Abstract
Mitochondria are specialized organelles, which serve as the “Power House” to generate energy for maintaining heart function. These organelles contain various enzymes for the oxidation of different substrates as well as the electron transport chain in the form of Complexes I to V for producing ATP through the process of oxidative phosphorylation (OXPHOS). Several studies have shown depressed OXPHOS activity due to defects in one or more components of the substrate oxidation and electron transport systems which leads to the depletion of myocardial high-energy phosphates (both creatine phosphate and ATP). Such changes in the mitochondria appear to be due to the development of oxidative stress, inflammation, and Ca2+-handling abnormalities in the failing heart. Although some investigations have failed to detect any changes in the OXPHOS activity in the failing heart, such results appear to be due to a loss of Ca2+ during the mitochondrial isolation procedure. There is ample evidence to suggest that mitochondrial Ca2+-overload occurs, which is associated with impaired mitochondrial OXPHOS activity in the failing heart. The depression in mitochondrial OXPHOS activity may also be due to the increased level of reactive oxygen species, which are formed as a consequence of defects in the electron transport complexes in the failing heart. Various metabolic interventions which promote the generation of ATP have been reported to be beneficial for the therapy of heart failure. Accordingly, it is suggested that depression in mitochondrial OXPHOS activity plays an important role in the development of heart failure.
1. Introduction
The heart is the most active organ in the body and requires a significant amount of energy in the form of adenosine triphosphate (ATP) to sustain its vital function continuously. Despite having a limited ATP storage capacity, the heart has a remarkably efficient and reliable energy production system facilitated by the abundant presence of mitochondria in cardiomyocytes. Mitochondria, known as the “powerhouse” in the heart, perform a crucial role in generating ATP through the process of oxidative phosphorylation (OXPHOS) [1,2,3,4,5]. Furthermore, these organelles are involved in the exchange and synthesis of metabolites, calcium storage, the production of reactive oxygen species (ROS), as well as cell survival and death signals, all of which are critical for regulating cardiac function in health and disease. Additionally, mitochondria have been shown to participate in various signaling pathways associated with the regulation of oxidative stress, inflammation, mitophagy, calcium handling, and apoptosis, which are fundamental to inducing cardiac dysfunction under diverse pathophysiological conditions [6,7,8,9,10,11]. Since mitochondria generate ATP upon the oxidation of different substrates, their function is dependent upon the type of substrate availability as well as the status of glycolysis, lipolysis and proteolysis in cardiomyocytes [12,13,14,15]. In particular, these organelles regulate metabolic pathways such as the tricarboxylic acid cycle and the beta-oxidation of free fatty acids, which contribute to the electron transport chain in the OXPHOS complex and impact processes like ROS production, redox state, and apoptosis [16,17,18,19].
Mitochondrial abnormalities associated with cardiac dysfunction include disruption in the process of energy generation, cation accumulation, ROS production, aldehydic load, and intracellular signaling. Since mitochondria play a fundamental role in sustaining cardiac contractility, the impairment of their function has been implicated in several cardiovascular diseases, in which a decrease in ATP production has been shown to cause a depletion of cardiac energy stores [19,20,21,22,23,24,25,26,27,28,29]. Risk factors of cardiovascular diseases, such as ischemia/reperfusion injury, hypertension, ventricular hypertrophy, cardiomyopathies, atherosclerosis, metabolic syndrome, and diabetic hyperglycemia, have been reported to produce mitochondrial dysfunction [30,31,32,33,34,35,36,37]. The role of mitochondria in producing ROS is critical because they function as redox messengers when generated within normal levels, but excessive ROS production can lead to oxidative stress and ultimately result in cell death. It has been indicated that oxidative stress, in addition to producing mitochondrial dysfunction, can cause the increased production of pro-inflammatory cytokines and the activation of fibroblasts in the extracellular matrix. These alterations result in interstitial fibrosis and passive stiffness of the myocardium. However, extensive work is required to establish the exact cause–effect relationship among myocardial oxidative stress, myocardial inflammation and mitochondrial dysfunction during the development of heart failure. Nonetheless, mitochondrial oxidative stress has been shown to increase Ca2+-influx, which worsens cardiomyocyte relaxation and elevates the left ventricle filling pressure, along with proteolytic damage to the heart [38,39,40,41,42]. The intrinsic compensatory mechanisms such as the level of intracellular Ca2+ and antioxidant system that typically control the oxidation of substrates and energy production in mitochondria also fail to offset the depletion of myocardial energy stores during the development of cardiac dysfunction [43,44,45,46].
Considering the high energy demand of the cardiac excitation–contraction and relaxation cycle, patients with abnormalities in the mitochondrial OXPHOS are at a higher risk of developing heart disease [34,35,44,47]. Different defects within the mitochondrial OXPHOS system, which can arise due to genetic or environmental factors, have been implicated in various cardiac disorders, including ischemia/reperfusion injury, hypertension, arrhythmias, cardiac hypertrophy, cardiomyopathies, and heart failure [3,30,35,36]. Thus, impaired OXPHOS plays a significant role in the onset of various cardiovascular diseases, which can manifest differently depending on the underlying cause and stage of heart disease [9,48,49,50]. Although abnormalities in mitochondrial OXPHOS have long been observed in cases of heart failure and other cardiac pathologies, the underlying causes of these abnormalities remain poorly understood. It is, therefore, the objective of this article to provide a comprehensive overview of the impact of impaired mitochondrial OXPHOS during the development and progression of heart failure as a consequence of various pathological conditions. Several components of the mitochondrial OXPHOS system as well as their functions in the heart will be described. In addition, some pharmacological and metabolic interventions aimed at OXPHOS pathway defects as targets, which prevent or treat heart failure and enhance patient survival, are also summarized.
2. Function of Mitochondrial OXPHOS in the Heart
Mitochondria are specialized organelles that feature two membranes and their own DNA system. Their inner membrane, characterized by numerous infoldings (cristae), hosts several proteins, including enzymes that facilitate mitochondrial OXPHOS. The controlling of this metabolic process depends on a variety of factors, including intrinsic kinetic parameters and the regulation of various enzymes, the architecture network as well as intermediate substrate concentrations under steady-state conditions [51,52]. OXPHOS complexes [53,54], namely complex I (NADH/ubiquinone oxidoreductase), complex II (succinate ubiquinone oxidoreductase), complex III (ubiquinol cytochrome c oxidoreductase), complex IV (cytochrome c oxidase), and complex V (ATP synthase) are located on the inner membrane [55,56,57,58,59,60]. These complexes, collectively known as the electron transport chain, transfer electrons from donors generated by the tricarboxylic acid cycle or fatty acid oxidation [61] for the generation of ATP. The nuclear and mitochondrial genomes regulate these complexes, with mitochondrial genes playing a major role in assembling the core complexes within the mitochondria. The cardiac cells need a lot of ATP and have mitochondria occupying approximately 20–40% of their volume [62]. These organelles produce about 6 kg of ATP daily through OXPHOS-associated electron transport mechanisms in the human heart [5]. In a normal heart, fatty acid, glucose, and a ketone body enter cardiomyocytes and are transported into mitochondria in various forms [14,15,63]. These are catabolized in the mitochondria to acetyl coenzyme A (acetyl-CoA) for entering the tricarboxylic cycle and then undergo a series of redox reactions in the electron transport chain. This process is associated with the production of NADH and FADH2, which are then oxidized by NADH dehydrogenase and succinate dehydrogenase in the inner membrane. OXPHOS complex I and complex II receive these electrons from the donors, after which Coenzyme Q (CoQ) transports them to complex III [57,64,65]. Eventually, the electrons are transferred to the hydrophilic heme protein cytochrome C and then to complex IV [57]. The electron transport chain then creates a proton-motive force and, simultaneously, protons are pumped into the mitochondrial intermembrane space against a concentration gradient (ΔpHm) [49,66].
The movement of electrons in the respiratory chain creates a negative charge inside the mitochondrial matrix and is termed as mitochondrial membrane potential (ΔΨm) [67]. Protons then re-enter the mitochondrial matrix via complex V due to the proton gradient to generate ATP from ADP [57]. Phosphocreatine functions as an energy buffer that facilitates intracellular ATP transfer, whereas the clusters of mitochondrial electron transport complexes combine to form supercomplexes, which are crucial in regulating electron flow within mitochondria [68,69,70]. It is noteworthy that the inner mitochondrial membrane is mostly impenetrable to cations and small molecules, thus making proton pumping a critical step in this conversion process [71]. During the OXPHOS process in mitochondria, superoxide anions are produced by about 2% of electrons passing through the electron transport chain in complexes I, II, and III [72,73], but these are rapidly altered or dismutated by superoxide dismutase (MnSOD and CuZnSOD) to form hydrogen peroxide, which is then broken down into water by antioxidant enzymes, including catalase, glutathione peroxidase and peroxiredoxins [74,75,76]. The ROS signaling molecules are the byproducts of oxygen metabolism and affect oxygen-sensing mechanisms like gene expression. It is also pointed out that cardiolipin is a critical phospholipid which is pivotal in stabilizing mitochondrial OXPHOS complexes and facilitating supercomplexes’ formation within the electron transport chain and thus precisely regulating ATP production in the mitochondria. The OXPHOS system complexes I-V and molecules like CoQ and cardiolipin work together to ensure the optimal functioning of the mitochondrial energy-producing system [77,78,79,80]. However, it is pointed out that in a study concerning the role of mitochondrial supercomplexes in maintaining OXPHOS activity, Milenkovic and coworkers [81] failed to demonstrate any change in the mitochondrial bioenergetic capacity under conditions associated with a major loss of respirasomes.
3. Impact of Impaired OXPHOS in the Pathogenesis of Heart Failure
3.1. Bioenergetics and OXPHOS Capacity
Several studies on both human and animal heart disease models have revealed a notable decline in the cellular ATP and phosphocreatine content, indicating an altered energy metabolism in the heart. As a result, different abnormalities observed in heart disease, including altered nutrient usage, reduced OXPHOS activity, increased oxidative stress, and aberrant calcium handling and mitochondrial dynamics, lead to alterations in energy metabolism, which ultimately result in the irreversible deterioration of heart function [82,83,84,85,86,87,88,89,90]. An imbalance in the ATP supply triggered by pathological stimuli under conditions such as left ventricular remodeling, chamber dilation, and hypertrophy can worsen the progression of heart disease. Such defects may increase the energy demand while reducing the energy supply, leading to altered bioenergetics in the diseased heart. These changes impair OXPHOS and associated activities, including the creatine kinase energy-transfer mechanism, elevating free adenosine diphosphate (ADP) levels, and decreasing the ATP content during the later stages of heart disease [44,45,91,92]. Various studies have referred to heart failure as an energy-deprived state characterized by a decline in ATP production and driven mainly by impaired OXPHOS. The appropriate functioning of the heart relies heavily on the efficient mitochondrial oxidative metabolism to maintain ATP production. Thus, any malfunction or disruption in the role of OXPHOS electron transport chain complexes for the production of energy in mitochondria may lead to an imbalance in cardiac cell metabolism.
