Abstract
Ozone (O3) is the predominant oxidant air pollutant associated with airway inflammation, lung dysfunction, and the worsening of preexisting respiratory diseases. We previously demonstrated the injurious roles of pulmonary immune receptors, tumor necrosis factor receptor (TNFR), and toll-like receptor 4, as well as a transcription factor NF-κB, in response to O3 in mice. In the current study, we profiled time-dependent and TNFR- and NF-κB-regulated lung transcriptome changes by subacute O3 to illuminate the underlying molecular events and downstream targets. Mice lacking Tnfr1/Tnfr2 (Tnfr-/-) or Nfkb1 (Nfkb1-/-) were exposed to air or O3. Lung RNAs were prepared for cDNA microarray analyses, and downstream and upstream mechanisms were predicted by pathway analyses of the enriched genes. O3 significantly altered the genes involved in inflammation and redox (24 h), cholesterol biosynthesis and vaso-occlusion (48 h), and cell cycle and DNA repair (48–72 h). Transforming growth factor-β1 was a predicted upstream regulator. Lack of Tnfr suppressed the immune cell proliferation and lipid-related processes and heightened epithelial cell integrity, and Nfkb1 deficiency markedly suppressed lung cell cycle progress during O3 exposure. Common differentially regulated genes by TNFR and NF-κB1 (e.g., Casp8, Il6, and Edn1) were predicted to protect the lungs from cell death, connective tissue injury, and inflammation. Il6-deficient mice were susceptible to O3-induced protein hyperpermeability, indicating its defensive role, while Tnf-deficient mice were resistant to overall lung injury caused by O3. The results elucidated transcriptome dynamics and provided new insights into the molecular mechanisms regulated by TNFR and NF-κB1 in pulmonary subacute O3 pathogenesis.
1. Introduction
Ozone (O3) is a highly reactive gaseous oxidant air pollutant. Elevated levels of ambient O3 have been associated with increased hospital visits and respiratory symptoms, including chest discomfort, breathing difficulties, coughs, and lung function decrement [1,2,3]. Subjects with pre-existing diseases such as asthma, rhinitis, and chronic obstructive pulmonary disorder are known to be particularly vulnerable to O3 and are at risk of hospitalization, exacerbations, or death [4,5,6].
Controlled O3 exposure to healthy volunteers and experimental animals elicit a number of pathophysiological effects, which include airway inflammation accompanied by airway hyperresponsiveness, chemokine/cytokine production, mucus overproduction and hypersecretion, reactive oxygen species production, decrements in pulmonary function, altered immune status, and epithelial damage and compensatory proliferation predominantly in ciliated cells of the upper respiratory tract and club cells in terminal bronchioles [7]. Pulmonary O3 responses were also augmented by metabolic disorders, including obesity and diabetes in humans, as well as in experimental animals [8,9,10], and association of air pollution and increased risk of diabetes was also reported in humans and mice [11,12]. Long term exposure to O3 may cause lung tumors in certain strains of mice [13].
Studies have investigated the roles of various inflammatory mediators in the pathogenic airway response to O3. Signal transducers, including epidermal growth factor receptor, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), mitogen activated kinases, and inflammasome proteins (e.g., Nlrp3) have been proposed to be downstream mechanisms of O3-induced airway inflammation [14,15,16]. Toll-like receptor 4 (Tlr4) has been identified as a O3-induced hyperpermeability susceptibility gene from murine genome-wide linkage analysis of subacute O3-induced airway hyperpermeability and injury [17,18]. Furthermore, tumor necrosis factor (Tnf) is a susceptibility gene for pulmonary inflammation induced by subacute O3 [19].
TNF is a master proinflammatory cytokine that causes diverse bioregulatory activities, including cell death, apoptosis, inflammation, and cell proliferation/differentiation [20]. TNF signaling activates NF-κB, as well as the mitogen activated kinase (MAPK), cascade/nuclear transactivation of activator protein (AP)-1, and receptor interacting serine/threonine kinase 1 [21,22]. Among the 27 TNF receptor (TNFR) superfamily, TNF binds to two distinct cellular membrane receptors, TNFR superfamily member 1A (TNF-R1p55) and 1B (TNF-R2p75) [23]. Inhibition or lack of TNF signaling significantly reduced O3-induced inflammation and airway hyperreactivity in rodent lungs [19,24,25,26]. Supporting the role for TNF in experimental O3 studies, lung functional changes were associated with a TNF -308G/A polymorphism in asthmatics [27]. NF-κB was proposed to play a key role in downstream of TNFR/TRAF-mediated lung injury caused by subacute O3 [14].
