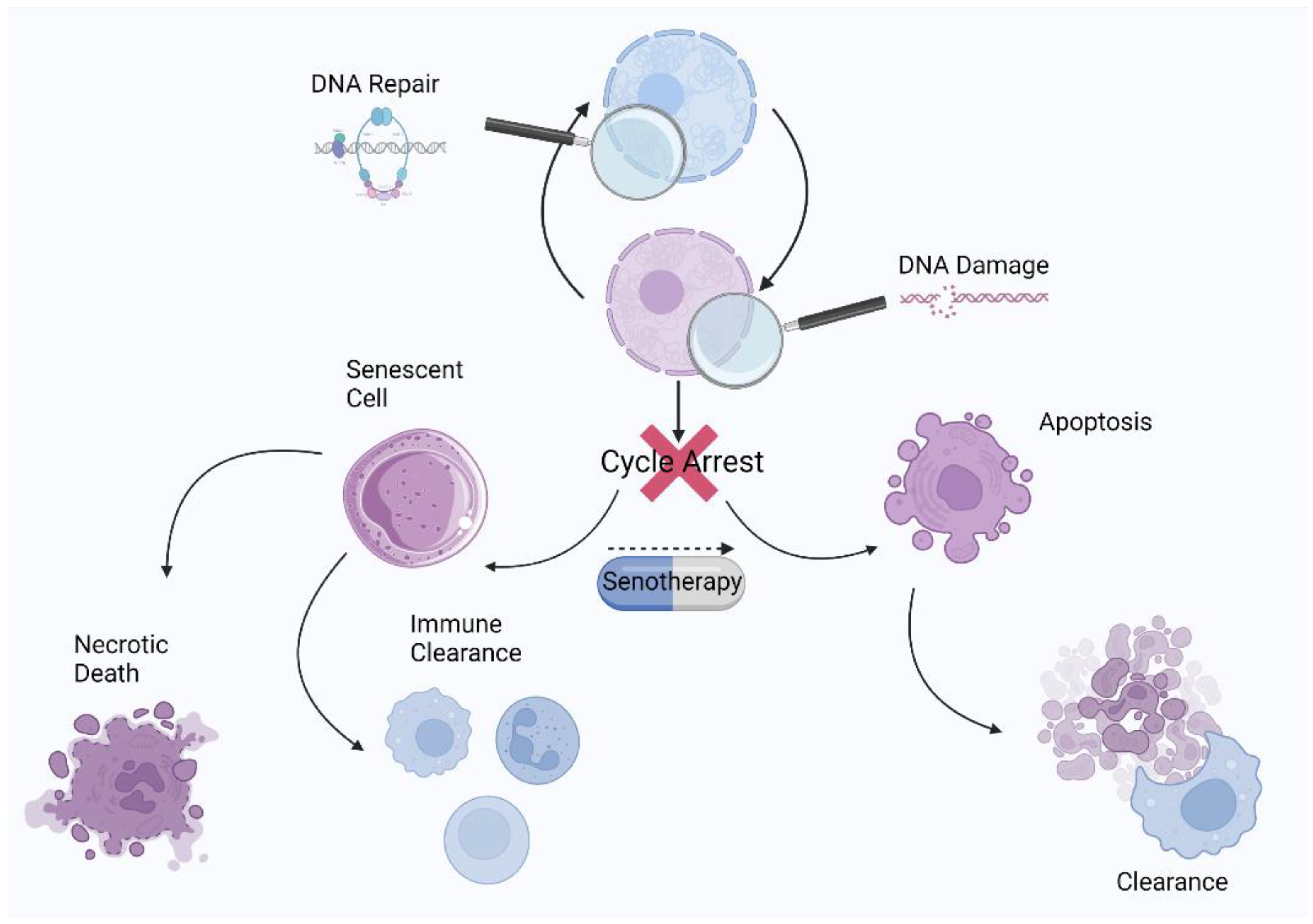
3. Cellular Senescence
4. Other Nonsenescent Forms of Cell Cycle Arrest
5. Hallmarks of Senescence
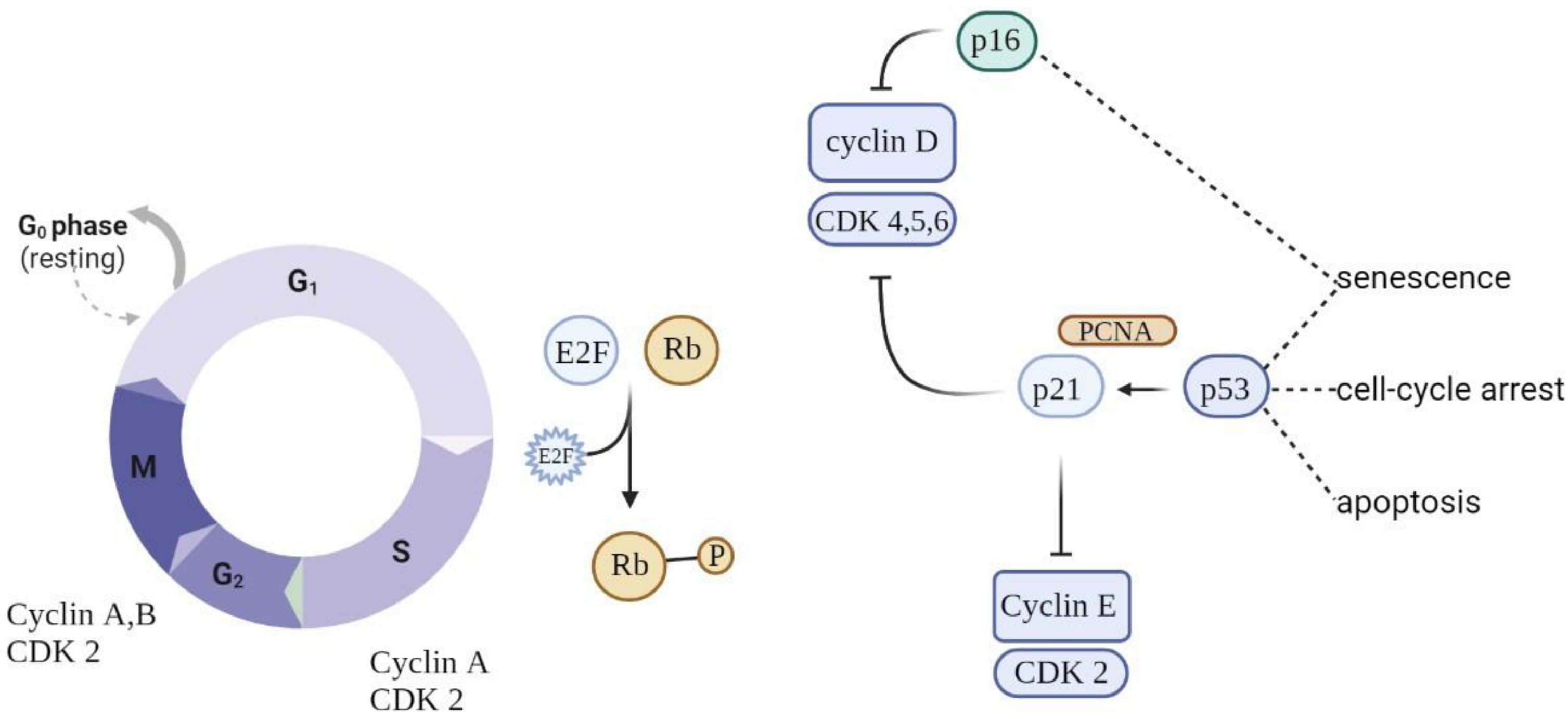
5.1. Stable Cell Cycle Arrest
5.2. Metabolic Changes
5.3. Morphological Changes
5.4. Epigenetic Alterations
5.5. DNA Damage and Persistent DNA Damage Response
5.6. Apoptosis Resistance
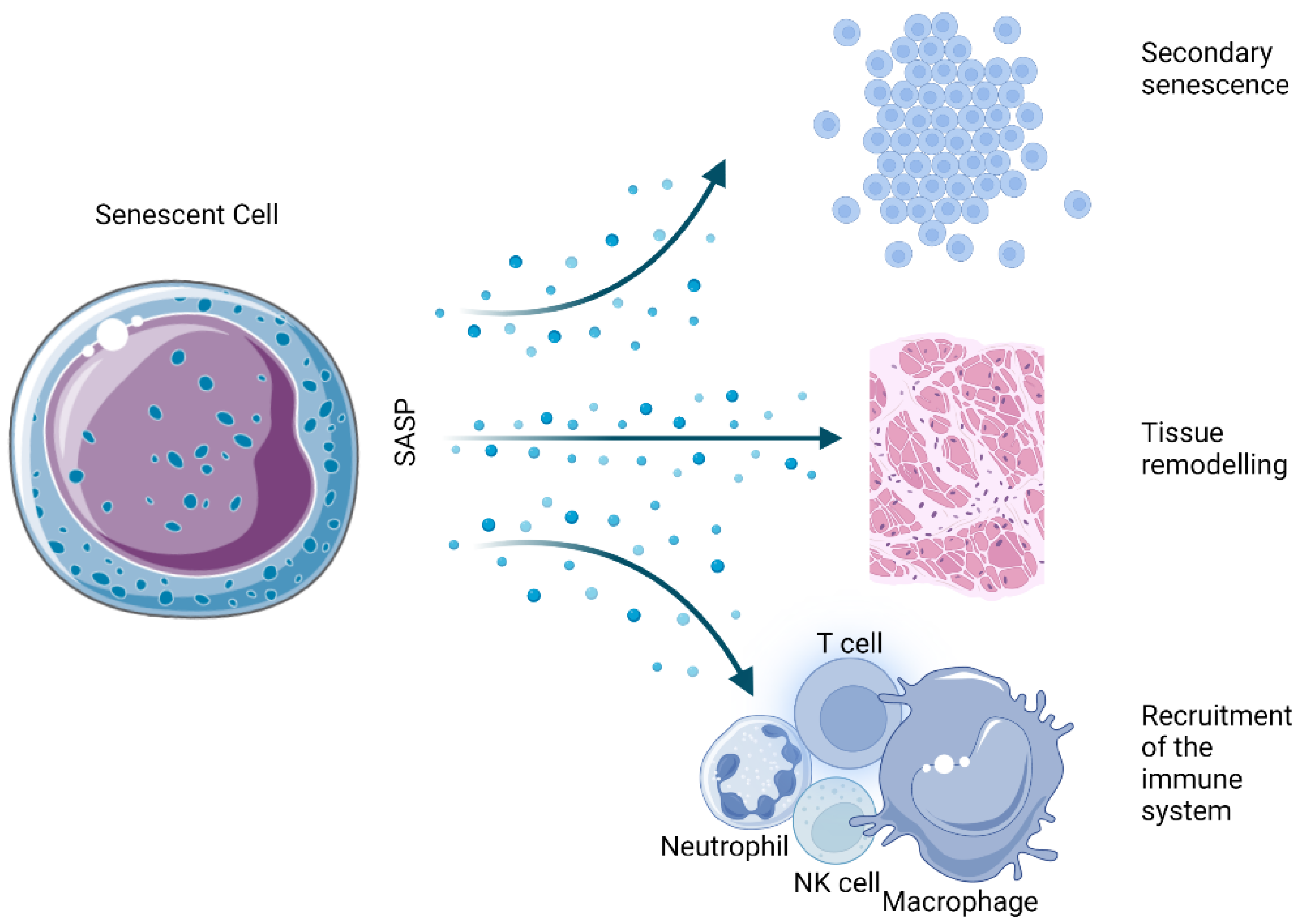
5.7. Secretory Phenotype
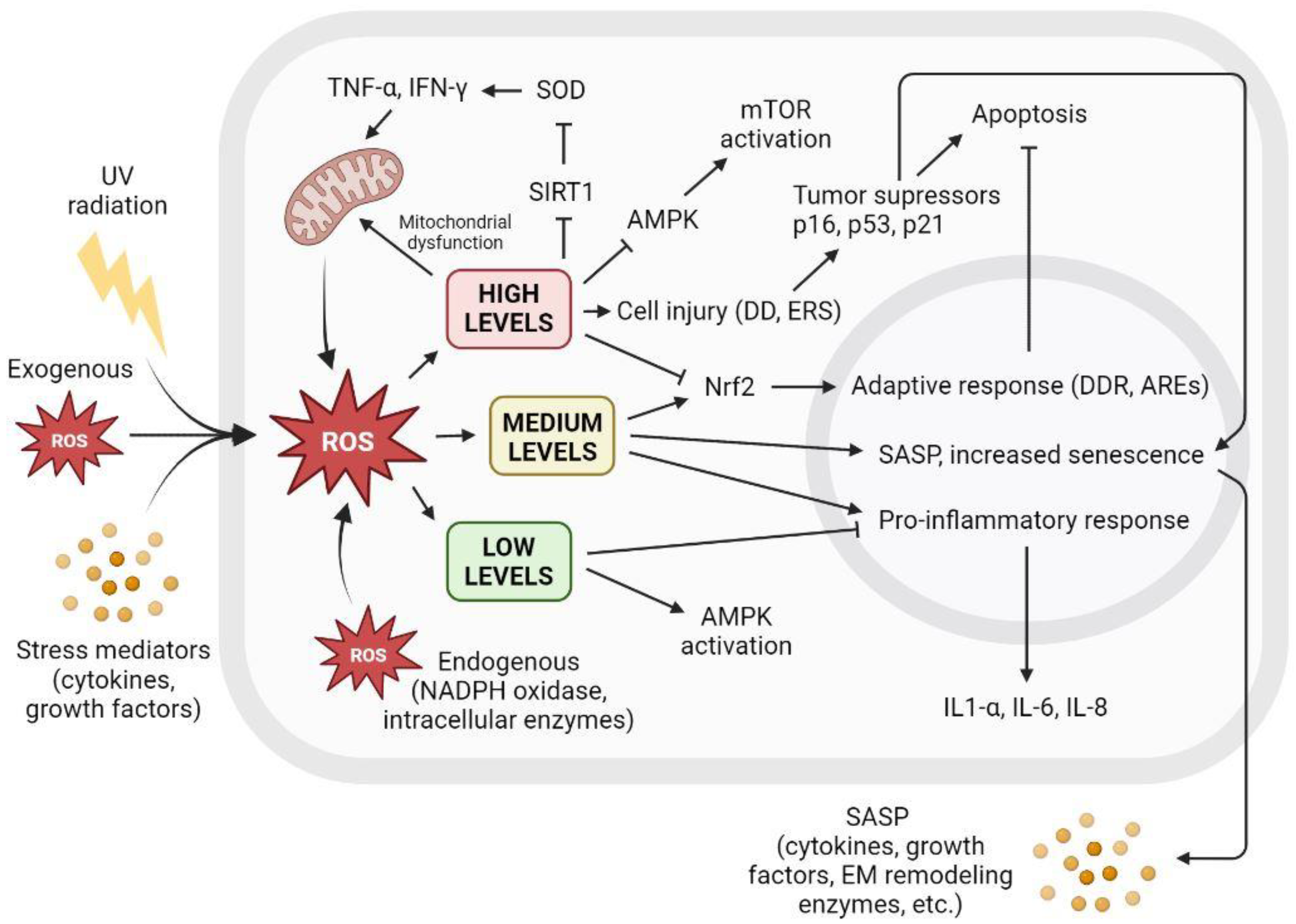
5.8. Reactive Oxygen Species
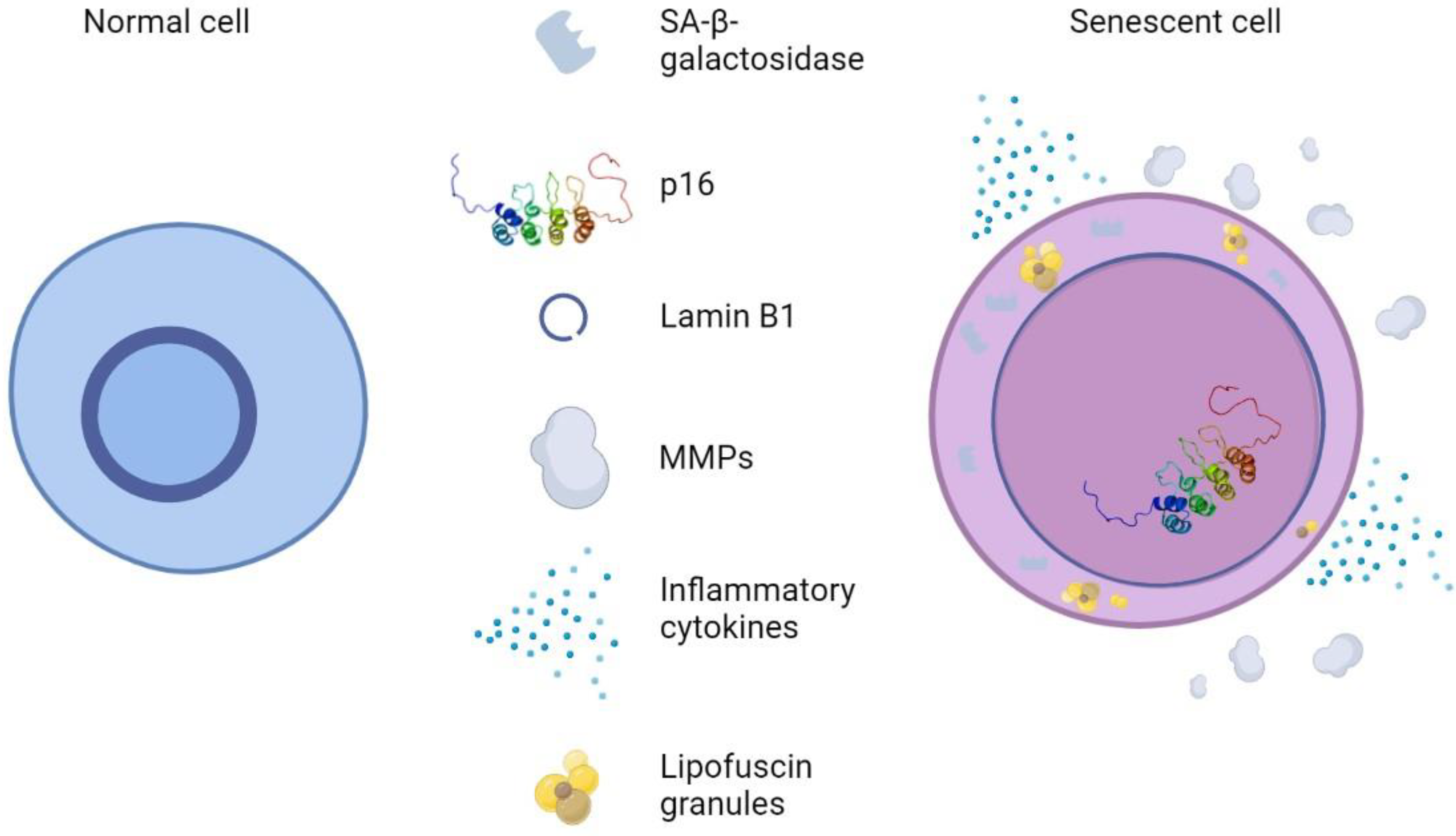
5.9. Biomarkers of Cellular Senescence
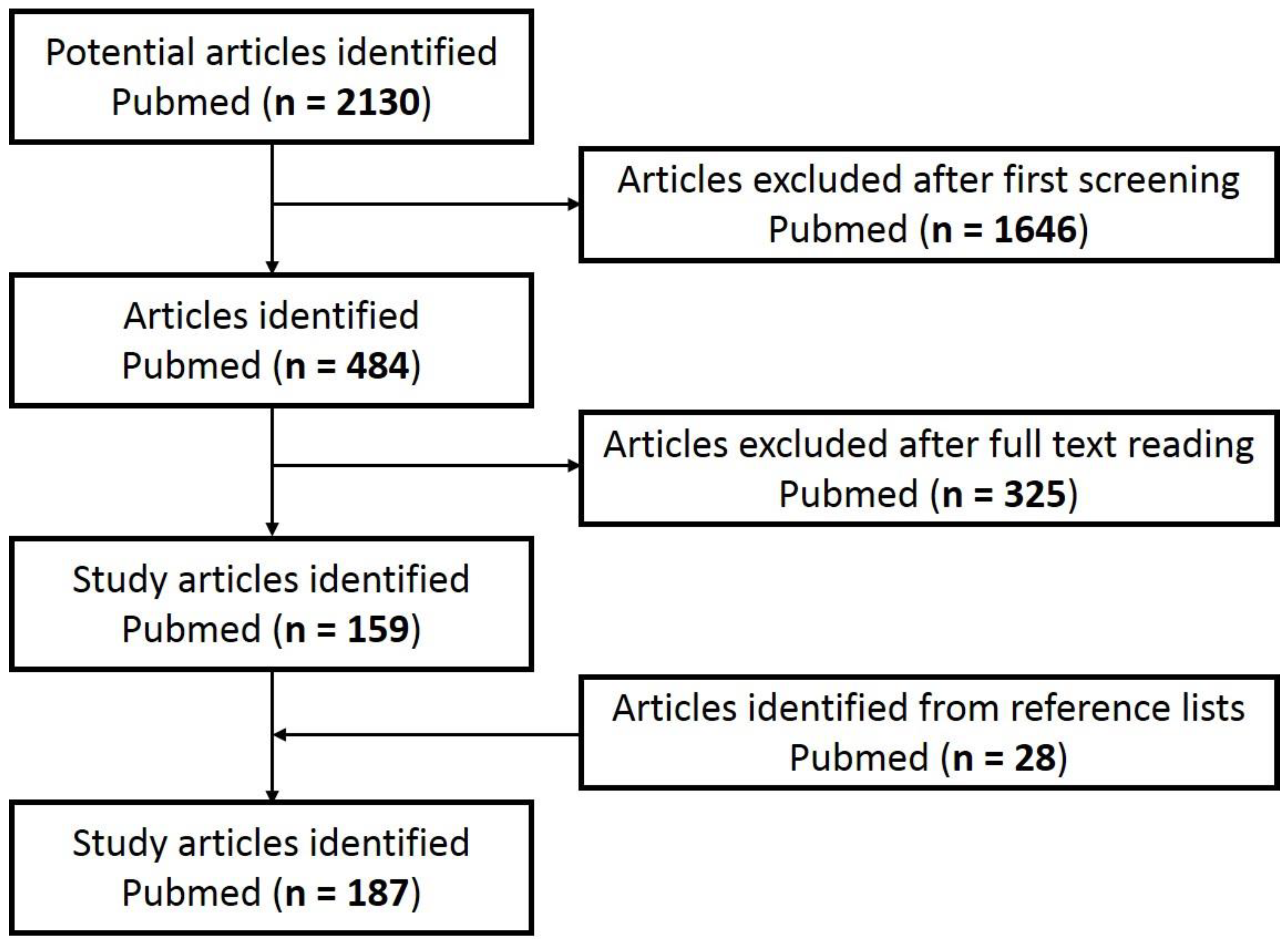
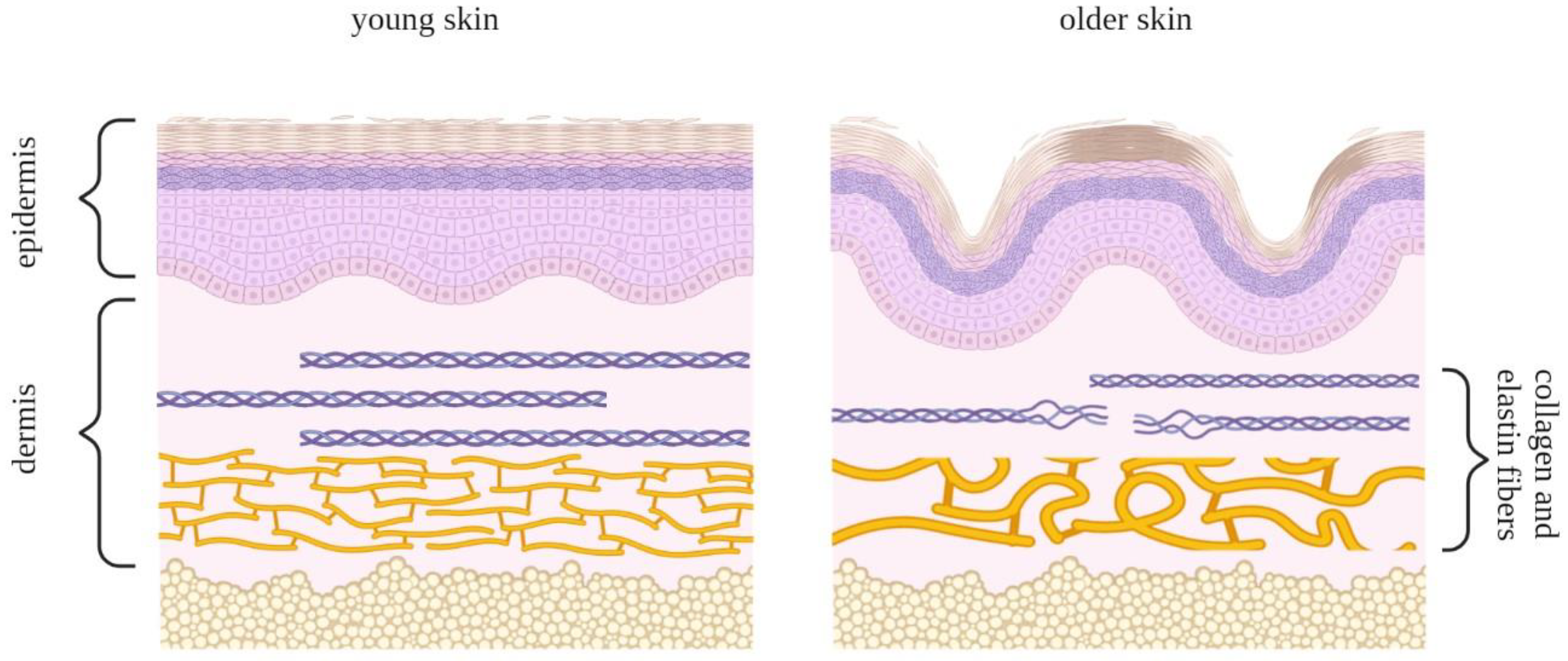
6. Hallmarks and Biomarkers of Skin Aging
7. Skin Senotherapy and Antioxidant Compounds
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- United Nations, Department of Economic and Social Affairs. Population Division World Population Prospects 2019: Highlights; United Nations: New York, NY, USA, 2019. [Google Scholar]
- United Nations, Department of Economic and Social Affairs. Population Division World Population Ageing 2017: Highlights; United Nations: New York, NY, USA, 2017. [Google Scholar]
- Burch, J.B.; Augustine, A.D.; Frieden, L.A.; Hadley, E.; Howcroft, T.K.; Johnson, R.; Khalsa, P.S.; Kohanski, R.A.; Li, X.L.; Macchiarini, F.; et al. Advances in geroscience: Impact on healthspan and chronic disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S1–S3. [Google Scholar] [CrossRef]
- Khokhlov, A.N. From carrel to hayflick and back or what we got from the 100 years of cytogerontological studies. Radiats. Biol. Radioecol. 2010, 50, 304–311. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Zinger, A.; Cho, W.C.; Ben-Yehuda, A. Cancer and aging—The inflammatory connection. Aging Dis. 2017, 8, 611–627. [Google Scholar] [CrossRef]
- Schmitt, R. Senotherapy: Growing old and staying young? Pflug. Arch. 2017, 469, 1051–1059. [Google Scholar] [CrossRef]
- Raffaele, M.; Vinciguerra, M. The costs and benefits of senotherapeutics for human health. Lancet Healthy Longev. 2022, 3, e67–e77. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Amaya-Montoya, M.; Pérez-Londoño, A.; Guatibonza-García, V.; Vargas-Villanueva, A.; Mendivil, C.O. Cellular senescence as a therapeutic target for age-related diseases: A review. Adv. Ther. 2020, 37, 1407–1424. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through wnt16b. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of pdgf-aa. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Merkt, W.; Zhou, Y.; Han, H.; Lagares, D. Myofibroblast fate plasticity in tissue repair and fibrosis: Deactivation, apoptosis, senescence and reprogramming. Wound Repair Regen. 2021, 29, 678–691. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein ccn1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Marescal, O.; Cheeseman, I.M. Cellular mechanisms and regulation of quiescence. Dev. Cell 2020, 55, 259–271. [Google Scholar] [CrossRef]
- Buttitta, L.A.; Edgar, B.A. Mechanisms controlling cell cycle exit upon terminal differentiation. Curr. Opin. Cell Biol. 2007, 19, 697–704. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Development 2019, 146, dev151837. [Google Scholar] [CrossRef]
- Yates, K.B.; Tonnerre, P.; Martin, G.E.; Gerdemann, U.; Al Abosy, R.; Comstock, D.E.; Weiss, S.A.; Wolski, D.; Tully, D.C.; Chung, R.T.; et al. Epigenetic scars of cd8. Nat. Immunol. 2021, 22, 1020–1029. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into t cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Ramos-Ibeas, P.; Gimeno, I.; Cañón-Beltrán, K.; Gutiérrez-Adán, A.; Rizos, D.; Gómez, E. Senescence and apoptosis during. Front. Cell Dev. Biol. 2020, 8, 619902. [Google Scholar] [CrossRef]
- Lemons, J.M.; Feng, X.J.; Bennett, B.D.; Legesse-Miller, A.; Johnson, E.L.; Raitman, I.; Pollina, E.A.; Rabitz, H.A.; Rabinowitz, J.D.; Coller, H.A. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010, 8, e1000514. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of t lymphocytes: Implications for enhancing human immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Tümpel, S.; Rudolph, K.L. Quiescence: Good and bad of stem cell aging. Trends Cell Biol. 2019, 29, 672–685. [Google Scholar] [CrossRef]
- Bellon, M.; Nicot, C. Telomere dynamics in immune senescence and exhaustion triggered by chronic viral infection. Viruses 2017, 9, 289. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Dewald, H.K.; Vasilopoulos, T.; Gittens-Williams, L.; Fitzgerald-Bocarsly, P.; Herbig, U. Senescence-associated β-galactosidase reveals the abundance of senescent cd8+ t cells in aging humans. Aging Cell 2021, 20, e13344. [Google Scholar] [CrossRef]