In the diseased heart, varying degrees of changes in substrate utilization, a reduction in the electron transport chain activity, depression in both ATP and phosphocreatine content, and attenuation in the rate of ATP transfer to phosphocreatine have been observed [93]. Mitochondrial dysfunction enhances ROS levels through electron transport chain-mediated ROS production due to the defective regeneration of NADPH and the excessive levels of ROS are known to cause detrimental effects in the myocardium. These ROS molecules are known to alter proteins, DNA, and lipids, leading to oxidative damage in the heart [37,72,94,95,96]. Additionally, an overload of ROS can result in the abnormal opening of the mPTP, releasing detrimental substances and leading to the swelling of the mitochondria, ruptured membranes, and triggering inflammation, apoptosis, and cell damage [97,98]. Furthermore, an overabundance of ROS can deplete the intracellular redox pool, impair cellular Ca2+ handling, cation channel activities, and ROS-mediated redox signaling pathways [99,100,101]. Although mitochondrial ROS signaling is important for the regulation of the oxidative metabolism, muscle contraction, and calcium transport [102,103,104,105], an imbalance between the production of ROS and the endogenous antioxidant system has been shown to produce oxidative stress. In fact, mitochondrial dysfunction increases oxidative stress through alterations in the tricarboxylic acid cycle and ATP synthase as well as cardiolipin degradation and mitochondrial electron leakage, which thus is considered to cause significant damage to the myocardium depending on the type and stage of heart disease [69]. Furthermore, heightened levels of mitochondrial oxidative stress markers have been reported to produce increased amount of oxidants in the electron transport chain at complex I in the diseased heart [106,107].
The extent of mitochondrial damage has been suggested as a key factor when determining myocardial injury due to myocardial infarction during progression to heart failure [36]. It is pointed out that acute changes such as cardiogenic shock and ischemia-reperfusion injury in myocardial infarction should be differentiated from chronic alterations associated with pathological hypertrophy and cardiac remodeling. Ischemia-reperfusion injury, as a consequence of coronary heart disease, dramatically increases mitochondrial permeability leading to the dissipation of electron and proton gradients, the dysregulation of mitochondrial calcium homeostasis, and the release of superoxide radicals which lead to myocardial cell death [108]. Furthermore, mitochondrial supercomplexes lose their integrity as the electron transport chain subunits degrade due to ischemia/reperfusion injury, leading to the impairment of mitochondrial function [109]. It was observed that a decrease in the complex I subunit and an increase in the complex II subunits occur, suggesting a redirection of the electron input through complex II [110]. During the reperfusion phase of the ischemia-reperfusion injury, an abrupt elevation in ROS levels can induce myocardial cell damage resulting in cellular death through necrosis [111]. The overproduction of ROS triggered by ischemia as well as ischemia-reperfusion has also been considered to initiate apoptosis in cardiac cells, which is a significant contributing factor for the development of heart failure [112]. It is noteworthy that mitochondrial respiration is a crucial determinant of the functional status of the OXPHOS system [113,114,115]. A study on dogs with chronic heart failure induced by intracoronary embolization revealed that the mitochondrial state-3 respiration and mitochondrial membrane potential in the failing heart were lower than those in the healthy heart. This decline in oxygen consumption by mitochondria and reductions in the membrane potential were associated with alterations in the OXPHOS complexes. In fact, dysfunction in the mitochondrial tricarboxylic acid cycle was observed to be closely linked to heart failure [116,117]. Another study involving myocardial infarction in mice demonstrated that chronic heart failure causes a decrease in the mitochondrial OXPHOS capacity due to decreased levels of succinyl-CoA in the myocardium. Furthermore, administering 5-aminolevulinic acid to infarcted mice was found to restore the succinyl-CoA levels and OXPHOS capacity by inducing excessive heme synthesis, potentially attenuating the progression of heart failure [118]. A recent study with cardiac tissue samples from heart failure patients has also revealed a dysfunction in succinyl-CoA metabolism [119].
3.2. Genetic Regulation of OXPHOS
It is pointed out that the DNA damage response and RNA polymerase II pausing pathway are significantly downregulated in failing human hearts, as well as primate and murine hearts, following myocardial infarction [120]. In a mouse model, the cardiac-specific inactivation of LARP7 (La ribonucleoprotein domain family member 7) resulted in decreased oxidative phosphorylation, mitochondrial biogenesis impairment and elevated oxidative stress, ultimately leading to heart failure. These irregularities, as well as the reduced deacetylase activity of SIRT1 (silent mating-type information regulation 2 homolog 1), which is responsible for the transcription of genes related to the mitochondrial OXPHOS system and energy metabolism, were shown to reduce cardiac function. The restoration of LARP7 expression in the infarcted heart through adenovirus-mediated LARP7 expression or by a small molecule ATM inhibitor has been shown to improve the function of the injured heart [65,121]. Decreased PGC1 (Peroxisomal proliferator-activated receptor gamma coactivator 1)-α levels and reduced nuclear genome-encoded OXPHOS complexes have been observed in animal models of heart failure [122,123]. Since heart failure with a preserved ejection fraction (HFpEF) and heart failure with a reduced ejection fraction (HFrEF) are metabolically distinct, changes in the OXPHOS gene transcripts have been shown to characterize these differences. The transcriptome analysis of ventricular tissue from patients with HFpEF and HFrEF has revealed the involvement of elevated genes in OXPHOS [124] and impaired complex1-mediated mitochondrial respiration in permeabilized cardiac fibers in the HFpEF condition [125]. Furthermore, the degradation of cardiolipin due to oxidative stress, a significant increase in mitochondrial electron leakage, and reduced levels of CoQ have been observed in patients and animals with heart failure, indicating malfunction in the electron transport chain and oxidative phosphorylation’s ability to produce sufficient ATP in the failing heart [126,127]. A sufficient intake of linoleic acid in heart failure has been demonstrated to increase cardiolipin levels, improve mitochondrial OXPHOS activity, and enhance left ventricular function [128].
It may be noted that the OXPHOS complexes are encoded by both the nuclear and mitochondrial genomes as various studies on gene expression have revealed that individuals afflicted with heart disease possess reduced levels of mitochondrial metabolic genes and proteins. In this regard, mutations in genes regulating mitochondrial proteins were found to adversely impact energy production, decrease mitochondrial function, increase ROS production, and result in the development of cardiomyopathy [129]. Patients diagnosed with dilated cardiomyopathy also exhibit significant alterations in their metabolic pathways, specifically, the enzymes related to OXPHOS and the tricarboxylic acid cycle were down-regulated for complex III at both transcriptional and proteomic levels. Furthermore, the activities of complex III and IV in the left ventricular tissue from these patients were depressed [129,130,131]. It has also been shown that dilated hypertrophy influences energy generation by the mitochondria, which can alter the transcript levels of nuclear DNA- and mitochondrial DNA (mtDNA)-encoded mitochondrial genes and result in the reduced production of new mitochondria, impaired mitochondrial OXPHOS, and increased ROS production [132]. On the other hand, hypertrophic cardiomyopathy was found to be associated with cytochrome C deficiency leading to death [132,133]. Mitochondrial defects in the electron transport chain have also been implicated in the pathogenesis of diabetic cardiomyopathy [134]. Since the dilated hypertrophy, hypertrophic cardiomyopathy, and diabetic cardiomyopathy have different oxidative stress patterns [135,136], it is likely that differences in the profiles of mitochondrial abnormalities may be a consequence of differences in ROS production in these pathological conditions.
3.3. Structural Changes in Mitochondrial Network
Alterations in the mitochondrial ultrastructure and function, including reductions in the activities of respiratory chain enzymes (complexes I to IV) and capacity for OXPHOS, are commonly observed in patients with heart failure, although these may not manifest until the later stages of the disease. In this regard, chronic hypertrophy without systolic dysfunction has been shown to be associated with normal or improved mitochondrial function in both animals and humans [130,137,138,139,140,141]. On the other hand, impaired OXPHOS in adverse ventricular remodeling due to volume overload was observed before any signs of systolic dysfunction or decompensation were detected [142]. Some studies have shown that OXPHOS rates tend to increase during the early stages of cardiac hypertrophy, but decline as the condition progresses towards heart failure [26,143]. A diminished expression of OXPHOS components was reported to result in a decline in mitochondrial respiration in heart failure and cardiomyopathies [82,144]. These changes in mitochondrial function may be a consequence of oxidative stress, which in heart failure arises from various sources of ROS such as the activation of NADPH and monoamine oxidase as well as mitochondrial complexes I, II, and III, which are considered to play a major role in ROS production. Compared to healthy hearts, cardiomyocytes exhibit a significant increase in ROS levels within the mitochondrial matrix in failing hearts [116,117,145]. The interaction between a small amount of ROS and mitochondrial components leads to mitochondrial dysfunction, which produces more ROS, and further damages the mitochondria, impairs contractile dysfunction, and worsens heart failure [146,147]. A marked increase in oxidative stress in heart failure may also be a consequence of the depletion of different antioxidant enzymes and antioxidants in the failing heart [148].
Malfunctions in the respiratory chain can initiate oxidative stress and prompt the emergence of cardiac hypertrophy. The loss of mitochondrial ribosomal protein S5 (MRPS5/uS5m) in the developing heart leads to cardiac defects and embryonic lethality, while postnatal loss impairs mitochondrial protein translation and OXPHOS during the development of cardiac hypertrophy and heart failure [149]. Since the structural and functional changes in the damaged mitochondrial network are critical, both fusion and fission processes are required in distributing protein and DNA [150,151]. The disruption of these processes can lead to mitochondrial damage and cell death. It should be pointed out that fusion is necessary for maintaining OXPHOS and energy levels, protecting against oxidizing molecules, and preserving the mitochondrial integrity [152,153]. Fusion proteins, Mfn-1, Mfn-2, and OPA-1 are essential for preserving mitochondrial integrity, while their suppression can lead to dilated cardiomyopathy and contractile abnormalities [154], and increase apoptosis and the fragmentation of the mitochondria [155,156,157,158,159]. Their deletion in a mouse heart was observed to result in an abnormal mitochondrial morphology and mitochondrial fragmentation leading to ventricular wall thickening and an increase in cardiac mass, accompanied by eccentric hypertrophy [160]. On the other hand, excessive fission leads to a loss of mitochondrial mass, impaired OXPHOS and ATP deficits, permeabilization, cytochrome C release, and apoptosis [161]. The deficiency of the fission protein, dynamin-related protein 1 (Drp1), which is highly expressed in the heart, exhibited lethal dilated cardiomyopathy with ventricular wall thinning and a reduced ejection fraction [162].
