
Ocular Hypertension Results in Hypoxia within Glia and Neurons throughout the Visual Projection


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Genotyping
2.3. Tamoxifen Injection
2.4. Intraocular Pressure (IOP) Measurements
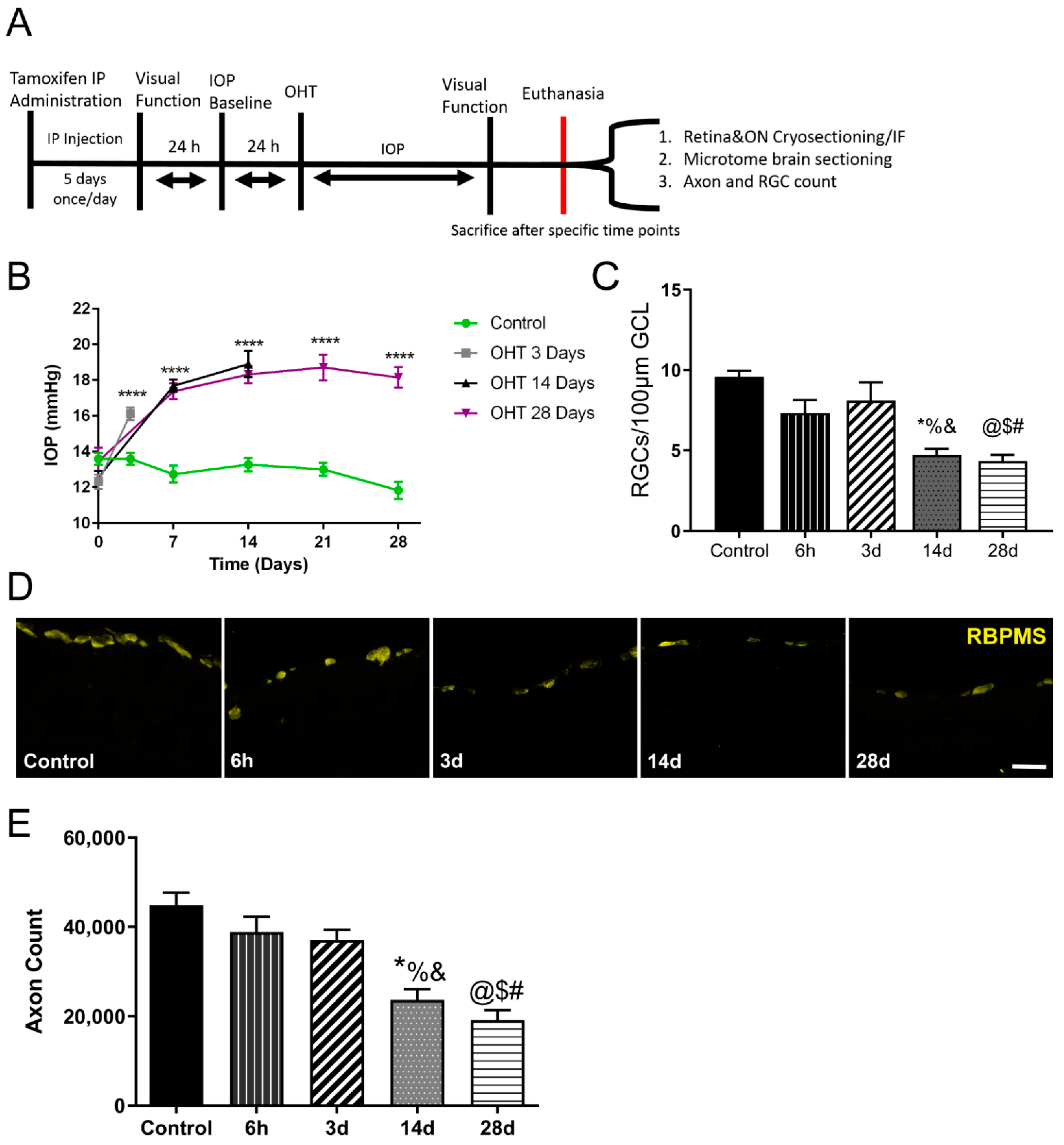
2.5. Ocular Hypertension (OHT) Model
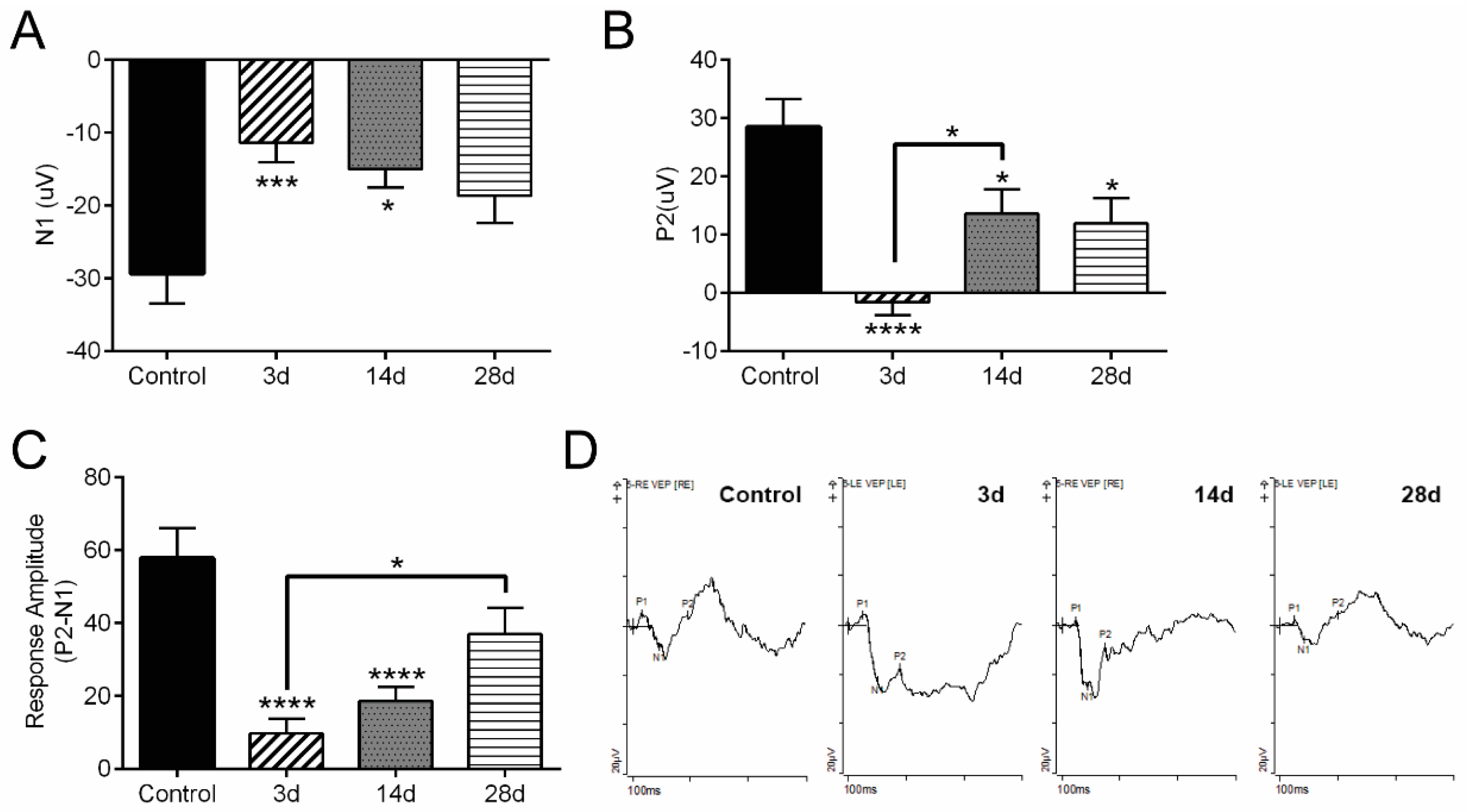
2.6. Determination of Visual Function
2.7. Hypoxia Detection by Pimonidazole
2.8. Perfusion and Tissue Preparation
2.9. Cryosectioning
2.10. Microtome Sectioning
2.11. Immunofluorescence (IF)
2.12. Histopathology for Light Microscopy
2.13. Quantification of Axons
2.14. Protein Extraction for Capillary-Based Electrophoresis (WES)
2.15. Statistical Analysis
3. Results
3.1. Visually Evoked Potential Significantly Decreased after OHT
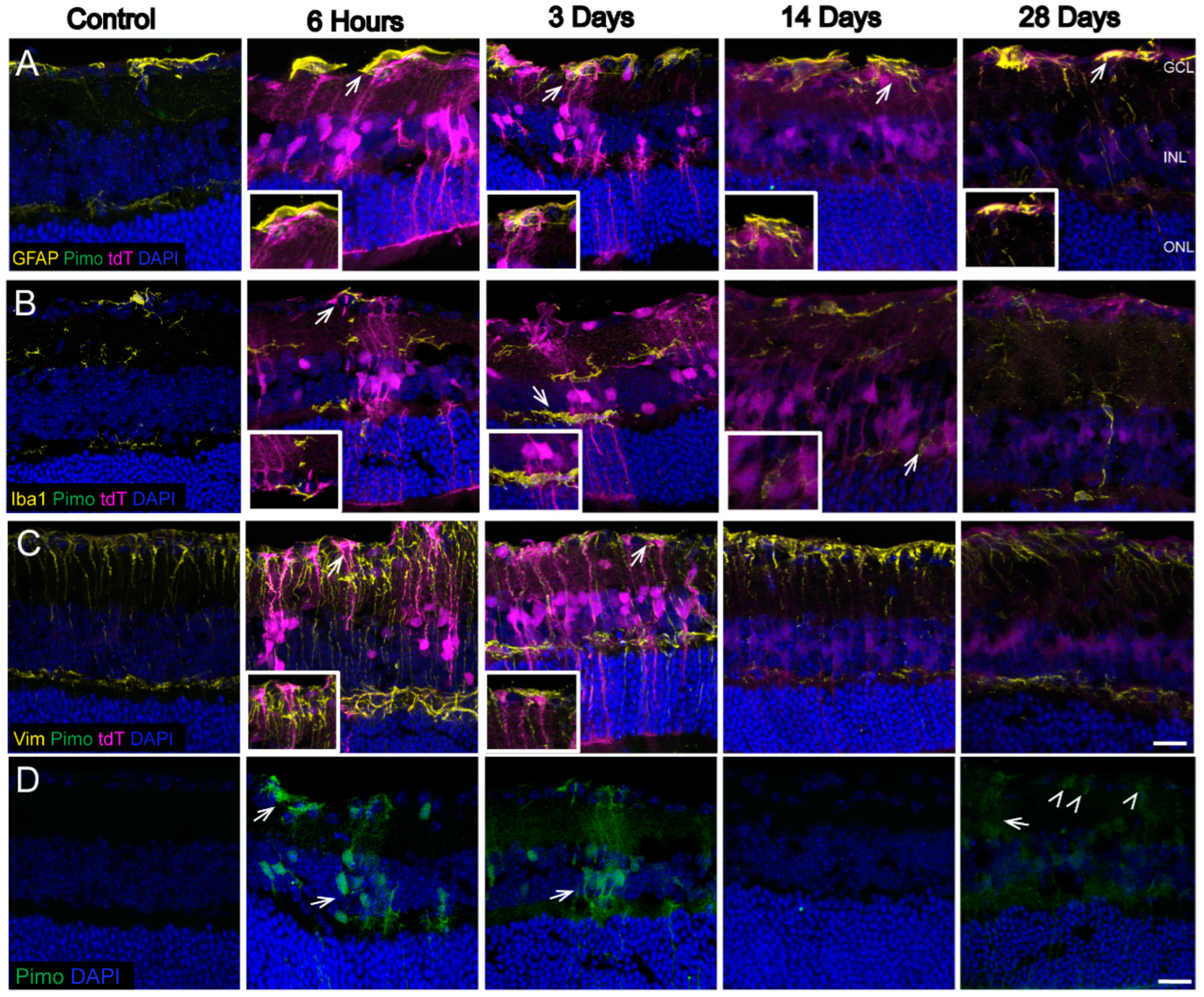
3.2. Hypoxic Glia and Neurons in Retina after OHT
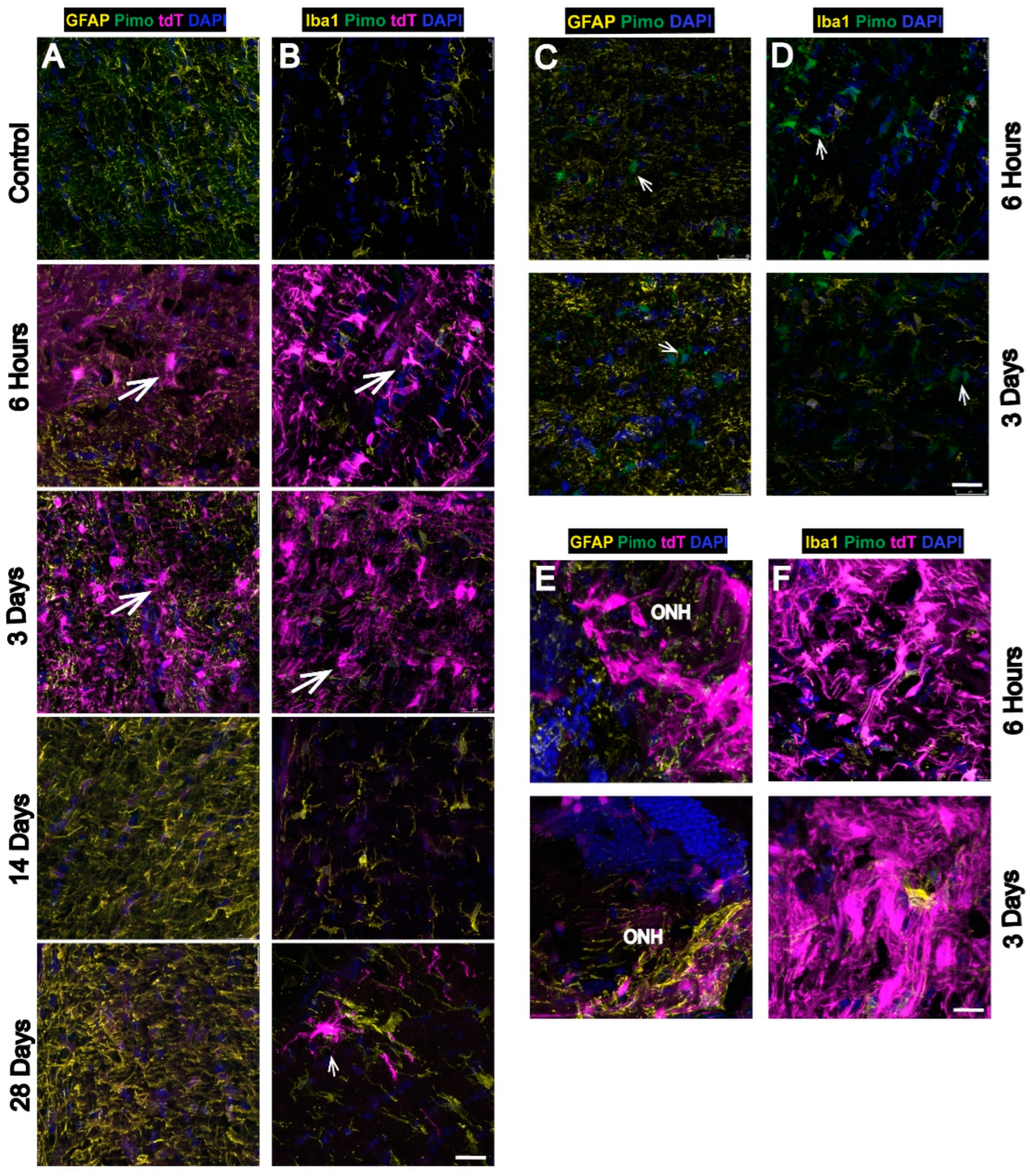
3.3. Strong Hypoxia in Optic Nerve (ON) Glia after OHT
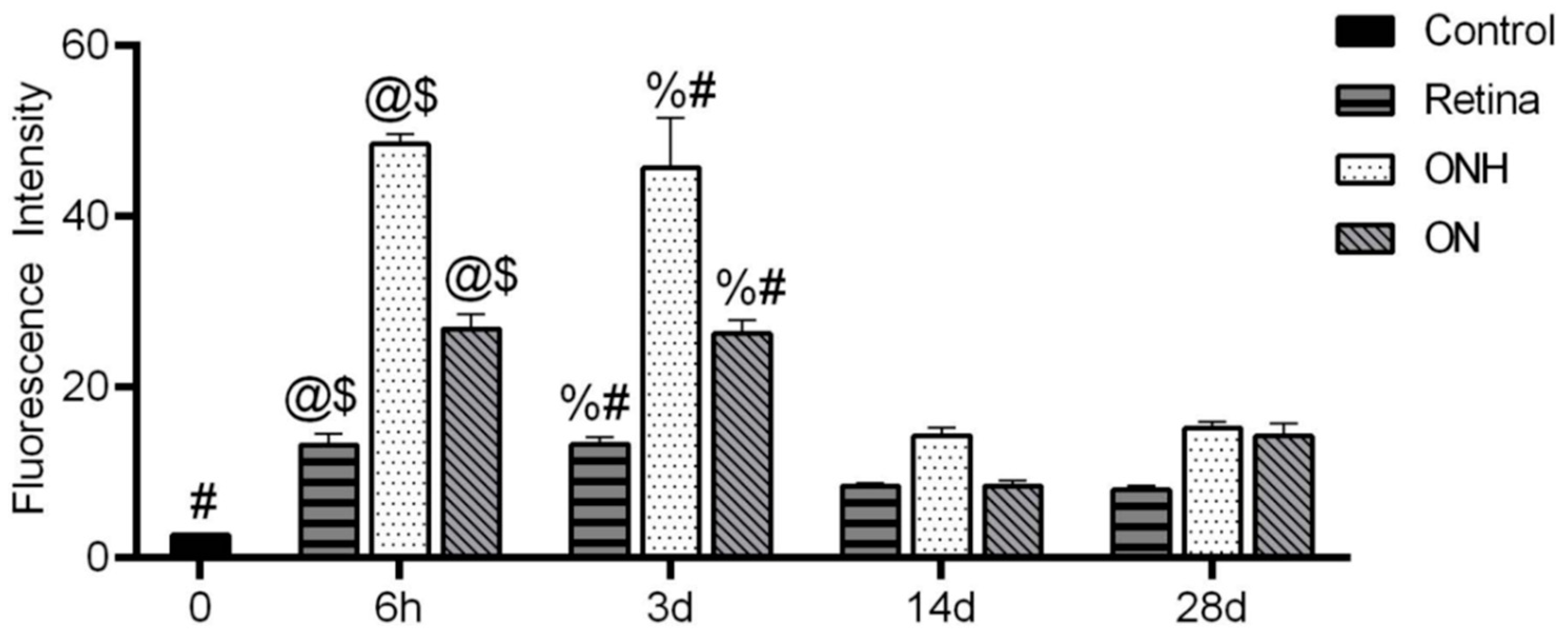
3.4. Hypoxia Is Highest in the Optic Nerve Head (ONH) after OHT
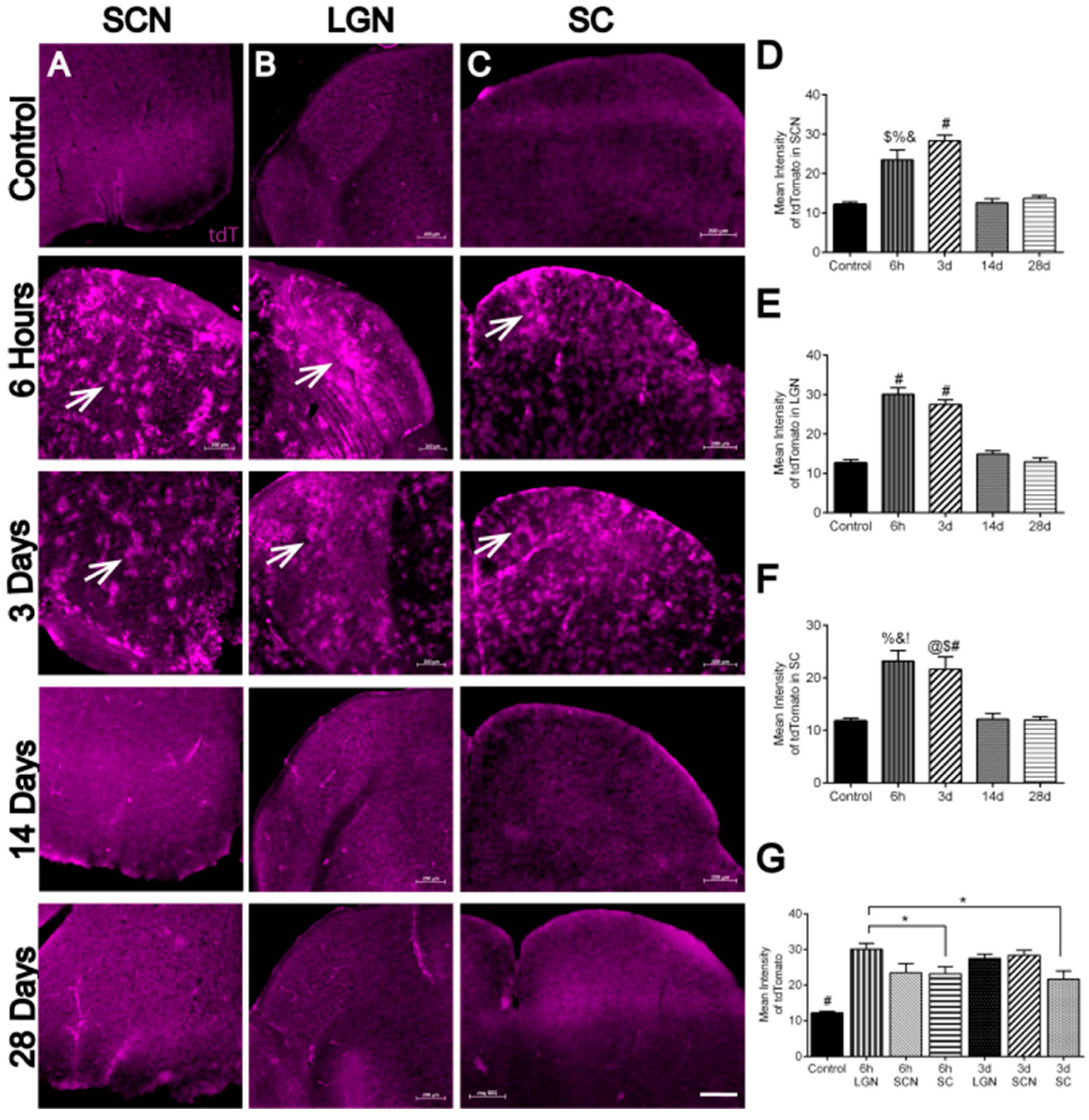
3.5. Hypoxia in Brain Visual Nuclei after OHT
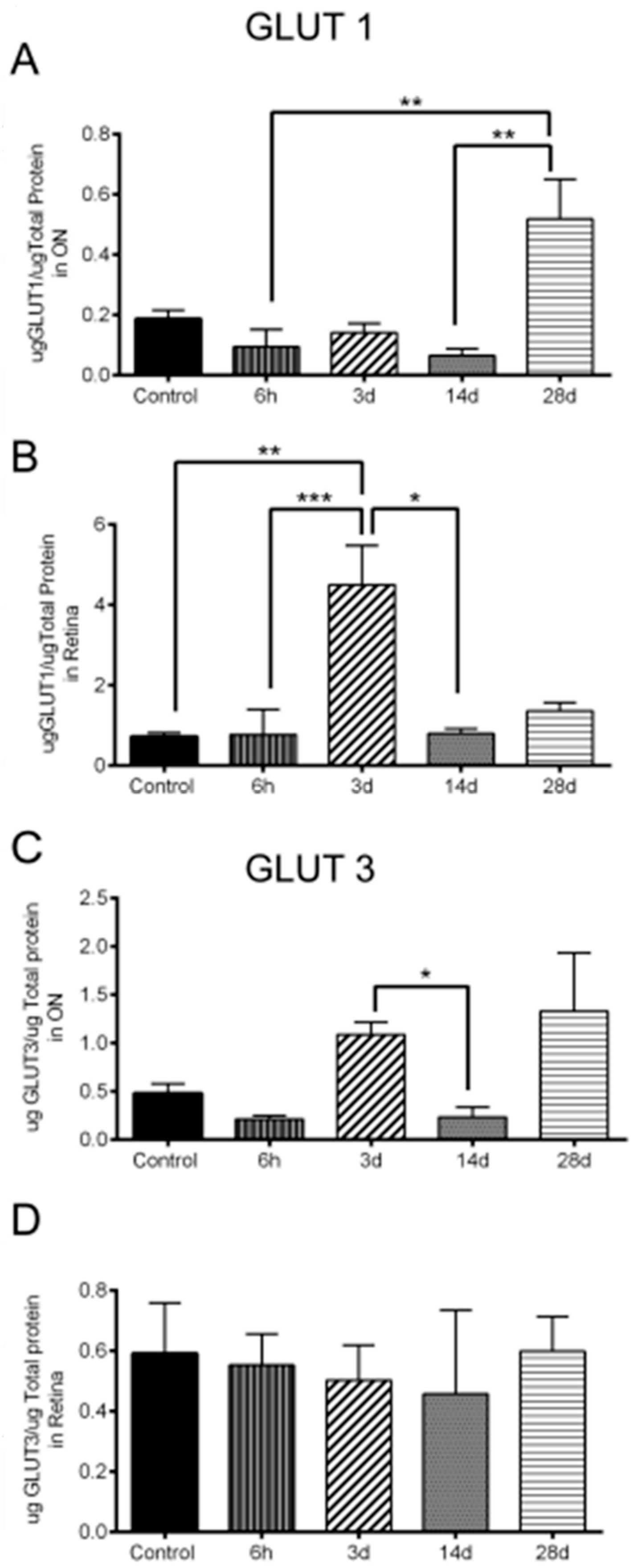
3.6. Significant Changes in GLUT1 and GLUT3 Levels after OHT
4. Discussion
4.1. Hypoxic Glia and Neurons in Glaucomatous Retina
4.2. Hypoxic Astrocytes and Microglia in Glaucomatous ON with the Highest Hypoxia in ONH
4.3. Hypoxia in Visual Centers of the Glaucomatous Brain
4.4. The CAG-ODD Reporter System