- Itahana, K.; Itahana, Y.; Dimri, G.P. Colorimetric detection of senescence-associated β galactosidase. Methods Mol. Biol. 2013, 965, 143–156. [Google Scholar]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘t cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where metabolism meets senescence: Focus on endothelial cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Valcourt, J.R.; Lemons, J.M.; Haley, E.M.; Kojima, M.; Demuren, O.O.; Coller, H.A. Staying alive: Metabolic adaptations to quiescence. Cell Cycle 2012, 11, 1680–1696. [Google Scholar] [CrossRef]
- Alrubayyi, A.; Moreno-Cubero, E.; Hameiri-Bowen, D.; Matthews, R.; Rowland-Jones, S.; Schurich, A.; Peppa, D. Functional restoration of exhausted cd8 t cells in chronic hiv-1 infection by targeting mitochondrial dysfunction. Front. Immunol. 2022, 13, 908697. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.Y.; Andrews, S.; Fraser, P.; Reik, W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of t cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef]
- Boland, M.J.; Nazor, K.L.; Loring, J.F. Epigenetic regulation of pluripotency and differentiation. Circ. Res. 2014, 115, 311–324. [Google Scholar] [CrossRef]
- Naylor, S. Biomarkers: Current perspectives and future prospects. Expert. Rev. Mol. Diagn. 2003, 3, 525–529. [Google Scholar] [CrossRef]
- Baker, G.T.; Sprott, R.L. Biomarkers of aging. Exp. Gerontol. 1988, 23, 223–239. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Kanaki, T.; Makrantonaki, E.; Zouboulis, C.C. Biomarkers of skin aging. Rev. Endocr. Metab. Disord. 2016, 17, 433–442. [Google Scholar] [CrossRef]
- Kim, W.Y.; Sharpless, N.E. The regulation of ink4/arf in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A code of mono-phosphorylation modulates the function of rb. Mol. Cell 2019, 73, 985–1000.e1006. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in health and disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C.M. The role of p53 in apoptosis. Discov. Med. 2010, 9, 145–152. [Google Scholar]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Akhter, F.; Chen, D.; Akhter, A.; Yan, S.F.; Yan, S.S. Age-dependent accumulation of dicarbonyls and advanced glycation endproducts (ages) associates with mitochondrial stress. Free Radic. Biol. Med. 2021, 164, 429–438. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- De Mera-Rodríguez, J.A.; Álvarez-Hernán, G.; Gañán, Y.; Martín-Partido, G.; Rodríguez-León, J.; Francisco-Morcillo, J. Is senescence-associated β-galactosidase a reliable. Front. Cell Dev. Biol. 2021, 9, 623175. [Google Scholar] [CrossRef]
- Gu, K.; Xu, Y.; Li, H.; Guo, Z.; Zhu, S.; Shi, P.; James, T.D.; Tian, H.; Zhu, W.H. Real-time tracking and in vivo visualization of β-galactosidase activity in colorectal tumor with a ratiometric near-infrared fluorescent probe. J. Am. Chem. Soc. 2016, 138, 5334–5340. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, D.G.; Lehle, S.; Borst, A.; Haferkamp, S.; Essmann, F.; Schulze-Osthoff, K. A-fucosidase as a novel convenient biomarker for cellular senescence. Cell Cycle 2013, 12, 1922–1927. [Google Scholar] [CrossRef]
- Koo, S.; Won, M.; Li, H.; Kim, W.Y.; Li, M.; Yan, C.; Sharma, A.; Guo, Z.; Zhu, W.H.; Sessler, J.L.; et al. Harnessing α-l-fucosidase for in vivo cellular senescence imaging. Chem. Sci. 2021, 12, 10054–10062. [Google Scholar] [CrossRef]
- Perše, M.; Injac, R.; Erman, A. Oxidative status and lipofuscin accumulation in urothelial cells of bladder in aging mice. PLoS ONE 2013, 8, e59638. [Google Scholar] [CrossRef]
- Vida, C.; de Toda, I.M.; Cruces, J.; Garrido, A.; Gonzalez-Sanchez, M.; De la Fuente, M. Role of macrophages in age-related oxidative stress and lipofuscin accumulation in mice. Redox Biol. 2017, 12, 423–437. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An overview of the role of lipofuscin in age-related neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef]
- Wolf, G. Lipofuscin, the age pigment. Nutr. Rev. 1993, 51, 205–206. [Google Scholar] [CrossRef]
- Crane, E.D.; Wong, W.; Zhang, H.; O’Neil, G.; Crane, J.D. Ampk inhibits mtor-driven keratinocyte proliferation after skin damage and stress. J. Investig. Dermatol. 2021, 141, 2170–2177.e2173. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. Ampk maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Neurohr, G.E.; Terry, R.L.; Lengefeld, J.; Bonney, M.; Brittingham, G.P.; Moretto, F.; Miettinen, T.P.; Vaites, L.P.; Soares, L.M.; Paulo, J.A.; et al. Excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell 2019, 176, 1083–1097.e1018. [Google Scholar] [CrossRef]
- Ott, C.; Jung, T.; Grune, T.; Höhn, A. Sips as a model to study age-related changes in proteolysis and aggregate formation. Mech. Ageing Dev. 2018, 170, 72–81. [Google Scholar] [CrossRef]