4. Stage- and Type-Dependent Changes in OXPHOS in Heart Failure
4.1. General Considerations
Various studies have found that defects in the electron transport complexes and other components of the mitochondrial OXPHOS system may vary depending on the cause (type) and severity (stage) of heart failure. When OXPHOS is impaired, it can result in the irregular production of ROS, which in turn leads to inflammation and oxidative stress, contributing to a range of cardiac abnormalities, such as cardiac hypertrophy, arrhythmias, and cardiomyopathy during the development of heart failure [26,27,48,49,163]. Reduced mitochondrial respiratory rates and changes in the OXPHOS function may signal the beginning of heart failure. These alterations commonly involve complex I-linked respiration, fatty acid oxidation, and the OXPHOS system in human hearts [113,114]. Further, it has been revealed that lower ADP-dependent respiratory rates are observed in dilated cardiomyopathy, pressure overload, or myocardial infarction [82,83,164,165]. It is also pointed out that some investigators have failed to detect any changes in heart failure [141], whereas others have demonstrated a depression in the OXPHOS activity at early stages [114]. It appears that changes in mitochondrial OXPHOS activities are dependent upon the type and stage of heart failure.
4.2. Cardiomyopathic Hamster Heart Failure
Since the patterns of oxidative stress have been shown to be different with respect to acute heart failure and chronic heart failure [166,167], it is likely that alterations in mitochondrial OXPHOS may also be dependent upon the stage of heart failure. In order to gain some information in this regard, we employed cardiomyopathic hamsters (UM-X7.1) for determining the mitochondrial function at various stages of heart failure [24,163]. On the basis of clinical observations and general characteristics such as the amount of abdominal fluid accumulation, lung and liver congestion, as well as the heart-to-body weight ratio, different age groups of cardiomyopathic animals were considered at prefailure, early failure, moderate failure, and severe stages of heart failure [24,163]. It can be seen from the data in Table 1 that there was a progressive depression in the high-energy phosphates (both creatine phosphate and ATP) content at early, moderate, and severe stages of heart failure without any significant change at the prefailure stage. However, when the OXPHOS activity, by using pyruvate-malate as a substrate, was examined in the mitochondria isolated from the hearts of cardiomyopathic hamsters at different stages of heart failure, the phosphorylation rate was depressed (without any changes in ADP/O ratio) only at the severe stages of heart failure (Figure 1). The depressed OXPHOS activity and respiratory rate at state 3 in mitochondrial preparations or whole-heart homogenates were also seen by using glutamate- pyruvate or glutamate alone as substrates at the severe stages of heart failure [163] in cardiomyopathic hamsters. Mitochondrial Ca2+ uptake activity, unlike mitochondrial ATPase activity, was also found to be decreased at the severe stage of heart failure in cardiomyopathic hamsters (Figure 2). Although these observations indicate a generalized defect in the mitochondrial function at severe stages of heart failure, the observed depression in the high-energy phosphate stores at early and moderate stages of heart failure cannot be explained on the basis of such changes in energy production [163]. It should be mentioned that the energy utilization systems due to myofibrillar ATPase and membrane ATPases at different stages of heart failure were either unaltered or depressed in this experimental model [24,163].

Table 1.
Creatine phosphate (CP) and adenosine triphosphate (ATP) content in the control and cardiomyopathic hamster (UM- X7.1) hearts at different stages of heart failure.
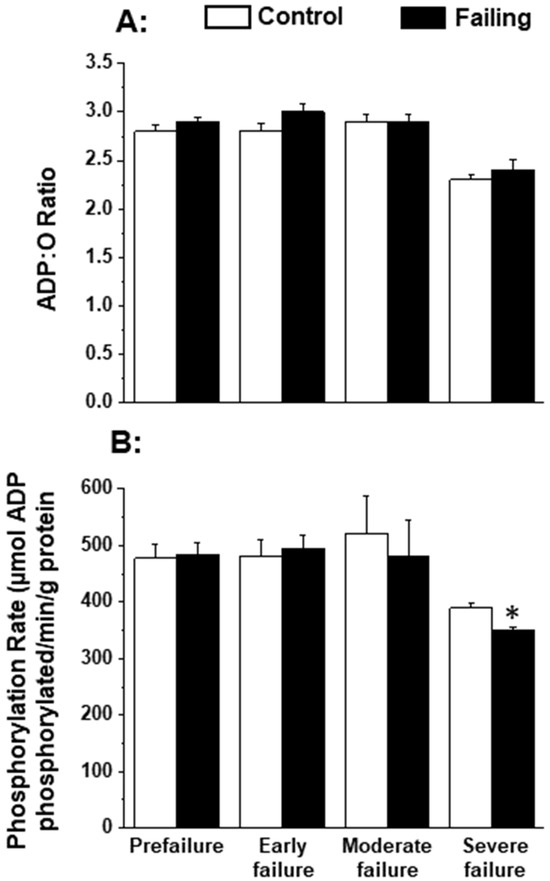
Figure 1.
Oxidative phosphorylation rate (B) and ADP:O ratio (A) in by heart mitochondria in cardiomyopathic hamsters (UM-X7.1) at different stages of congestive heart failure. Data are based on the results in our articles [24,163]. The isolation of mitochondria was carried out using 10 mM ethylenediaminetetra-acetic acid (EDTA) [163]. Each value is mean of ± SE of four to six experiments. Mitochondria were isolated by pooling 4 hearts for each experiment. The substrate employed was 1.5 mM pyruvate plus 0.3 nM malate. Different groups of cardimyopathyic hamsters were selected on the basis of their age: Prefailure (90 to 100 days), Early failure (120 to 160 days), Moderate failure (160 to 200 days), and Severe failure (200 to 280 days). Age-matched control hamsters were used for each group. The depression in phosphorylation rate at severe stages of heart failure was due to a significant decrease in the state 3 respiratory rate without any changes in the state 4 respiration [163]. * p < 0.05. ADP, adenosine diphosphate.
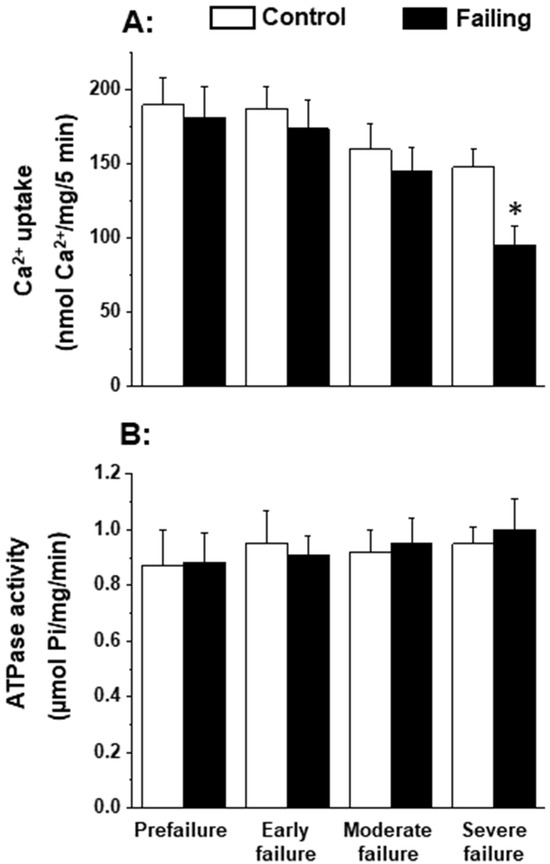
Figure 2.
Mitochondrial Ca2+-uptake (A) and ATPase (B) activities in cardiomyopathic hamsters (UM-X7.1) at different stages of congestive heart failure. Data are based on the results in our articles [24,163]. It is pointed out that mitochondria were isolated using a medium containing 10 mM ethylenediaminetetra-acetic acid (EDTA) [163]. Each value is mean of ± SE of four to six experiments. Mitochondria were isolated by pooling 4 hearts for each experiment. Different groups of cardimyopathyic hamsters were selected on the basis of their age: Prefailure (90 to 100 days), Early failure (120 to 160 days), Moderate failure (160 to 200 days), and Severe failure (200 to 280 days). Age-matched control hamsters were used for each group. * p < 0.05.
Our inability to demonstrate depression mitochondrial OXPHOS activity at early and moderate stages of heart failure was found to be due to a loss of Ca2+ from the mitochondria during the process of isolation because the medium employed for this procedure contained ethylenediamine-tetra acetic acid (EDTA). It should also be mentioned that the mitochondrial Ca2+ content (About 10 umol/mg protein), in preparations obtained from the failing hearts were not different from that of the controls [24]. However, when the mitochondria were prepared using an isolation medium in the absence of EDTA, the mitochondrial Ca2+ content in the failing hearts were 5 to 7 times higher than that in the control and mitochondrial OXPHOS activity was significantly depressed at the early, moderate, and severe stages of heart failure [24]. These results can be seen to explain the variable defects in mitochondrial OXPHOS activity in heart failure as reported by several investigators. Furthermore, these observations also support the role of mitochondrial Ca2+-overload in depressing OXPHOS activity in these organelles [23]. Since ROS are formed due to defects in mitochondrial electron transport in disease hearts [117,145], it is likely that depressed OXPHOS activity observed in cardiomyopathic hamsters at severe stages of heart failure may also be partially due to changes in the electron transport chain complexes. In view of the findings that oxidative stress, myocardial inflammation, and Ca2+-handling abnormalities are generally associated with cardiac dysfunction [86,103,104,137], it is proposed that these pathogenic factors may serve as mechanisms for inducing mitochondrial Ca2+-overload and the subsequent depression of mitochondrial OXPHOS activity during the development of heart failure (Figure 3). However, it is pointed out that the earliest changes in the mitochondria may be of a compensatory nature for removing cellular stress due to cardiac inflammation and alterations in the substrate metabolism whereas delayed changes including depressed mitochondrial OXPHOS in heart failure may be of an adaptive nature for lowering the ROS production.