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Sivakumar, V.; Foulds, W. Early Response of Neurons and Glial Cells to Hypoxia in the Retina. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations into Hypoxia and Oxidative Stress at the Optic Nerve Head in a Rat Model of Glaucoma. Front. Neurosci. 2017, 11, 478. [Google Scholar] [CrossRef] [PubMed]
- Koshiji, M.; To, K.K.; Hammer, S.; Kumamoto, K.; Harris, A.L.; Modrich, P.; Huang, L.E. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol. Cell 2005, 17, 793–803. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Jassim, A.H.; Inman, D.M. Evidence of Hypoxic Glial Cells in a Model of Ocular Hypertension. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1–15. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Lin, M.; Chen, Y.; Jin, J.; Hu, Y.; Zhou, K.K.; Zhu, M.; Le, Y.Z.; Ge, J.; Johnson, R.S.; Ma, J.X. Ischaemia-induced retinal neovascularisation and diabetic retinopathy in mice with conditional knockout of hypoxia-inducible factor-1 in retinal Muller cells. Diabetologia 2011, 54, 1554–1566. [Google Scholar] [CrossRef]
- Mowat, F.M.; Luhmann, U.F.; Smith, A.J.; Lange, C.; Duran, Y.; Harten, S.; Shukla, D.; Maxwell, P.H.; Ali, R.R.; Bainbridge, J.W. HIF-1alpha and HIF-2alpha are differentially activated in distinct cell populations in retinal ischaemia. PLoS ONE 2010, 5, e11103. [Google Scholar] [CrossRef]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [PubMed]
- Bricker-Anthony, C.; D’Surney, L.; Lunn, B.; Hines-Beard, J.; Jo, M.; Bernardo-Colon, A.; Rex, T.S. Erythropoietin either Prevents or Exacerbates Retinal Damage from Eye Trauma Depending on Treatment Timing. Optom. Vis. Sci. 2017, 94, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Ergorul, C.; Ray, A.; Huang, W.; Wang, D.Y.; Ben, Y.; Cantuti-Castelvetri, I.; Grosskreutz, C.L. Hypoxia inducible factor-1alpha (HIF-1alpha) and some HIF-1 target genes are elevated in experimental glaucoma. J. Mol. Neurosci. 