- Moujaber, O.; Fishbein, F.; Omran, N.; Liang, Y.; Colmegna, I.; Presley, J.F.; Stochaj, U. Cellular senescence is associated with reorganization of the microtubule cytoskeleton. Cell. Mol. Life Sci. 2019, 76, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Swanson, E.C.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013, 203, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Kondo, T. Identification of the lamina-associated-polypeptide-2-binding domain of b-type lamin. Eur. J. Biochem. 1998, 251, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin b1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.U.; Soujanya, M.; Mishra, R.K. Deterioration of nuclear morphology and architecture: A hallmark of senescence and aging. Ageing Res. Rev. 2021, 67, 101264. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef] [PubMed]
- Boumendil, C.; Hari, P.; Olsen, K.C.F.; Acosta, J.C.; Bickmore, W.A. Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev. 2019, 33, 144–149. [Google Scholar] [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of e2f target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.H.; Ryeom, S. Detection of oncogene-induced senescence in vivo. Methods Mol. Biol. 2017, 1534, 185–198. [Google Scholar] [PubMed]
- Pérez, R.F.; Tejedor, J.R.; Bayón, G.F.; Fernández, A.F.; Fraga, M.F. Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell 2018, 17, e12744. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Boisvert, F.M.; Hendzel, M.J.; Bazett-Jones, D.P. Promyelocytic leukemia (pml) nuclear bodies are protein structures that do not accumulate rna. J. Cell Biol. 2000, 148, 283–292. [Google Scholar] [CrossRef]
- Guan, D.; Kao, H.Y. The function, regulation and therapeutic implications of the tumor suppressor protein, pml. Cell Biosci. 2015, 5, 60. [Google Scholar] [CrossRef]
- Rodier, F.; Muñoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppé, J.P.; Campeau, E.; Beauséjour, C.M.; Kim, S.H.; et al. DNA-scars: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 2011, 124, 68–81. [Google Scholar] [CrossRef]
- Malaquin, N.; Carrier-Leclerc, A.; Dessureault, M.; Rodier, F. Ddr-mediated crosstalk between DNA-damaged cells and their microenvironment. Front. Genet. 2015, 6, 94. [Google Scholar] [CrossRef]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef]
- Muñoz-Lorente, M.A.; Cano-Martin, A.C.; Blasco, M.A. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 2019, 10, 4723. [Google Scholar] [CrossRef]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar] [PubMed]
- Fan, Y.; Cheng, J.; Zeng, H.; Shao, L. Senescent cell depletion through targeting bcl-family proteins and mitochondria. Front. Physiol. 2020, 11, 593630. [Google Scholar] [CrossRef] [PubMed]
- Parihar, A.; Eubank, T.D.; Doseff, A.I. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J. Innate Immun. 2010, 2, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of survival networks in senescent cells: From mechanisms to interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic ras and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The senescence-associated secretory phenotype (sasp) in the challenging future of cancer therapy and age-related diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Antonangeli, F.; Zingoni, A.; Soriani, A.; Santoni, A. Senescent cells: Living or dying is a matter of nk cells. J. Leukoc. Biol. 2019, 105, 1275–1283. [Google Scholar] [CrossRef]
- Lu, S.Y.; Chang, K.W.; Liu, C.J.; Tseng, Y.H.; Lu, H.H.; Lee, S.Y.; Lin, S.C. Ripe areca nut extract induces g1 phase arrests and senescence-associated phenotypes in normal human oral keratinocyte. Carcinogenesis 2006, 27, 1273–1284. [Google Scholar] [CrossRef]
- Suzuki, Y.; Takaya, K.; Watanabe, S.; Otaki, M.; Kono, H.; Kishi, K. Evaluation of the effect of age of the younger mice on the rejuvenation of the older mice by heterochronic parabiosis. Aging 2022, 14, 2507–2512. [Google Scholar] [CrossRef]
- Özcan, S.; Alessio, N.; Acar, M.B.; Mert, E.; Omerli, F.; Peluso, G.; Galderisi, U. Unbiased analysis of senescence associated secretory phenotype (sasp) to identify common components following different genotoxic stresses. Aging 2016, 8, 1316–1329. [Google Scholar] [CrossRef]
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. Mir-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via zeb1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Borodkina, A.; Shatrova, A.; Abushik, P.; Nikolsky, N.; Burova, E. Interaction between ros dependent DNA damage, mitochondria and p38 mapk underlies senescence of human adult stem cells. Aging 2014, 6, 481–495. [Google Scholar] [CrossRef]