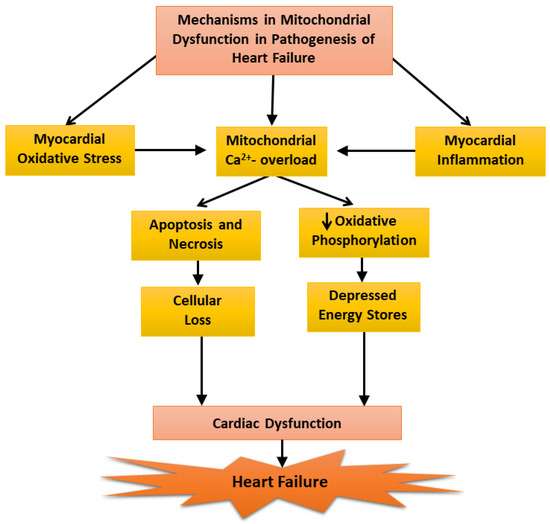
Figure 3.
Mechanisms of mitochondrial dysfunction in the pathogenesis of heart failure.
5. Mitochondrial Targets for Potential Therapeutic Interventions
Since the heart depends heavily on mitochondrial OXPHOS, which accounts for 90% of cellular ATP production, strategies that target defects in this pathway are essential for preserving energy production, enhancing cardiac function, and the survival of heart failure patients. Several therapeutic approaches in this regard involve targeting the mitochondria within the failing heart to modulate the organization of the respiratory complexes into supercomplexes for oxidative phosphorylation [168,169,170,171,172,173,174,175,176]. In coronary artery disease patients, the activities of complexes I, II, and III are depressed in the failing heart, despite an upregulation in protein expression, indicating a functional deficit in OXPHOS-related proteins [177]. Empagliflozin has shown promise as a treatment option to improve cardiac function by increasing OXPHOS, enhancing glucose and fatty acid oxidation as well as promoting cardiac efficiency [178,179,180]. Furthermore, elamipretide associated with cardiolipin has been observed to restore mitochondrial bioenergetics [181] because mitochondrial cardiolipin is essential for the proper assembly and stability of the electron transport chain to ensure the function of OXPHOS [182]. Studies in animal models of chronic heart failure have shown that elamipretide elicited a normalization of mitochondrial function as this agent improved respiration, restored the membrane potential, reduced ROS formation, and enhanced the maximum rate of ATP synthesis. Since the mutation of ribosomal protein S5 (MRPS5/uS5m) in the mitochondria has been observed to result in impaired mitochondrial protein translation and a depressed level of K1f15 protein in the OXPHOS pathway, exogenous Klf15 was found to rescue defects and restore balance to the cardiac metabolome [149]. The impairment of OXPHOS leads to a decline in the function of complexes I, II, and III in individuals with coronary artery disease and those with failing hearts [177,183]. The overexpression of PFK (phosphofructokinase) or the administration of PFKM has been demonstrated to inhibit doxorubicin-induced cardiotoxicity by enhancing glycolysis and the OXPHOS system, and thus PFKM may be considered for developing new treatment for heart failure [184].
There is also experimental evidence to suggest sodium-glucose cotransporter-2 (SGLT2) inhibitors restore the balance between glycolysis and OXPHOS [180], providing significant cardiac protection to patients suffering from heart failure [185]. On the other hand, metformin, which is known to promote glucose uptake and exert beneficial actions in various non-diabetic malignant diseases [186,187], did not show conclusive beneficial effects in non-diabetic patients with coronary heart disease [188]. Nonetheless, it is noteworthy that the mitochondrial antioxidant system can be selectively activated to prevent or treat mitochondrial dysfunction and, in this context, coenzyme Q10, a natural antioxidant, is known to activate the mitochondrial antioxidant system. It is pointed out that a CoQ10 deficiency has been linked to electron transport chain dysfunction and oral supplementation has been reported to reverse this trend [189,190,191,192] as CoQ10 supplementation was observed to improve cardiac function, reduce cardiovascular mortality, and enhance survival rates in heart failure [193]. MitoQ, a compound that mimics Coenzyme Q10, has also shown to be highly effective in protecting against oxidative damage. It is capable of preventing lipid peroxidation and the mitochondrial damage caused by superoxide radicals [194,195]. The therapeutic benefits of MitoQ have been demonstrated in various animals and humans with diabetes, hypertension, and inflammation, in addition to offering protection against oxidative stress and improving the integrity of the cardiac mitochondrial network in heart failure [196,197,198]. Thus, it would be worthwhile to undertake a large double-blind clinical trial to establish the beneficial effects of MitoQ in heart failure. It may also be noted that the inhibition of Drp1 (dynamin-related protein 1) maintains mitochondrial integrity and improves OXPHOS, playing a cardioprotective role during cardiac stress circumstances such as ischemia-reperfusion injury and cardiac arrest in cells by hindering excessive fission at the onset of reperfusion in animal models [199,200,201,202]. Thus, the development of appropriate inhibitors of Drp1 may prove valuable for preserving mitochondrial function in heart failure.
6. Conclusions and Perspectives
By virtue of their ability to generate energy as ATP, mitochondria play an important role in maintaining the cardiac structure and function. These organelles produce ATP upon the oxidation of different substrates as well as the OXPHOS system, involving a specialized electron transport chain organized in the form of complexes I to V. While some ATP is transformed into creatine phosphate for the storage of energy in the myocardium, most of ATP is utilized for the contraction–relaxation cycle and maintaining cation homeostasis by myofibrillar ATPase, as well as sarcoplasmic reticulum and sarcolemmal ATPases in cardiomyocytes, respectively. Thus any abnormality in the process of substrate oxidation or any component of the mitochondrial OXPHOS system can be seen to decrease high-energy phosphate stores in the myocardium, resulting in cardiac dysfunction and progression to heart failure. Such a view is not intended to de-emphasize the contribution of other subcellular and molecular defects in the pathogenesis for the development of heart failure.
It is noteworthy that mitochondria have a remarkable ability to accumulate Ca2+ and serve as a Ca2+-sink to maintain cellular integrity. However, several studies have shown that mitochondrial Ca2+-overload is one of the major causative factors for inducing defects in the OXPHOS system under a wide variety of pathological conditions. In fact, defects in the sarcoplasmic and sarcolemmal Ca2+-transport systems in the failing hearts have been shown to elicit mitochondrial Ca2+-overload. Furthermore, it should be noted that mitochondrial dysfunction is also associated with impaired electron transport for the production of ROS due to changes in any one or all of the complexes in the mitochondrial electron transport chain for the induction of abnormalities in OXPHOS. In addition, the activation of enzymes such as NADPH and monoamine oxidase by vasoactive hormones, which become accumulated in the mitochondria during the development of heart failure, has also been reported to generate ROS. Both mitochondrial Ca2+ and ROS not only depress the antioxidant reserve within mitochondria and open mitochondrial pores for the leakage of cytotoxic substances such as cytochrome C, but also provide signals for the development of apoptosis, cellular damage, and subsequent heart failure.
In view of the role of mitochondrial Ca2+-overload and mitochondrial ROS generation for depressing the OXPHOS system, it is evident that mitochondrial dysfunction plays a critical role in the depletion of myocardial high-energy phosphate stores during the development of heart failure. Accordingly, several pharmacological and metabolic interventions, which promote the mitochondrial OXPHOS function, have been reported to produce beneficial effects in heart failure. Likewise, different antioxidants, which prevent the generation and effectiveness of mitochondrial ROS, have been shown to delay the progression of heart failure. Thus, there is real challenge to develop specific and safe metabolic and antioxidants interventions (either alone or in combination) targeting the mitochondrial OXPHOS system for the improved therapy of heart failure.