2010, 42, 183–191. [Google Scholar] [CrossRef][Green Version]
- Kimura, W.; Xiao, F.; Canseco, D.C.; Muralidhar, S.; Thet, S.; Zhang, H.M.; Abderrahman, Y.; Chen, R.; Garcia, J.A.; Shelton, J.M.; et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015, 523, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Rojas, B.; Gallego, B.I.; Ramírez, A.I.; Salazar, J.J.; de Hoz, R.; Valiente-Soriano, F.; Avilés-Trigueros, M.; Villegas-Perez, M.; Vidal-Sanz, M.; Triviño, A.; et al. Microglia in mouse retina contralateral to experimental glaucoma exhibit multiple signs of activation in all retinal layers. J. Neuroinflamm. 2014, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, L.; Morrison, R.S.; Horner, P.J.; Inman, D.M. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1437–1446. [Google Scholar] [CrossRef]
- Kleesattel, D.; Crish, S.D.; Inman, D.M. Decreased Energy Capacity and Increased Autophagic Activity in Optic Nerve Axons With Defective Anterograde Transport. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8215–8227. [Google Scholar] [CrossRef]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. alpha-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS ONE 2013, 8, e65389. [Google Scholar] [CrossRef]
- Wilson, G.N.; Smith, M.A.; Inman, D.M.; Dengler-Crish, C.M.; Crish, S.D. Early Cytoskeletal Protein Modifications Precede Overt Structural Degeneration in the DBA/2J Mouse Model of Glaucoma. Front. Neurosci. 2016, 10, 494. [Google Scholar] [CrossRef]