- Lawless, C.; Jurk, D.; Gillespie, C.S.; Shanley, D.; Saretzki, G.; von Zglinicki, T.; Passos, J.F. A stochastic step model of replicative senescence explains ros production rate in ageing cell populations. PLoS ONE 2012, 7, e32117. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- McCarthy, D.A.; Clark, R.R.; Bartling, T.R.; Trebak, M.; Melendez, J.A. Redox control of the senescence regulator interleukin-1α and the secretory phenotype. J. Biol. Chem. 2013, 288, 32149–32159. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, E.; Cox, L.S. Interconnections between inflammageing and immunosenescence during ageing. Cells 2022, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, G.R.; Almeida, P.P.; de Oliveira Santos, L.; Rodrigues, L.P.; de Carvalho, J.L.; Boroni, M. Hallmarks of aging in macrophages: Consequences to skin inflammaging. Cells 2021, 10, 1323. [Google Scholar] [CrossRef] [PubMed]
- Aumayr, K.; Susani, M.; Horvat, R.; Wrba, F.; Mazal, P.; Klatte, T.; Koller, A.; Neudert, B.; Haitel, A. P16ink4a immunohistochemistry for detection of human papilloma virus-associated penile squamous cell carcinoma is superior to in-situ hybridization. Int. J. Immunopathol. Pharmacol. 2013, 26, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. P16ink4a is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef]
- Santiago-Cardona, P.G.; Pérez-Morales, J.; González-Flores, J. Detection of retinoblastoma protein phosphorylation by immunoblot analysis. Methods Mol. Biol. 2018, 1726, 49–64. [Google Scholar]
- Pérez-Morales, J.; Núñez-Marrero, A.; Santiago-Cardona, P.G. Immunohistochemical detection of retinoblastoma protein phosphorylation in human tumor samples. Methods Mol. Biol. 2018, 1726, 77–84. [Google Scholar]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving atm, p53, and p21(cip1), but not p16(ink4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Jurk, D.; Passos, J.F. Robust multiparametric assessment of cellular senescence. Methods Mol. Biol. 2013, 965, 409–419. [Google Scholar]
- Mohamad Kamal, N.S.; Safuan, S.; Shamsuddin, S.; Foroozandeh, P. Aging of the cells: Insight into cellular senescence and detection methods. Eur. J. Cell Biol. 2020, 99, 151108. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Ma, X.; Cui, C.; Deenik, P.R.; Henderson, P.K.P.; Sigler, A.L.; Cui, L. Real-time imaging of senescence in tumors with DNA damage. Sci. Rep. 2019, 9, 2102. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, S.; Yamashita, Y.; Kase, M.; Maruyama, M.; Sugimoto, T. Overview and assessment of the histochemical methods and reagents for the detection of β-galactosidase activity in transgenic animals. Anat. Sci. Int. 2016, 91, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Heo, C.H.; Sen, D.; Byun, H.O.; Kwak, I.H.; Yoon, G.; Kim, H.M. Ratiometric two-photon fluorescent probe for quantitative detection of β-galactosidase activity in senescent cells. Anal. Chem. 2014, 86, 10001–10005. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ren, X.; Meng, X.; Zhang, Y.; Chen, D.; Tang, F. Novel fluorescence method for detection of α-l-fucosidase based on cdte quantum dots. Anal. Chem. 2012, 84, 4077–4082. [Google Scholar] [CrossRef]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef]
- Salmonowicz, H.; Passos, J.F. Detecting senescence: A new method for an old pigment. Aging Cell 2017, 16, 432–434. [Google Scholar] [CrossRef]
- Faragher, R.G.A. Simple detection methods for senescent cells: Opportunities and challenges. Front. Aging 2021, 2, 686382. [Google Scholar] [CrossRef]
- Zhao, H.; Darzynkiewicz, Z. Biomarkers of cell senescence assessed by imaging cytometry. Methods Mol. Biol. 2013, 965, 83–92. [Google Scholar]
- Vergnes, L.; Péterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin b1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef]
- Spector, D.L. Immunofluorescence localization of nuclear proteins. Cold Spring Harb. Protoc. 2011, 2011, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Canela, A.; Vera, E.; Klatt, P.; Blasco, M.A. High-throughput telomere length quantification by fish and its application to human population studies. Proc. Natl. Acad. Sci. USA 2007, 104, 5300–5305. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Smith, D.L.; Esteves, K.; Drury, S. Telomere length measurement by qpcr—Summary of critical factors and recommendations for assay design. Psychoneuroendocrinology 2019, 99, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Klement, K.; Melle, C.; Murzik, U.; Diekmann, S.; Norgauer, J.; Hemmerich, P. Accumulation of annexin a5 at the nuclear envelope is a biomarker of cellular aging. Mech. Ageing Dev. 2012, 133, 508–522. [Google Scholar] [CrossRef]