Author Contributions
S.K.B. searched the literature and wrote the first draft; N.S.D. conceived the concept and participated in preparing and revising the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This project did not receive any grant funding. The infrastructure support for this project was provided by the St. Boniface Hospital Albrechtsen Research Center, Winnipeg. Thanks are due to Khushman Kaur for help in preparing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Davis, R.E.; Williams, M. Mitochondrial Function and Dysfunction: An Update. J. Pharmacol. Exp Ther. 2012, 342, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Van Der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871, Erratum in Genetics 2018, 208, 1673. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial function, biology, and role in disease: A scientific statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial Control of Cellular Life, Stress, and Death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R. Mitochondrial heart function. Ann. Rev. Physiol. 1979, 41, 485–506. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Lopez-Crisosto, C.; Pennanen, C.; Vasquez-Trincado, C.; Morales, P.E.; Bravo-Sagua, R.; Quest, A.F.G.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum–mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 2017, 14, 342–360. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Budde, H.; Hassoun, R.; Tangos, M.; Zhazykbayeva, S.; Herwig, M.; Varatnitskaya, M.; Sieme, M.; Delalat, S.; Sultana, I.; Kolijn, D.; et al. The Interplay between S-Glutathionylation and Phosphorylation of Cardiac Troponin I and Myosin Binding Protein C in End-Stage Human Failing Hearts. Antioxidants 2021, 10, 1134. [Google Scholar] [CrossRef]
- Leichert, L.I.; Gehrke, F.; Gudiseva, H.V.; Blackwell, T.; Ilbert, M.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Jakob, U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 8197–8202. [Google Scholar] [CrossRef]
- Delbridge, L.M.D.; Mellor, K.M.; Taylor, D.J.; Gottlieb, R.A. Myocardial stress and autophagy: Mechanisms and potential therapies. Nat. Rev. Cardiol. 2017, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated Analysis of Protein Composition, Tissue Diversity, and Gene Regulation in Mouse Mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef]
- Glatz, J.F.; Nabben, M.; Young, M.E.; Schulze, P.C.; Taegtmeyer, H.; Luiken, J.J. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165579. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.W.; Fahy, E.; Zhang, B.; Glenn, G.M.; Warnock, D.E.; Wiley, S.; Murphy, A.N.; Gaucher, S.P.; Capaldi, R.A.; Gibson, B.W.; et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 2003, 21, 281–286. [Google Scholar] [CrossRef]
- Zhao, Q.; Sun, Q.; Zhou, L.; Liu, K.; Jiao, K. Complex Regulation of Mitochondrial Function During Cardiac Development. J. Am. Heart Assoc. 2019, 8, e012731. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Rutter, J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013, 27, 2615–2627. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.S.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef]
- Stein, L.R.; Imai, S.-I. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Birsoy, K.; Mihaylova, M.M.; Snitkin, H.; Stasinski, I.; Yucel, B.; Bayraktar, E.C.; Carette, J.E.; Clish, C.B.; Brummelkamp, T.R.; et al. Inhibition of ATPIF1 Ameliorates Severe Mitochondrial Respiratory Chain Dysfunction in Mammalian Cells. Cell Rep. 2014, 7, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wrogemann, K.; Nylen, E.G. Mitochondrial calcium overloading in cardiomyopathic hamsters. J. Mol. Cell. Cardiol. 1978, 10, 185–195. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Lee, S.-L.; Shah, K.R.; Elimban, V.; Suzuki, S.; Jasmin, G. Behaviour of subcellular organelles during the development of congestive heart failure in cardiomyopathic hamsters (UM-X7. 1). In Cardiomyopathic Heart; New York Raven Press Ltd.: New York, NY, USA, 1994; pp. 1–14. [Google Scholar]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases—From pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Zhang, Y.; Marcillat, O.; Giulivi, C.; Ernster, L.; Davies, K.J. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J. Biol. Chem. 1990, 265, 16330–16336. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W. Mitochondrial dysfunction in cardiovascular disease. Free Radic. Biol. Med. 2005, 38, 1278–1295. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Q.; Feng, X.; Liu, Y.; Zhou, Y. Mitochondrial Dysfunction in Cardiovascular Diseases: Potential Targets for Treatment. Front. Cell Dev. Biol. 2022, 10, 841523. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Calbet, J.A.L.; Martín-Rodríguez, S.; Martin-Rincon, M.; Morales-Alamo, D. An integrative approach to the regulation of mitochondrial respiration during exercise: Focus on high-intensity exercise. Redox Biol. 2020, 35, 101478. [Google Scholar] [CrossRef]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2013, 18, 607–622. [Google Scholar] [CrossRef]
- Schwarzer, M.; Rohrbach, S.; Niemann, B. Heart and Mitochondria: Pathophysiology and Implications for Cardiac Surgeons. Thorac. Cardiovasc. Surg. 2018, 66, 011–019. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Borghetti, G.; von Lewinski, D.; Eaton, D.M.; Sourij, H.; Houser, S.R.; Wallner, M. Diabetic Cardiomyopathy: Current and Future Therapies. Beyond Glycemic Control. Front. Physiol. 2018, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of different ectopic fat depots on cardiovascular and metabolic diseases. J. Cell. Physiol. 2019, 234, 21630–21641. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, P.; Bergamaschi, L.; Saturi, G.; D’Angelo, E.C.; Magnani, I.; Toniolo, S.; Stefanizzi, A.; Rinaldi, A.; Bartoli, L.; Angeli, F.; et al. Secondary Prevention Medical Therapy and Outcomes in Patients with Myocardial Infarction with Non-Obstructive Coronary Artery Disease. Front. Pharmacol. 2020, 10, 1606. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.; Dhalla, N.S. Heart mitochondrial function in chronic experimental diabetes in rats. Can. J. Cardiol. 1985, 1, 48–54. [Google Scholar] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225, Erratum in Nature 2011, 475, 122. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J.; Goldenthal, M.J. Mitochondrial centrality in heart failure. Heart Fail. Rev. 2008, 13, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart-an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; Enríquez, J.A.; Pérez-Martos, A.; Montoya, J.; Fernández-Silva, P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 2011, 11, 207–213. [Google Scholar] [CrossRef]
- Rosca, M.G.; Hoppel, C.L. New aspects of impaired mitochondrial function in heart failure. J. Bioenerg. Biomembr. 2009, 41, 107–112. [Google Scholar] [CrossRef]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res. 2010, 88, 40–50. [Google Scholar] [CrossRef]
- Murray, A.J.; Cole, M.A.; Lygate, C.A.; Carr, C.A.; Stuckey, D.J.; Little, S.E.; Neubauer, S.; Clarke, K. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J. Mol. Cell. Cardiol. 2008, 44, 694–700. [Google Scholar] [CrossRef]
- Stoldt, S.; Wenzel, D.; Kehrein, K.; Riedel, D.; Ott, M.; Jakobs, S. Spatial orchestration of mitochondrial translation and OXPHOS complex assembly. Nat. Cell Biol. 2018, 20, 528–534. [Google Scholar] [CrossRef]
- Benard, G.; Faustin, B.; Passerieux, E.; Galinier, A.; Rocher, C.; Bellance, N.; Delage, J.-P.; Casteilla, L.; Letellier, T.; Rossignol, R. Physiological diversity of mitochondrial oxidative phosphorylation. Am. J. Physiol. Physiol. 2006, 291, C1172–C1182. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal structure of the entire respiratory complex I. Nature 2013, 494, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Uno, S.; Kimura, H.; Murai, M.; Miyoshi, H. Exploring the quinone/inhibitor-binding pocket in mitochondrial respiratory complex I by chemical biology approaches. J. Biol. Chem. 2019, 294, 679–696. [Google Scholar] [CrossRef]
- Tocilescu, M.A.; Fendel, U.; Zwicker, K.; Kerscher, S.; Brandt, U.; Moloney, D.J.; Shair, L.H.; Lu, F.M.; Xia, J.; Locke, R.; et al. Exploring the Ubiquinone Binding Cavity of Respiratory Complex I. J. Biol. Chem. 2007, 282, 29514–29520. [Google Scholar] [CrossRef]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhai, Y.; Lou, J.; Liu, M.; Pang, X.; Sun, F. Thiabendazole inhibits ubiquinone reduction activity of mitochondrial respiratory complex II via a water molecule mediated binding feature. Protein Cell 2011, 2, 531–542. [Google Scholar] [CrossRef]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of mammalian respiratory supercomplex I1III2IV1. Cell 2016, 167, 1598–1609. [Google Scholar] [CrossRef]