- Coleman, D.J.; Trokel, S. Direct-recorded intraocular pressure variations in a human subject. Arch. Ophthalmol. 1969, 82, 637–640. [Google Scholar] [CrossRef]
- Turner, D.C.; Miranda, M.; Morris, J.S.; Girkin, C.A.; Downs, J.C. Acute Stress Increases Intraocular Pressure in Nonhuman Primates. Ophthalmol. Glaucoma 2019, 2, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Z.; Cepurna, W.O.; Johnson, E.C.; Morrison, J.C.; Wang, R.K. Impact of intraocular pressure on changes of blood flow in the retina, choroid, and optic nerve head in rats investigated by optical microangiography. Biomed. Opt. Express 2012, 3, 2220–2233. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Konieczka, K.; Flammer, A.J. The primary vascular dysregulation syndrome: Implications for eye diseases. EPMA J. 2013, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Hannon, B.G.; Feola, A.J.; Gerberich, B.G.; Read, A.T.; Prausnitz, M.R.; Ethier, C.R.; Pardue, M.T. Using retinal function to define ischemic exclusion criteria for animal models of glaucoma. Exp. Eye. Res. 2021, 202, 108354. [Google Scholar] [CrossRef] [PubMed]
- Danhier, P.; Krishnamachary, B.; Bharti, S.; Kakkad, S.; Mironchik, Y.; Bhujwalla, Z.M. Combining Optical Reporter Proteins with Different Half-lives to Detect Temporal Evolution of Hypoxia and Reoxygenation in Tumors. Neoplasia 2015, 17, 871–881. [Google Scholar] [CrossRef]
- Park, H.Y.; Park, J.H.K.a.C.K. Alterations of the synapse of the inner retinal layers after chronic intraocular pressure elevation in glaucoma animal model. Mol. Brain 2014, 7, 53. [Google Scholar] [CrossRef]