- Balaji, N.; Devy, A.S.; Sumathi, M.K.; Vidyalakshmi, S.; Kumar, G.S.; D’Silva, S. Annexin v–Affinity assay–Apoptosis detection system in granular cell ameloblastoma. J. Int. Oral Health 2013, 5, 25–30. [Google Scholar]
- Bressenot, A.; Marchal, S.; Bezdetnaya, L.; Garrier, J.; Guillemin, F.; Plénat, F. Assessment of apoptosis by immunohistochemistry to active caspase-3, active caspase-7, or cleaved parp in monolayer cells and spheroid and subcutaneous xenografts of human carcinoma. J. Histochem. Cytochem. 2009, 57, 289–300. [Google Scholar] [CrossRef]
- Liu, P.F.; Hu, Y.C.; Kang, B.H.; Tseng, Y.K.; Wu, P.C.; Liang, C.C.; Hou, Y.Y.; Fu, T.Y.; Liou, H.H.; Hsieh, I.C.; et al. Expression levels of cleaved caspase-3 and caspase-3 in tumorigenesis and prognosis of oral tongue squamous cell carcinoma. PLoS ONE 2017, 12, e0180620. [Google Scholar] [CrossRef]
- McComb, S.; Chan, P.K.; Guinot, A.; Hartmannsdottir, H.; Jenni, S.; Dobay, M.P.; Bourquin, J.P.; Bornhauser, B.C. Efficient apoptosis requires feedback amplification of upstream apoptotic signals by effector caspase-3 or -7. Sci. Adv. 2019, 5, eaau9433. [Google Scholar] [CrossRef]
- Kyrylkova, K.; Kyryachenko, S.; Leid, M.; Kioussi, C. Detection of apoptosis by tunel assay. Methods Mol. Biol. 2012, 887, 41–47. [Google Scholar]
- Rodier, F. Detection of the senescence-associated secretory phenotype (sasp). Methods Mol. Biol. 2013, 965, 165–173. [Google Scholar]
- Hari, P.; Acosta, J.C. Detecting the senescence-associated secretory phenotype (sasp) by high content microscopy analysis. Methods Mol. Biol. 2017, 1534, 99–109. [Google Scholar] [PubMed]
- Rolt, A.; Nair, A.; Cox, L.S. Correction to: Optimisation of a screening platform for determining il-6 inflammatory signalling in the senescence-associated secretory phenotype (sasp). Biogerontology 2019, 20, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Kitamura, M. Sensitive detection and monitoring of senescence-associated secretory phenotype by sasp-rap assay. PLoS ONE 2012, 7, e52305. [Google Scholar] [CrossRef] [PubMed]
- Shehat, M.G.; Tigno-Aranjuez, J. Flow cytometric measurement of ros production in macrophages in response to fcγr cross-linking. J. Vis. Exp. 2019, 145, e59167. [Google Scholar]
- Nosaka, Y.; Nosaka, A.Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef]
- Choi, S.I.; Jung, T.D.; Cho, B.Y.; Choi, S.H.; Sim, W.S.; Han, X.; Lee, S.J.; Kim, Y.C.; Lee, O.H. Anti-photoaging effect of fermented agricultural by-products on ultraviolet b-irradiated hairless mouse skin. Int. J. Mol. Med. 2019, 44, 559–568. [Google Scholar] [CrossRef]
- Letsiou, S. Tracing skin aging process: A mini- review of in vitro approaches. Biogerontology 2021, 22, 261–272. [Google Scholar] [CrossRef]
- Khavkin, J.; Ellis, D.A.F. Aging skin: Histology, physiology, and pathology. Facial Plast. Surg. Clin. N. Am. 2011, 19, 229–234. [Google Scholar] [CrossRef]
- Reilly, D.M.; Lozano, J. Skin collagen through the lifestages: Importance for skin health and beauty. Plast. Aesthetic Res. 2021, 8, 2. [Google Scholar] [CrossRef]
- Puizina-Ivić, N. Skin aging. Acta Derm. Alp. Pannonica Adriat. 2008, 17, 47–54. [Google Scholar]
- Sárdy, M. Role of matrix metalloproteinases in skin ageing. Connect. Tissue Res. 2009, 50, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Dreesen, O. Faces of cellular senescence in skin aging. Mech. Ageing Dev. 2021, 198, 111525. [Google Scholar] [CrossRef] [PubMed]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Trost, A.; Richter, K. Oxidative stress in aging human skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.Y.; Lee, J.W.; Papaccio, F.; Bellei, B.; Picardo, M. Alterations of the pigmentation system in the aging process. Pigment. Cell Melanoma Res. 2021, 34, 800–813. [Google Scholar] [CrossRef]
- Goorochurn, R.; Viennet, C.; Granger, C.; Fanian, F.; Varin-Blank, N.; Roy, C.L.; Humbert, P. Biological processes in solar lentigo: Insights brought by experimental models. Exp. Dermatol. 2016, 25, 174–177. [Google Scholar] [CrossRef]
- Varesi, A.; Chirumbolo, S.; Campagnoli, L.I.M.; Pierella, E.; Piccini, G.B.; Carrara, A.; Ricevuti, G.; Scassellati, C.; Bonvicini, C.; Pascale, A. The role of antioxidants in the interplay between oxidative stress and senescence. Antioxidants 2022, 11, 1224. [Google Scholar] [CrossRef]
- Song, S.; Tchkonia, T.; Jiang, J.; Kirkland, J.L.; Sun, Y. Targeting senescent cells for a healthier aging: Challenges and opportunities. Adv. Sci. 2020, 7, 2002611. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef]
- Wissler Gerdes, E.O.; Misra, A.; Netto, J.M.E.; Tchkonia, T.; Kirkland, J.L. Strategies for late phase preclinical and early clinical trials of senolytics. Mech. Ageing Dev. 2021, 200, 111591. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Boccardi, V.; Mecocci, P. Senotherapeutics: Targeting senescent cells for the main age-related diseases. Mech. Ageing Dev. 2021, 197, 111526. [Google Scholar] [CrossRef] [PubMed]