- Hong, S.; Pedersen, P.L. ATP Synthase and the Actions of Inhibitors Utilized To Study Its Roles in Human Health, Disease, and Other Scientific Areas. Microbiol. Mol. Biol. Rev. 2008, 72, 590–641. [Google Scholar] [CrossRef]
- Ong, S.-B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial Dynamics in Cardiovascular Health and Disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C., Jr.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 1, e84897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ji, R.; Liao, X.; Castillero, E.; Kennel, P.J.; Brunjes, D.L.; Franz, M.; Möbius-Winkler, S.; Drosatos, K.; George, I.; et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 2018, 137, 2052–2067. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Logan, A.; Pell, V.R.; Shaffer, K.J.; Evans, C.; Stanley, N.J.; Robb, E.L.; Prime, T.A.; Chouchani, E.T.; Cochemé, H.M.; Fearnley, I.M.; et al. Assessing the Mitochondrial Membrane Potential in Cells and In Vivo using Targeted Click Chemistry and Mass Spectrometry. Cell Metab. 2016, 23, 379–385. [Google Scholar] [CrossRef]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Chapa-Dubocq, X.R.; Khuchua, Z.; Camara, A.K. Mitochondrial respiratory supercomplexes in mammalian cells: Structural versus functional role. J. Mol. Med. 2021, 99, 57–73. [Google Scholar] [CrossRef]
- Gadicherla, A.K.; Stowe, D.F.; Antholine, W.E.; Yang, M.; Camara, A.K. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 419–429. [Google Scholar] [CrossRef]
- Törnroth-Horsefield, S.; Neutze, R. Opening and closing the metabolite gate. Proc. Natl. Acad. Sci. USA 2008, 105, 19565–19566. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-R.; Zweier, J.L. Cardiac Mitochondria and Reactive Oxygen Species Generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Muraguchi, T.; Hoshii, T.; Hirao, A. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid. Redox Signal. 2008, 10, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Schwall, C.T.; Greenwood, V.L.; Alder, N.N. The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim. Biophys Acta (BBA)-Bioenerg. 2012, 1817, 1588–1596. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 2013, 179, 42–48. [Google Scholar] [CrossRef]
- Bazán, S.; Mileykovskaya, E.; Mallampalli, V.K.P.S.; Heacock, P.; Sparagna, G.C.; Dowhan, W. Cardiolipin-dependent re-constitution of respiratory supercomplexes from purified Saccharomyces cerevisiae complexes III and IV. J. Biol. Chem. 2013, 288, 401–411. [Google Scholar] [CrossRef]
- Milenkovic, D.; Misic, J.; Hevler, J.F.; Molinié, T.; Chung, I.; Atanassov, I.; Li, X.; Filograna, R.; Mesaros, A.; Mourier, A.; et al. Preserved respiratory chain capacity and physiology in mice with profoundly reduced levels of mitochondrial respirasomes. Cell Metab. 2023, 35, 1799–1813.e7. [Google Scholar] [CrossRef]
- Sharov, V.G.; Goussev, A.; Lesch, M.; Goldstein, S.; Sabbah, H.N. Abnormal Mitochondrial Function in Myocardium of Dogs with Chronic Heart Failure. J. Mol. Cell. Cardiol. 1998, 30, 1757–1762. [Google Scholar] [CrossRef]
- Sharov, V.G.; Todor, A.V.; Silverman, N.; Goldstein, S.; Sabbah, H.N. Abnormal Mitochondrial Respiration in Failed Human Myocardium. J. Mol. Cell. Cardiol. 2000, 32, 2361–2367. [Google Scholar] [CrossRef]
- Song, M.; Chen, Y.; Gong, G.; Murphy, E.; Rabinovitch, P.S.; Dorn, G.W. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ. Res. 2014, 115, 348–353. [Google Scholar] [CrossRef]
- Ghadially, F. Intramitochondrial lipidic inclusions. In Ultrastructural Pathology of the Cell and Matrix; Butter-worth-Heinemann: Boston, MA, USA, 1997; pp. 310–313. [Google Scholar]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W. Mitochondrial Fusion is Essential for Organelle Function and Cardiac Homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Narendra, D.P.; Youle, R.J.; de Oliveira, M.R.; Manayi, A.; Daglia, M.; Hajheydari, Z.; Nabavi, S.M.; Pernas, L.; Scorrano, L.; Akbar, M.; et al. Targeting Mitochondrial Dysfunction: Role for PINK1 and Parkin in Mitochondrial Quality Control. Antioxid. Redox Signal. 2011, 14, 1929–1938. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef]
- Koene, S.; Smeitink, J. Mitochondrial medicine: Entering the era of treatment. J. Intern. Med. 2009, 265, 193–209. [Google Scholar] [CrossRef]
- Sharov, V.G.; Todor, A.; Khanal, S.; Imai, M.; Sabbah, H.N. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J. Mol. Cell. Cardiol. 2007, 42, 150–158. [Google Scholar]
- Osterholt, M.; Nguyen, T.D.; Schwarzer, M.; Doenst, T. Alterations in mitochondrial function in cardiac hypertrophy and heart failure. Heart Fail. Rev. 2013, 18, 645–656. [Google Scholar] [CrossRef]
- Lygate, C.A.; Neubauer, S.; Van Dobbenburgh, J.O.; Lahpor, J.R.; Woolley, S.R.; de Jonge, N.; Klöpping, C.; Van Echteld, C.J.; Ye, Y.; Gong, G.; et al. Metabolic Flux as a Predictor of Heart Failure Prognosis. Circ. Res. 2014, 114, 1228–1230. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial Oxidative Stress: Implications for Cell Death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Altamirano, F.; Szweda, P.A.; Elnwasany, A.; Lee, D.I.; Yoo, H.; Kass, D.A.; Szweda, L.I.; et al. NAD + Repletion Reverses Heart Failure with Preserved Ejection Fraction. Circ. Res. 2021, 128, 1629–1641. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar]
- Hüttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1701–1720. [Google Scholar] [CrossRef]
- Li, Q.; Su, D.; O’Rourke, B.; Pogwizd, S.M.; Zhou, L. Mitochondria-derived ROS bursts disturb Ca2+ cycling and induce abnormal automaticity in guinea pig cardiomyocytes: A theoretical study. Am. J. Physiol. Circ. Physiol. 2015, 308, H623–H636. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Makazan, Z.; Saini, H.K.; Dhalla, N.S. Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1986–H1994. [Google Scholar] [CrossRef] [PubMed]
- Makazan, Z.; Saini-Chohan, H.K.; Dhalla, N.S. Mitochondrial oxidative phosphorylation in hearts subjected to Ca2+ depletion and Ca2+ repletion. Can. J. Physiol. Pharmacol. 2009, 87, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Aon, M.A.; Liu, T.; O’Rourke, B. Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J. Mol. Cell. Cardiol. 2011, 51, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Okonko, D.O.; Shah, A.M. Mitochondrial dysfunction and oxidative stress in CHF. Nat. Rev. Cardiol. 2015, 12, 6–8. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.-I.; Egashira, K.; Takeshita, A. Mitochondrial Electron Transport Complex I Is a Potential Source of Oxygen Free Radicals in the Failing Myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef]
- Jang, S.; Lewis, T.S.; Powers, C.; Khuchua, Z.; Baines, C.P.; Wipf, P.; Javadov, S.; Strader, M.B.; Alayash, A.I.; Rohlenova, K.; et al. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia–Reperfusion. Antioxid. Redox Signal. 2017, 27, 57–69. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; Alburquerque-Béjar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodríguez-Sinovas, A.; García-Dorado, D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef]
- Kadenbach, B.; Ramzan, R.; Moosdorf, R.; Vogt, S. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion 2011, 11, 700–706. [Google Scholar] [CrossRef]
- Loor, G.; Schumacker, P.T. Role of hypoxia-inducible factor in cell survival during myocardial ischemia–reperfusion. Cell Death Differ. 2008, 15, 686–690. [Google Scholar] [CrossRef]
- Gong, G.; Liu, J.; Liang, P.; Guo, T.; Hu, Q.; Ochiai, K.; Hou, M.; Ye, Y.; Wu, X.; Mansoor, A.; et al. Oxidative capacity in failing hearts. Am. J. Physiol. Circ. Physiol. 2003, 285, H541–H548. [Google Scholar] [CrossRef]
- Lemieux, H.; Semsroth, S.; Antretter, H.; Höfer, D.; Gnaiger, E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 2011, 43, 1729–1738. [Google Scholar] [CrossRef]
- Savchenko, L.; Martinelli, I.; Marsal, D.; Zhdan, V.; Tao, J.; Kunduzova, O. Myocardial capacity of mitochondrial oxidative phosphorylation in response to prolonged electromagnetic stress. Front. Cardiovasc. Med. 2023, 10, 1205893. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic Therapy with Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs with Advanced Heart Failure. Circ. Heart Fail. 2016, 9, e002206. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Stowe, D.F.; Abdel-Kawi, S.H.; Hashem, K.S.; Abd-Allah, S.; Chen, C.; Cao, J.; Ma, X.; Wang, X.; Yan, S.; et al. Potential Therapeutic Benefits of Strategies Directed to Mitochondria. Antioxid. Redox Signal. 2010, 13, 279–347. [Google Scholar] [CrossRef]