- Kanamori, A.; Nakamura, M.; Nakanishi, Y.; Yamada, Y.; Negi, A. Long-term glial reactivity in rat retinas ipsilateral and contralateral to experimental glaucoma. Exp. Eye Res. 2005, 81, 48–56. [Google Scholar] [CrossRef]
- Woldemussie, E.; Wijono, M.; Ruiz, G. Muller cell response to laser-induced increase in intraocular pressure in rats. Glia 2004, 47, 109–119. [Google Scholar] [CrossRef]
- Wu, H.; Chen, Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid. Redox Signal. 2015, 22, 1032–1046. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Poitry-Yamate, C.; Poitry, S.; Tsacopoulos, M. Lactate Released by Miiller Glial Cells Is Metabolized by Photoreceptors from Mammalian Retina. J. Neurosci. 1995, 15, 5179–5191. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Starnes, C.A.; Sauer, M.W.; Firouzgan, Z.; Chen, S.C. Cultured retinal neuronal cells and Muller cells both show net production of lactate. Neurochem. Int. 2004, 45, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Uga, S.; Smelser, G.K. Comparative study of the fine structure of retinal Müller cells in various vertebrates. Investig. Ophthalmol. 1973, 12, 434–448. [Google Scholar]
- Lindsay, K.J.; Du, J.; Sloat, S.R.; Contreras, L.; Linton, J.D.; Turner, S.J.; Sadilek, M.; Satrustegui, J.; Hurley, J.B. Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc. Natl. Acad. Sci. USA 2014, 111, 15579–15584. [Google Scholar] [CrossRef]
- Eichler, W.; Kuhrt, H.; Hoffmann, S.; Wiedemann, P.; Reichenbach, A. VEGF release by retinal glia depends on both oxygen and glucose supply. Neuroreport 2000, 11, 3533–3537. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Palmer, P.G.; Smith, M.A.; Gevorgyan, V.; Wilson, G.N.; Crish, S.D.; Inman, D.M. Structural and Functional Rescue of Chronic Metabolically Stressed Optic Nerves through Respiration. J. Neurosci. 2018, 38, 5122–5139. [Google Scholar] [CrossRef]