- Domaszewska-Szostek, A.; Puzianowska-Kuźnicka, M.; Kuryłowicz, A. Flavonoids in skin senescence prevention and treatment. Int. J. Mol. Sci. 2021, 22, 6814. [Google Scholar] [CrossRef]
- Borchers, A.; Pieler, T. Programming pluripotent precursor cells derived from xenopus embryos to generate specific tissues and organs. Genes 2010, 1, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Tryniszewski, W.; Sarniak, A.; Wlodarczyk, A.; Nowak, P.J.; Nowak, D. Concentration dependence of anti- and pro-oxidant activity of polyphenols as evaluated with a light-emitting Fe2+-egta-H2O2 system. Molecules 2022, 27, 3453. [Google Scholar] [CrossRef] [PubMed]
- Csekes, E.; Račková, L. Skin aging, cellular senescence and natural polyphenols. Int. J. Mol. Sci. 2021, 22, 12641. [Google Scholar] [CrossRef]
- Britto, S.M.; Shanthakumari, D.; Agilan, B.; Radhiga, T.; Kanimozhi, G.; Prasad, N.R. Apigenin prevents ultraviolet-b radiation induced cyclobutane pyrimidine dimers formation in human dermal fibroblasts. Mutat. Res. Genet. Toxicol. Environ. Mutagen 2017, 821, 28–35. [Google Scholar] [CrossRef]
- Das, S.; Das, J.; Paul, A.; Samadder, A.; Khuda-Bukhsh, A.R. Apigenin, a bioactive flavonoid from lycopodium clavatum, stimulates nucleotide excision repair genes to protect skin keratinocytes from ultraviolet b-induced reactive oxygen species and DNA damage. J. Acupunct. Meridian Stud. 2013, 6, 252–262. [Google Scholar] [CrossRef]
- Wang, S.-C.; Chen, S.-F.; Lee, Y.-M.; Chuang, C.-L.; Bau, D.-T.; Lin, S.-S. Baicalin scavenges reactive oxygen species and protects human keratinocytes against uvc-induced cytotoxicity. In Vivo 2013, 27, 707. [Google Scholar]
- Hahn, H.J.; Kim, K.B.; Bae, S.; Choi, B.G.; An, S.; Ahn, K.J.; Kim, S.Y. Pretreatment of ferulic acid protects human dermal fibroblasts against ultraviolet a irradiation. Ann. Dermatol. 2016, 28, 740–748. [Google Scholar] [CrossRef]
- Seo, S.H.; Jeong, G.S. Fisetin inhibits tnf-α-induced inflammatory action and hydrogen peroxide-induced oxidative damage in human keratinocyte hacat cells through pi3k/akt/nrf-2-mediated heme oxygenase-1 expression. Int. Immunopharmacol. 2015, 29, 246–253. [Google Scholar] [CrossRef]
- Chiang, H.M.; Chan, S.Y.; Chu, Y.; Wen, K.C. Fisetin ameliorated photodamage by suppressing the mitogen-activated protein kinase/matrix metalloproteinase pathway and nuclear factor-κb pathways. J. Agric. Food Chem. 2015, 63, 4551–4560. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.; Park, S.-Y.; Lee, H.J.; Lee, T.Y.; Sun, Z.-w.; Yi, T.H. Gallic acid regulates skin photoaging in uvb-exposed fibroblast and hairless mice. Phytother. Res. 2014, 28, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Reeve, V.E.; Widyarini, S.; Domanski, D.; Chew, E.; Barnes, K. Protection against photoaging in the hairless mouse by the isoflavone equol. Photochem. Photobiol. 2005, 81, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.J.; Alshetaili, A.; Aldayel, I.A.; Alablan, F.M.; Alsulays, B.; Alshahrani, S.; Alalaiwe, A.; Ansari, M.N.; Ur Rehman, N.; Shakeel, F. Formulation, characterization, in vitro and in vivo evaluations of self-nanoemulsifying drug delivery system of luteolin. J. Taibah Univ. Sci. 2020, 14, 1386–1401. [Google Scholar] [CrossRef]
- Martinez, R.M.; Pinho-Ribeiro, F.A.; Steffen, V.S.; Caviglione, C.V.; Vignoli, J.A.; Barbosa, D.S.; Baracat, M.M.; Georgetti, S.R.; Verri, W.A., Jr.; Casagrande, R. Naringenin inhibits uvb irradiation-induced inflammation and oxidative stress in the skin of hairless mice. J. Nat. Prod. 2015, 78, 1647–1655. [Google Scholar] [CrossRef]
- Martinez, R.M.; Pinho-Ribeiro, F.A.; Steffen, V.S.; Silva, T.C.; Caviglione, C.V.; Bottura, C.; Fonseca, M.J.; Vicentini, F.T.; Vignoli, J.A.; Baracat, M.M.; et al. Topical formulation containing naringenin: Efficacy against ultraviolet b irradiation-induced skin inflammation and oxidative stress in mice. PLoS ONE 2016, 11, e0146296. [Google Scholar] [CrossRef]
- Jang, H.-J.; Yang, K.E.; Oh, W.K.; Lee, S.-I.; Hwang, I.-H.; Ban, K.-T.; Yoo, H.-S.; Choi, J.-S.; Yeo, E.-J.; Jang, I.-S. Nectandrin b-mediated activation of the ampk pathway prevents cellular senescence in human diploid fibroblasts by reducing intracellular ros levels. Aging 2019, 11, 3731–3749. [Google Scholar] [CrossRef]
- Maruki-Uchida, H.; Kurita, I.; Sugiyama, K.; Sai, M.; Maeda, K.; Ito, T. The protective effects of piceatannol from passion fruit (Passiflora edulis) seeds in uvb-irradiated keratinocytes. Biol. Pharm. Bull. 2013, 36, 845–849. [Google Scholar] [CrossRef]