- Takada, S.; Maekawa, S.; Furihata, T.; Kakutani, N.; Setoyama, D.; Ueda, K.; Nambu, H.; Hagiwara, H.; Handa, H.; Fumoto, Y.; et al. Succinyl-CoA-based energy metabolism dysfunction in chronic heart failure. Proc. Natl. Acad. Sci. USA 2022, 119, e2203628119. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.R.; Michel, C.R.; Lin, Y.H.; McKinsey, T.A.; Jeong, M.Y.; Ambardekar, A.V.; Cleveland, J.C.; Reisdorph, R.; Reisdorph, N.; Woulfe, K.C.; et al. Defining decreased protein succinylation of failing human cardiac myofibrils in ischemic cardiomyopathy. J. Mol. Cell. Cardiol. 2020, 138, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Wu, Q.; Ni, C.; Zhang, P.; Zhong, Z.; Wu, Y.; Wang, Y.; Xu, Y.; Kong, M.; Cheng, H.; et al. Lack of Remuscularization Following Transplantation of Human Embryonic Stem Cell-Derived Cardiovascular Progenitor Cells in Infarcted Nonhuman Primates. Circ. Res. 2018, 122, 958–969. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, F.; Yan, P.; Zhang, S.; Lou, Y.; Geng, Z.; Li, Z.; Zhang, Y.; Xu, Y.; Lu, Y.; et al. LARP7 Protects Against Heart Failure by Enhancing Mitochondrial Biogenesis. Circulation 2021, 143, 2007–2022. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of Mitochondrial Dynamics in the Failing Heart: Normalization Following Long-Term Therapy with Elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328. [Google Scholar] [CrossRef]
- Zhuang, L.; Jia, K.; Chen, C.; Li, Z.; Zhao, J.; Hu, J.; Zhang, H.; Fan, Q.; Huang, C.; Xie, H.; et al. DYRK1B-STAT3 drives cardiac hypertrophy and heart failure by impairing mito-chondrial bioenergetics. Circulation 2022, 145, 829–846. [Google Scholar] [CrossRef]
- Hahn, V.S.; Knutsdottir, H.; Luo, X.; Bedi, K.; Margulies, K.B.; Haldar, S.M.; Stolina, M.; Yin, J.; Khakoo, A.Y.; Vaishnav, J.; et al. Myocardial Gene Expression Signatures in Human Heart Failure with Preserved Ejection Fraction. Circulation 2021, 143, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Silva, D.; Wüst, R.C.I.; Conceição, G.; Gonçalves-Rodrigues, P.; Gonçalves, N.; Gonçalves, A.; Kuster, D.W.D.; Leite-Moreira, A.F.; van der Velden, J.; Beleza, J.M.d.S.; et al. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol. 2020, 228, e13378. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Hartmann, M.; Rehling, P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 810–821. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Dunselman, P.; Wedel, H.; Cleland, J.G.; Lindberg, M.; Hjalmarson, Å.; Kjekshus, J.; Waagstein, F.; Apetrei, E.; Barrios, V.; et al. Coenzyme Q10, Rosuvastatin, and Clinical Outcomes in Heart Failure: A Pre-Specified Substudy of CORONA (Controlled Rosuvastatin Multinational Study in Heart Failure). J. Am. Coll. Cardiol. 2010, 56, 1196–1204. [Google Scholar] [CrossRef]
- Maekawa, S.; Takada, S.; Nambu, H.; Furihata, T.; Kakutani, N.; Setoyama, D.; Ueyanagi, Y.; Kang, D.; Sabe, H.; Kinugawa, S. Linoleic acid improves assembly of the CII subunit and CIII2/CIV complex of the mitochondrial oxidative phosphorylation system in heart failure. Cell Commun. Signal. 2019, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- Hammer, E.; Goritzka, M.; Ameling, S.; Darm, K.; Steil, L.; Klingel, K.; Trimpert, C.; Herda, L.R.; Dörr, M.; Kroemer, H.K.; et al. Characterization of the Human Myocardial Proteome in Inflammatory Dilated Cardiomyopathy by Label-free Quantitative Shotgun Proteomics of Heart Biopsies. J. Proteome Res. 2011, 10, 2161–2171. [Google Scholar] [CrossRef]
- Jarreta, D.; Orús, J.; Barrientos, A.; Miró, O.; Roig, E.; Heras, M.; Moraes, C.T.; Cardellach, F.; Casademont, J. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc. Res. 2000, 45, 860–865. [Google Scholar] [CrossRef]
- Buchwald, A.; Till, H.; Unterberg, C.; Oberschmidt, R.; Figulla, H.R.; Wiegand, V. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur. Heart J. 1990, 11, 509–516. [Google Scholar] [CrossRef]
- Abel, E.D.; Doenst, T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 234–242. [Google Scholar] [CrossRef]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol. 2017, 7, 170007. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD+/NADH redox state and diabetic cardiomyopathy. Antioxid. Redox Signal. 2019, 30, 375–398. [Google Scholar] [CrossRef] [PubMed]
- Mongirdienė, A.; Liuizė, A.; Karčiauskaitė, D.; Mazgelytė, E.; Liekis, A.; Sadauskienė, I. Relationship between Oxidative Stress and Left Ventricle Markers in Patients with Chronic Heart Failure. Cells 2023, 12, 803. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.K.; Bhullar, S.K.; Elimban, V.; Dhalla, N.S. Oxidative Stress as A Mechanism for Functional Alterations in Cardiac Hypertrophy and Heart Failure. Antioxidants 2021, 10, 931. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Chen, D.; Watkins, S.C.; Feldman, A.M. Mitochondrial Abnormalities in Tumor Necrosis Factor-α–Induced Heart Failure Are Associated with Impaired DNA Repair Activity. Circulation 2001, 104, 2492–2497. [Google Scholar] [CrossRef]
- Ozcan, C.; Bienengraeber, M.; Hodgson, D.M.; Mann, D.L.; Terzic, A. Mitochondrial tolerance to stress impaired in failing heart. J. Mol. Cell. Cardiol. 2003, 35, 1161–1166. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Sharov, V.; Riddle, J.M.; Kono, T.; Lesch, M.; Goldstein, S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J. Mol. Cell. Cardiol. 1992, 24, 1333–1347. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.-I.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA Damage and Dysfunction Associated with Oxidative Stress in Failing Hearts After Myocardial Infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Sobel, B.E.; Spann, J.F., Jr.; Pool, P.E.; Sonnenblick, E.H.; Braunwald, E. Normal oxidative phosphorylation in mitochondria from the failing heart. Circ. Res. 1967, 21, 355–364. [Google Scholar] [CrossRef]
- Marcil, M.; Ascah, A.; Matas, J.; Belanger, S.; Deschepper, C.; Burelle, Y. Compensated volume overload increases the vulnerability of heart mitochondria without affecting their functions in the absence of stress. J. Mol. Cell. Cardiol. 2006, 41, 998–1009. [Google Scholar] [CrossRef]
- Sordahl, L.; McCollum, W.; Wood, W.; Schwartz, A.; Peterzan, M.A.; Lygate, C.A.; Neubauer, S.; Rider, O.J.; Gong, G.; Liu, J.; et al. Mitochondria and sarcoplasmic reticulum function in cardiac hypertrophy and failure. Am. J. Physiol. Content 1973, 224, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty Acid Oxidation Enzyme Gene Expression Is Downregulated in the Failing Heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; De Mazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Stowe, D.F.; Camara, A.K.S. Mitochondrial reactive oxygen species production in excitable cells: Modulators of mitochondrial and cell function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival anti-oxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Gao, F.; Liang, T.; Lu, Y.W.; Fu, X.; Dong, X.; Pu, L.; Hong, T.; Zhou, Y.; Zhang, Y.; Liu, N.; et al. A defect in mitochondrial protein translation influences mitonuclear communication in the heart. Nat. Commun. 2023, 14, 1595. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H.V. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy 2011, 7, 243–245. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Ong, S.-B.; Kalkhoran, S.B.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur. J. Pharmacol. 2015, 763, 104–114. [Google Scholar] [CrossRef]
- Müller, M.; Donhauser, E.; Maske, T.; Bischof, C.; Dumitrescu, D.; Rudolph, V.; Klinke, A. Mitochondrial Integrity Is Critical in Right Heart Failure Development. Int. J. Mol. Sci. 2023, 24, 11108. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W.; Clark, C.F.; Eschenbacher, W.H.; Kang, M.Y.; Engelhard, J.T.; Warner, S.J.; Matkovich, S.J.; Jowdy, C.C. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ. Res. 2011, 108, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 Are Essential for Postnatal Metabolic Remodeling in Heart. Circ. Res. 2012, 111, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Langford, D.; Amini, S.; Ahooyi, T.M.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Chen, Y.; Csordás, G.; Jowdy, C.; Schneider, T.G.; Csordás, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 Maintains Mitochondrial Structure and Contributes to Stress-Induced Permeability Transition in Cardiac Myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W., 2nd. Mitochondrial Fission and Fusion Factors Reciprocally Orchestrate Mitophagic Culling in Mouse Hearts and Cultured Fibroblasts. Cell Metab. 2015, 21, 273–286. [Google Scholar] [CrossRef]
- Große, L.; Wurm, C.A.; Brüser, C.; Neumann, D.; Jans, D.C.; Jakobs, S. Bax assembles into large ring-like structures re-modeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016, 35, 402–413. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Brady, C.; Zablocki, D.; Ohishi, M.; Sadoshima, J. Molecular mechanisms mediating mitochondrial dynamics and mitophagy and their functional roles in the cardiovascular system. J. Mol. Cell. Cardiol. 2015, 78, 116–122. [Google Scholar] [CrossRef]