- Sivakumar, V.; Foulds, W.S.; Luu, C.D.; Ling, E.A.; Kaur, C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J. Pathol. 2011, 224, 245–260. [Google Scholar] [CrossRef]
- Cserep, C.; Posfai, B.; Lenart, N.; Fekete, R.; Laszlo, Z.I.; Lele, Z.; Orsolits, B.; Molnar, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef]
- Kaur, C.; Rathnasamy, G.; Ling, E.A. Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J. Neuroimmune Pharmacol. Off. J. Soc. Neuroimmune Pharmacol. 2013, 8, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Barron, M.J.; Griffiths, P.; Turnbull, D.M.; Bates, D.; Nichols, P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br. J. Ophthalmol. 2004, 88, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Cwerman-Thibault, H.; Lechauve, C.; Augustin, S.; Roussel, D.; Reboussin, E.; Mohammad, A.; Degardin-Chicaud, J.; Simonutti, M.; Liang, H.; Brignole-Baudouin, F.; et al. Neuroglobin Can Prevent or Reverse Glaucomatous Progression in DBA/2J Mice. Mol. Ther. Methods Clin. Dev. 2017, 5, 200–220. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Zhang, S.H.; Nickerson, J.M.; Gao, F.J.; Sun, Z.; Chen, X.Y.; Zhang, S.J.; Gao, F.; Chen, J.Y.; Luo, Y.; et al. Cumulative mtDNA damage and mutations contribute to the progressive loss of RGCs in a rat model of glaucoma. Neurobiol. Dis. 2015, 74, 167–179. [Google Scholar] [CrossRef]