- Sohn, E.-J.; Kim, J.M.; Kang, S.-H.; Kwon, J.; An, H.J.; Sung, J.-S.; Cho, K.A.; Jang, I.-S.; Choi, J.-S. Restoring effects of natural anti-oxidant quercetin on cellular senescent human dermal fibroblasts. Am. J. Chin. Med. 2018, 46, 853–873. [Google Scholar] [CrossRef]
- Sánchez-Marzo, N.; Pérez-Sánchez, A.; Barrajón-Catalán, E.; Castillo, J.; Herranz-López, M.; Micol, V. Rosemary diterpenes and flavanone aglycones provide improved genoprotection against uv-induced DNA damage in a human skin cell model. Antioxidants 2020, 9, 255. [Google Scholar] [CrossRef]
- Pérez-Sánchez, A.; Barrajón-Catalán, E.; Herranz-López, M.; Castillo, J.; Micol, V. Lemon balm extract (Melissa officinalis, L.) promotes melanogenesis and prevents uvb-induced oxidative stress and DNA damage in a skin cell model. J. Dermatol. Sci. 2016, 84, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Agulló-Chazarra, L.; Borrás-Linares, I.; Lozano-Sánchez, J.; Segura-Carretero, A.; Micol, V.; Herranz-López, M.; Barrajón-Catalán, E. Sweet cherry byproducts processed by green extraction techniques as a source of bioactive compounds with antiaging properties. Antioxidants 2020, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, A.; Manzione, L. Dasatinib: A new step in molecular target therapy. Ann. Oncol. 2007, 18 (Suppl. 6), vi42–vi46. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M.V. Long-term treatment with senolytic drugs dasatinib and quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 2021, 12, 5213. [Google Scholar] [CrossRef] [PubMed]
- La Rosée, P.; Martiat, P.; Leitner, A.; Klag, T.; Müller, M.C.; Erben, P.; Schenk, T.; Saussele, S.; Hochhaus, A. Improved tolerability by a modified intermittent treatment schedule of dasatinib for patients with chronic myeloid leukemia resistant or intolerant to imatinib. Ann. Hematol. 2013, 92, 1345–1350. [Google Scholar] [CrossRef]
- Saccon, T.D.; Nagpal, R.; Yadav, H.; Cavalcante, M.B.; Nunes, A.D.C.; Schneider, A.; Gesing, A.; Hughes, B.; Yousefzadeh, M.; Tchkonia, T.; et al. Senolytic combination of dasatinib and quercetin alleviates intestinal senescence and inflammation and modulates the gut microbiome in aged mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2021, 76, 1895–1905. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Senescence | Quiescence | Terminal Differentiation | T-Cell Exhaustion | References | |
|---|---|---|---|---|---|
| Type of cell cycle arrest | Generally irreversible | Reversible | Generally irreversible | Largely irreversible | [26,27,29,30] |
| Cause | Repetitive stimulation; DNA damage agents; stress signals | Signals of mitogen deprivation; contact inhibition | Genetically preprogramed | Continuous antigenic stimulation | [26,27,31,32] |
| Typical features | Large flat cells | Reduced cell size | n/a | n/a | [33,34] |
| Cell cycle arrest driver: ↑ p16, p21, p53 | CDK inhibitors: ↑ p21,27, 57 | p21, p27, and p57 | ↑ p27, p15; ↓ cyclin E-Cdk2, Cdc25A | [26,27,31,35] | |
| ↑ Macromolecular damage ↓ Telomere length, telomerase activity | Does not exhibit macromolecular damage | Does not exhibit macromolecular damage | ↓ Telomere length, telomerase activity | [36,37,38] | |
| ↑ SA-β-gal activity | Does not result in the upregulation of SA-β-gal activity | Does not result in the upregulation of SA-β-gal activity | Does not result in the upregulation of SA-β-gal activity | [39,40] | |
| n/a | n/a | n/a | ↑ Inhibitory receptors: PD1, TIM3, LAG3, CTLA4, TIGIT | [41] | |
| ↑ Glycolysis | ↓↑ Glycolysis (depending on cell type) | n/a | ↓ Glycolysis | [42,43,44] | |
| Cytokine pattern | SASP, proinflammatory cytokines: ↑ IL-1, IL-6, IL-8, IFN-γ, TNF | n/a | n/a | ↓ IL-2 ↓ TNF ↓ IFN-γ, β-chemokines | [45,46] |
| Epigenetic changes | ↑ SAHF Abnormal DNA methylation | ↑ H3K27me3 chromatin modifications; expression level of several histones is strongly reduced | ↑ H3K9me3 and H3K27me3; reduced levels of global DNA methylation; enhancers are enriched for H3K27me3 and DNA methylation, which is associated with the lower expression of their target genes | Exhaustion-associated DNA methylation patterns | [37,47,48,49] |
| Hallmarks of Senescence | Biomarker | Observation | Detection Method | References |
|---|---|---|---|---|
| Cell cycle arrest | p16/pRB axis | ↑ p16 ↑ pRb ↓ Phospho-pRb | WB, IHC, IF | [56,115,116,117,118,119] |
| p53/p21 axis | ↑ p21 ↑ p53 ↑ Phospho-p53 | |||
| Absence of proliferation | ↓ Ki67 ↓ PCNA | IHC, IF | [24,120,121] | |
| Decrease in/absence of DNA synthesis | ↓ BrdU, EdU | Staining incorporation, immunofluorescence | [122] | |
| Metabolic adaptations | SA-β-gal | ↑ | NIR Fluorescence Enzymatic staining | [123,124,125] |
| α-fucosidase | ↑ | Fluorescence Enzymatic staining | [64,126] | |