- Panagia, V.; Lee, S.L.; Singh, A.; Pierce, G.N.; Jasmin, G.; Dhalla, N.S. Impairment of mitochondrial and sarcoplasmic reticular functions during the development of heart failure in cardiomyopathic (UM-X7. 1) hamsters. Can. J. Cardiol. 1986, 2, 236–247. [Google Scholar]
- Sanbe, A.; Tanonaka, K.; Niwano, Y.; Takeo, S. Improvement of cardiac function and myocardial energy metabolism of rats with chronic heart failure by long-term coenzyme Q10 treatment. J. Pharmacol. Exp. Ther. 1994, 269, 51–56. [Google Scholar] [PubMed]
- Pierce, G.N.; Tuana, B.S.; Moffat, M.P.; Singal, P.K.; Panagia, V.; Dhalla, N.S. Mitochondrial oxidative phosphorylation and calcium transport in cardiac hypertrophy due to pressure overload in pigs. In Functional Aspects of the Normal, Hypertrophied, and Failing Heart; Springer: Berlin/Heidelberg, Germany, 1984; pp. 316–325. [Google Scholar]
- Tomandlova, M.; Parenica, J.; Lokaj, P.; Ondrus, T.; Kala, P.; Miklikova, M.; Helanova, K.; Helan, M.; Malaska, J.; Benesova, K.; et al. Prognostic value of oxidative stress in patients with acute myocardial infarction complicated by cardiogenic shock: A prospective cohort study. Free Radic. Biol. Med. 2021, 174, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Aimo, A.; Castiglione, V.; Borrelli, C.; Saccaro, L.F.; Franzini, M.; Masi, S.; Emdin, M.; Giannoni, A. Oxidative stress and inflammation in the evolution of heart failure: From pathophysiology to therapeutic strategies. Eur. J. Prev. Cardiol. 2020, 27, 494–510. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; Smeitink, J. Metabolic manipulators: A well founded strategy to combat mitochondrial dysfunction. J. Inherit. Metab. Dis. 2011, 34, 315–325. [Google Scholar] [CrossRef]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef]
- Alehagen, U.; Johansson, P.; Björnstedt, M.; Rosén, A.; Dahlström, U. Cardiovascular mortality and N-terminal-proBNP reduced after combined selenium and coenzyme Q10 supplementation: A 5-year prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. Int. J. Cardiol. 2013, 167, 1860–1866. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef]
- Wang, W.; Karamanlidis, G.; Tian, R. Novel targets for mitochondrial medicine. Sci. Transl. Med. 2016, 8, 326rv3. [Google Scholar] [CrossRef]
- Mishra, J.; Jhun, B.S.; Hurst, S.; O-Uchi, J.; Csordás, G.; Sheu, S.-S. The mitochondrial Ca2+ uniporter: Structure, function, and pharmacology. In Pharmacology of Mitochondria; Singh, H., Sheu, S.-S., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; Volume 240, pp. 129–156. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Z.; Zhang, W.; Liu, X. Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol. Res. 2022, 175, 106038. [Google Scholar] [CrossRef] [PubMed]
- Zanini, G.; Selleri, V.; Malerba, M.; Solodka, K.; Sinigaglia, G.; Nasi, M.; Mattioli, A.V.; Pinti, M. The Role of Lonp1 on Mitochondrial Functions during Cardiovascular and Muscular Diseases. Antioxidants 2023, 12, 598. [Google Scholar] [CrossRef] [PubMed]
- Ait-Aissa, K.; Blaszak, S.C.; Beutner, G.; Tsaih, S.-W.; Morgan, G.; Santos, J.H.; Flister, M.J.; Joyce, D.L.; Camara, A.K.S.; Gutterman, D.D.; et al. Mitochondrial Oxidative Phosphorylation defect in the Heart of Subjects with Coronary Artery Disease. Sci. Rep. 2019, 9, 7623. [Google Scholar] [CrossRef]
- Li, X.; Flynn, E.R.; Carmo, J.M.D.; Wang, Z.; da Silva, A.A.; Mouton, A.J.; Omoto, A.C.M.; Hall, M.E.; Hall, J.E. Direct Cardiac Actions of Sodium-Glucose Cotransporter 2 Inhibition Improve Mitochondrial Function and Attenuate Oxidative Stress in Pressure Overload-Induced Heart Failure. Front. Cardiovasc. Med. 2022, 9, 859253. [Google Scholar] [CrossRef] [PubMed]
- Byrne, N.J.; Parajuli, N.; Levasseur, J.L.; Boisvenue, J.; Beker, D.L.; Masson, G.; Fedak, P.W.; Verma, S.; Dyck, J.R. Empagliflozin Prevents Worsening of Cardiac Function in an Experimental Model of Pressure Overload-Induced Heart Failure. JACC Basic Transl. Sci. 2017, 2, 347–354. [Google Scholar] [CrossRef]
- Li, X.; Lu, Q.; Qiu, Y.; do Carmo, J.M.; Wang, Z.; da Silva, A.A.; Mouton, A.; Omoto, A.C.; Hall, M.E.; Li, J.; et al. Direct cardiac actions of the sodium glucose co-transporter 2 inhibitor empagliflozin im-prove myocardial oxidative phosphorylation and attenuate pressure-overload heart failure. J. Am. Heart Assoc. 2021, 10, e018298. [Google Scholar] [CrossRef]
- Szeto, H.H.; Birk, A.V. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef]
- Luévano-Martínez, L.A.; Forni, M.F.; dos Santos, V.T.; Souza-Pinto, N.C.; Kowaltowski, A.J. Cardiolipin is a key determinant for mtDNA stability and segregation during mitochondrial stress. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1847, 587–598. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Zhou, M.; Sun, X.; Wang, C.; Wang, F.; Fang, C.; Hu, Z. PFKM inhibits doxorubicin-induced cardiotoxicity by enhancing oxidative phosphorylation and glycolysis. Sci. Rep. 2022, 12, 11684. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Braunwald, E. Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Abdelgadir, E.; Ali, R.; Rashid, F.; Bashier, A. Effect of Metformin on Different Non-Diabetes Related Conditions, a Special Focus on Malignant Conditions: Review of Literature. J. Clin. Med. Res. 2017, 9, 388–395. [Google Scholar] [CrossRef]
- Preiss, D.; Lloyd, S.M.; Ford, I.; McMurray, J.J.; Holman, R.R.; Welsh, P.; Fisher, M.; Packard, C.J.; Sattar, N. Metformin for non-diabetic patients with coronary heart disease (the CAMERA study): A randomised controlled trial. Lancet Diabetes Endocrinol. 2014, 2, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P.; Q-SYMBIO study investigators. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef] [PubMed]
- FL, C.; Hatefi, Y.; RL, L.; Widmer, C. Isolation of a quinone from beef heart mitochondria. Biochim. Biophys. Acta. 1957, 25, 220–221. [Google Scholar]
- Rötig, A.; Appelkvist, E.-L.; Geromel, V.; Chretien, D.; Kadhom, N.; Edery, P.; Lebideau, M.; Dallner, G.; Munnich, A.; Ernster, L.; et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000, 356, 391–395. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Larriva, A.P.A.-D.; Limia-Perez, L.; Romero-Cabrera, J.L.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q10 Supplementation for the Reduction of Oxidative Stress: Clinical Implications in the Treatment of Chronic Diseases. Int. J. Mol. Sci. 2020, 21, 7870. [Google Scholar] [CrossRef]
- Baggio, E.; Gandini, R.; Plancher, A.C.; Passeri, M.; Carmosino, G. Italian multicenter study on the safety and efficacy of coenzyme Q10 as adjunctive therapy in heart failure. Mol. Asp. Med. 1994, 15, s287–s294. [Google Scholar] [CrossRef]
- Botting, K.J.; Skeffington, K.L.; Niu, Y.; Allison, B.J.; Brain, K.L.; Itani, N.; Beck, C.; Logan, A.; Murray, A.J.; Murphy, M.P.; et al. Translatable mitochondria-targeted protection against programmed cardiovascular dysfunction. Sci. Adv. 2020, 6, eabb1929. [Google Scholar] [CrossRef]
- Goh, K.Y.; He, L.; Song, J.; Jinno, M.; Rogers, A.J.; Sethu, P.; Halade, G.V.; Rajasekaran, N.S.; Liu, X.; Prabhu, S.D.; et al. Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biol. 2019, 21, 101100. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.I.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F.; et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2018, 51, 1701024. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Song, J.; Ernst, P.; Latimer, M.N.; Ha, C.-M.; Goh, K.Y.; Ma, W.; Rajasekaran, N.-S.; Zhang, J.; Liu, X.; et al. MitoQ regulates redox-related noncoding RNAs to preserve mitochondrial network integrity in pressure-overload heart failure. Am. J. Physiol. Circ. Physiol. 2020, 318, H682–H695. [Google Scholar] [CrossRef] [PubMed]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2013, 28, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Sharp, W.W.; Beiser, D.G.; Fang, Y.H.; Han, M.; Piao, L.; Varughese, J.; Archer, S.L. Inhibition of the mitochondrial fission protein Drp1 improves survival in a murine cardiac arrest model. Crit. Care Med. 2015, 43, e38. [Google Scholar] [CrossRef]
- Tian, L.; Neuber-Hess, M.; Mewburn, J.; Dasgupta, A.; Dunham-Snary, K.; Wu, D.; Chen, K.-H.; Hong, Z.; Sharp, W.W.; Kutty, S.; et al. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J. Mol. Med. 2017, 95, 381–393. [Google Scholar] [CrossRef]
- Disatnik, M.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute Inhibition of Excessive Mitochondrial Fission After Myocardial Infarction Prevents Long-term Cardiac Dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef]
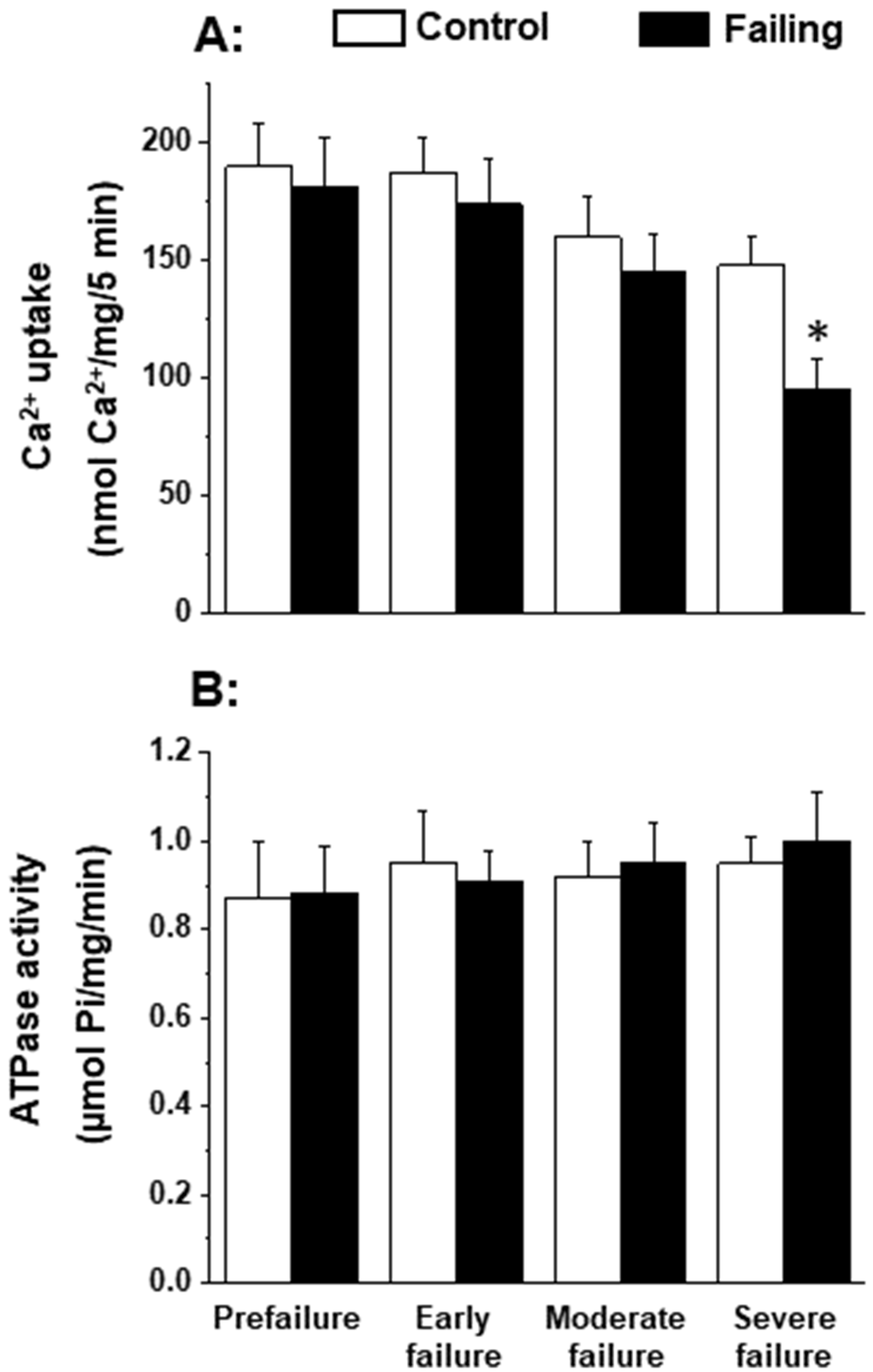
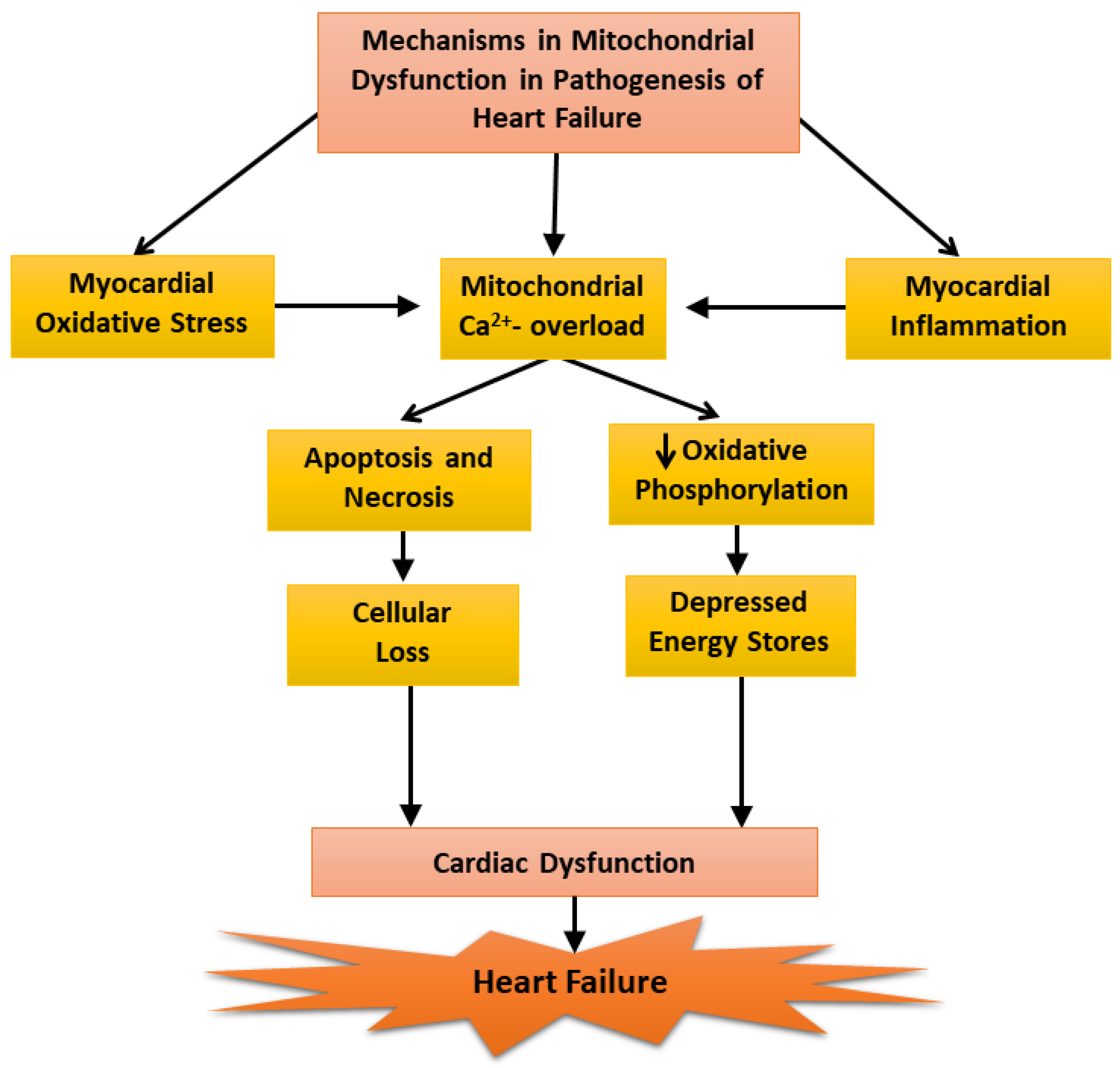
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).