- Chidlow, G.; Ebneter, A.; Wood, J.P.; Casson, R.J. The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathol. 2011, 121, 737–751. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Figure | Experiment | Eye/ON Number |
|---|---|---|
| 1/B | IOP measurement | n = 16 eyes at 3 d, 14 eyes at 14 d, 12 eyes at 28 d, and 30 control eyes |
| 1/C | RGC count | n = 12 eyes at 6 h, 12 eyes at 3 d, 15 eyes at 14 d, 21 eyes at 28 d, and 20 control eyes. |
| 1/E | Axon count | n = 7 ONs at 6 h, 6 ONs at 3 d, 8 ONs at 14 d, 7 ONs at 28 d, and 13 control ONs. |
| 2/A | N1 amplitude | n = 14 eyes at 3 d, 14 eyes at 14 d, 8 eyes at 28 d, and 12 control eyes. |
| 2/B | P2 amplitude | n = 14 eyes at 3 d, 14 eyes at 14 d, 8 eyes at 28 d, and 12 control eyes |
| 2/C | Response amplitude | n = 14 eyes at 3 d, 14 eyes at 14 d, 8 eyes at 28 d, and 12 control eyes. |
| 3 | Retinal hypoxic neurons | n = 6 eyes at 6 h, 6 eyes at 3 d, 6 eyes at 14 d, 6 eyes at 28 d, and 10 control eyes |
| 4 | Retinal hypoxic glia | n = 6 eyes at 6 h, 6 eyes at 3 d, 6 eyes at 14 d, 6 eyes at 28 d, and 10 control eyes |
| 5 | Hypoxic glia in ON/ONH | n = 6 ONHs and ONs at 6 h, 6 ONHs and ONs at 3 d, 6 ONHs and ONs at 14 d, 6 ONHs and ONs at 28 d, and 6 control ONHs and ONs |
| 6 | Highest hypoxia in ONH/ON | n = 6 ONHs and ONs at 6 h, 6 ONHs and ONs at 3 d, 6 ONHs and ONs at 14 d, 6 ONHs and ONs at 28 d, and 6 control ONHs and ONs |
| 7/A–D | Hypoxia in SCN | n = 4 SCNs at 6 h, 5 SCNs at 3 d, 4 SCNs at 14 d, 4 SCNs at 28 d, and 4 control SCNs. |
| 7/E | Hypoxia in LGN | n = 4 LGNs at 6 h, 4 LGNs at 3 d, 4 LGNs at 14 d, 5 LGNs at 28 d, and 4 control LGNs |
| 7/F | Hypoxia in SC | n = 5 SCs at 6 h, 4 SCs at 3 d, 4 SCs at 14 d, 5 SCs at 28 d, and 4 control SCs |