| Lipofuscin | ↑ | Dye incorporation (SSB, GL13) Fluorescence | [127,128] | |
| Morphological changes | Wide and flattened cells High vacuolization | n/a | IF Scanning electron microscopy Light microscopy Flow cytometry | [129,130] |
| Lamin B1 | ↓ | qPCR, IF, WB | [131] | |
| Plasma membrane proteins | ↑ ICAM-1, DEP1 | Immunohistochemistry IF, WB, flow cytometry | [132] | |
| Epigenetic alterations | SAHF | ↑ PML bodies ↑ H3K9 methylation | IF | [133] |
| DNA damage | γH2AX 53BPI ATM ATR TIF | ↑ | IF | [89] |
| Telomere shortening | ↓ | qPCR, FISH | [134,135] | |
| Apoptosis resistance | Annexin V Cleaved caspases Cleaved PARP | ↓/absent | IF IHC WB | [136,137,138,139,140] |
| Blunt ends of double-stranded DNA breaks | - | TUNEL assay | [141] | |
| Secretory phenotype | SASPs | ↑ IL-1, IL-6, IL-8, ↑ TNF-α, GROα/β, ↑ MMP-1, MMP-3, MMP-9 ↑ IGFBPs ↑ SERPINs ↑ TIMPs | ELISA Immunofluorescence WB SASP-responsive alkaline phosphatase assay | [142,143,144,145] |
| ROS | O2, H2O2, O2•−, HO• | ↑ | Chemiluminescent oxygen detection, fluorometry, flow cytometry | [146,147] |
| Polyphenol | Route of Administration | Aging Inductor | Research Model | Mechanism | Main Senotherapeutic Effects | Reference |
|---|---|---|---|---|---|---|
| Apigenin | In vitro | UVA and UVB | Human dermal fibroblasts | ↓ ROS ↓ NF-kB pathway ↓ MAPK ↓ MMP-1 | ↑ Viability ↑ Collagen synthesis ↑ DNA repair | [148,168] |
| Topical | UVA | Mice | ↓ ROS ↓ NF-kB pathway ↓ MAPK ↓ MMP-1 | ↑ Dermal thickness ↑ Collagen deposition | [169] | |
| Baicalin | In vitro | UVB | Human dermal fibroblasts, human skin samples | ↓ ROS ↓ MMP-1, MMP-3 ↓ p16, p21, p53 | ↑ Collagen synthesis ↑ Viability ↓ DNA damage ↓ Apoptosis | [81] |
| In vitro | UVC | Human keratinocytes | ↓ ROS | ↓ DNA damage | [170] | |
| Ferulic acid | In vitro | UVA | Human dermal fibroblasts | ↓ ROS ↑ SOD1 ↑ CAT ↓ p16 ↓ MMP-1, -3 | ↑ Proliferation and cell cycle ↑ ECM reconstruction | [171] |
| Fisetin | In vitro | Hydrogen peroxide | Human keratinocytes | ↓ ROS ↓ NF-kB ↓ iNOS ↓ COX-2 ↓ IL-1β, -6, TNF-α | ↓ SASP secretion ↑ Viability | [172] |
| In vitro | UVB | Human dermal fibroblasts | ↓ ROS ↓ MAPK/AP-1/MMP | ↓ SASP secretion ↓ Collagen degradation | [173] | |
| Gallic acid | In vitro | UVB | Human dermal fibroblasts | ↓ ROS ↓ MMP-1 ↓ IL-6 | ↑ Procollagen type I | [174] |
| Topical and oral | UVB | Mice | ↑ TGF-β1 ↓ MMP-1 ↓ IL-6 | ↓ Wrinkle formation ↓ Skin dryness ↑ Procollagen type I ↑ Elastin | ||
| Genistein | Topical | UVB | Mice | ↓ ROS ↓ DNA pyrimidine dimer formation | ↓ DNA damage | [175] |
| Luteolin | In vitro and topical | UVA | Human dermal fibroblasts, human keratinocytes, and human skin explants | ↓ ROS ↓ MMP-1 ↓ IL-6, -20 ↓ p38/MAPK | ↓ SASP secretion ↓ Collagen degradation ↓ Hyaluronic acid degradation | [176] |
| Naringenin | Intraperitoneal | UVB | Mice | ↓ ROS ↓ MMP-9 ↓ TNF-α, IFN-γ ↓ IL-1β, -4, -5, -6, -12, -13, -17, -22, -23 | ↓ SASP secretion ↓ Inflammatory infiltrations | [177] |
| Topical | UVB | Mice | ↓ ROS ↓ IL-1β, -6, -10, TNF-α | ↓ SASP secretion | [178] | |
| Nectandrin B | In vitro | Cell passage ≥72 | Human diploid fibroblasts | ↓ ROS ↑ AMPK ↓ p16, p21, p27, p53 ↓ Cyclin D1 ↓ SA-β-gal ↓ Caveolin-1 | ↓ Senescence ↓ Apoptosis | [179] |
| Piceatannol | In vitro | UVB | Human keratinocytes | ↓ ROS ↑ GSH ↓ NF-kB ↓ MMP-1 | ↓ Melanogenesis ↑ Collagen synthesis ↓ Photoaging | [180] |
| Quercetin | In vitro | Cell passage ≥17 | Human dermal fibroblasts | ↓ ROS ↑ SOD2, -3 ↑ CAT ↓ p16, p53 | ↓ Senescence ↑ Mitochondrial membrane potential | [181] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bulbiankova, D.; Díaz-Puertas, R.; Álvarez-Martínez, F.J.; Herranz-López, M.; Barrajón-Catalán, E.; Micol, V. Hallmarks and Biomarkers of Skin Senescence: An Updated Review of Skin Senotherapeutics. Antioxidants 2023, 12, 444. https://doi.org/10.3390/antiox12020444
Bulbiankova D, Díaz-Puertas R, Álvarez-Martínez FJ, Herranz-López M, Barrajón-Catalán E, Micol V. Hallmarks and Biomarkers of Skin Senescence: An Updated Review of Skin Senotherapeutics. Antioxidants. 2023; 12(2):444. https://doi.org/10.3390/antiox12020444
Chicago/Turabian StyleBulbiankova, Darya, Rocío Díaz-Puertas, Francisco Javier Álvarez-Martínez, María Herranz-López, Enrique Barrajón-Catalán, and Vicente Micol. 2023. "Hallmarks and Biomarkers of Skin Senescence: An Updated Review of Skin Senotherapeutics" Antioxidants 12, no. 2: 444. https://doi.org/10.3390/antiox12020444
APA StyleBulbiankova, D., Díaz-Puertas, R., Álvarez-Martínez, F. J., Herranz-López, M., Barrajón-Catalán, E., & Micol, V. (2023). Hallmarks and Biomarkers of Skin Senescence: An Updated Review of Skin Senotherapeutics. Antioxidants, 12(2), 444. https://doi.org/10.3390/antiox12020444