| 8 | Hypoxia in SC | n = 5 SCs at 6 h, 3 d, 14 d, 28 d; 10 sections per SC |
| 9 | Hypoxia in SC | n = 5 SCs at 6 h, 3 d, 14 d, 28 d; 10 sections per SC |
| 10/A | GLUT1 increase in ON | n = 6 ONs at 6 h, 7 ONs at 3 d, 6 ONs at 14 d, 4 ONs at 28 d, and 18 control ONs |
| 10/B | GLUT1 increase in retina | n = 6 retinas at 6 h, 6 retinas at 3 d, 6 retinas at 14 d, 4 retinas at 28 d, and 18 control retinas |
| 10/C | GLUT3 increase in ON | n = 5 ONs at 6 h, 7 ONs at 3 d, 6 ONs at 14 d, 5 ONs at 28 d, and 19 control ONs |
| 10/D | No change of GLUT3 in retina | n = 9 retinas at 6 h, 11 retinas at 3 d, 6 retinas at 14 d, 4 retinas at 28 d, and 18 control retinas |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jassim, A.H.; Nsiah, N.Y.; Inman, D.M. Ocular Hypertension Results in Hypoxia within Glia and Neurons throughout the Visual Projection. Antioxidants 2022, 11, 888. https://doi.org/10.3390/antiox11050888
Jassim AH, Nsiah NY, Inman DM. Ocular Hypertension Results in Hypoxia within Glia and Neurons throughout the Visual Projection. Antioxidants. 2022; 11(5):888. https://doi.org/10.3390/antiox11050888
Chicago/Turabian StyleJassim, Assraa Hassan, Nana Yaa Nsiah, and Denise M. Inman. 2022. "Ocular Hypertension Results in Hypoxia within Glia and Neurons throughout the Visual Projection" Antioxidants 11, no. 5: 888. https://doi.org/10.3390/antiox11050888
APA StyleJassim, A. H., Nsiah, N. Y., & Inman, D. M. (2022). Ocular Hypertension Results in Hypoxia within Glia and Neurons throughout the Visual Projection. Antioxidants, 11(5), 888. https://doi.org/10.3390/antiox11050888