The current study was designed to identify the transcriptome events underlying pulmonary O3 pathogenesis and downstream targets of the TNFR and NF-κB signaling pathways. We determined time-dependent lung gene expression profiles changed by subacute O3 in wild-type mice and in Tnfr-deficient mice. We also identified NF-κB-dependent transcriptome changes using p50 NF-κB (NF-κB1)-deficient and -sufficient mice.
2. Materials and Methods
2.1. Animals and Inhalation Exposure
Male mice (6–8 weeks) deficient in TNF-specific TNFR1 and TNFR2 (B6.129S-Tnfrsf1atm1ImxTnfrsf1btm1Imx/J; Tnfr-/-), NF-kB p50/p105 subunit (B6;129P-Nfkb1tm1Bal/J; Nfkb1-/-), TNF-a (B6.129S-Tnftm1Gkl/J; Tnf-/-), and interleukin (IL)-6 (B6;129S2-Il6tm1Kopf/J; Il6-/-), and their respective wild-type mice (C57BL/6J for Tnfr+/+ and Tnf+/+; B6129SF2/J for Il6+/+; B6129PF2/J for Nfkb1+/+), were purchased from Jackson Laboratories (Bar Harbor, ME, USA). On arrival in the National Institute of Environmental Health Sciences (NIEHS)/ALION animal facility, the mice were provided diet (NIH_31) and water ad libitum. After acclimation, the mice were placed in individual stainless-steel wire cages within a Hazelton 1000 chamber (Lab Products, Maywood, NJ, USA) equipped with a charcoal and high-efficiency particulate air-filtered air supply. The mice had free access to water and diet during exposure. Tnfr+/+ and Tnfr-/- mice were exposed continuously for 6, 24, 48, or 72 h to 0.3-parts per million (ppm) O3. The other mice were exposed to 0.3-ppm O3 for 48 h. The O3 dosage used in the current study is a reasonable exposure level from which to make comparisons with humans, as rodents require 4−5-fold higher doses of O3 than humans in order to create an equal deposition and pulmonary inflammatory response, as indicated previously [14]. O3 was generated from ultra-high purity air (<1 ppm total hydrocarbons; National Welders, Inc., Raleigh, NC, USA) using a silent arc discharge O3 generator (Model L-11, Pacific Ozone Technology, Benicia, CA, USA). Constant chamber air temperature (72 ± 3° F) and relative humidity (50 ± 15%) were maintained. The O3 concentration was continually monitored (Dasibi model 1008-PC, Dasibi Environmental Corp., Austin, TX, USA). Parallel exposure to filtered air was done in a separate chamber. Immediately following the end of exposure, the mice were euthanized by sodium pentobarbital overdose (104 mg/kg). All animal use was approved by the NIEHS Animal Care and Use Committee.
2.2. Bronchoalveolar Lavage (BAL) Analyses and Lung Histopathology
The right lungs from each mouse were lavaged in situ with HBSS, and the BAL returns were analyzed for the total protein content and cell differentials, as described previously [24]. Left lung tissues from each mouse were inflated gently with 10% neutrally buffered formalin, fixed under constant pressure for 30 min, and proximal (around generation 5) and distal (approximately generation 11) levels of the main axial airway were sectioned for paraffin embedding. Tissue sections (5-μm thick) were stained with hematoxylin and eosin (H&E). The tissues were also processed for immunohistochemical staining using a rat monoclonal (IgG1) anti-macrophage receptor with collagenous structure (MARCO; 1:50 dilution of clone ED31, Hycult Biotech, Wayne, PA, U.S.A.). Briefly, deparaffinized and hydrated tissue sections on microscope slides were treated sequentially with antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA), 0.1% proteinase K, and endogenous peroxidase quenching solution (5% H2O2) before blocking with 1.5% serum (Vectastain ABC kits). Tissue sections were then incubated overnight at 4 °C with the anti-MARCO antibody. After incubation with biotinylated rat secondary antibody (1:200, Vectastain ABC kits) and Avidin/Biotin solution, the antigens were detected by a 3,3′-diaminobenzidine-peroxidase substrate solution (10 min), and the slides were mounted with cover glasses after dehydration.
2.3. Lung RNA Isolation and cDNA Microarray Analysis
Lung tissues from Tnfr+/+ and Tnfr-/- mice were homogenized in 2 mL Trizol (Thermo Fisher Scientific, Waltham, MA, USA) and the isolated total lung RNA was processed for Affymetrix GeneChip array analyses using mouse MOE430A arrays (Affymetrix, Inc., Santa Clara, CA, U.S.A.) in George Washington University (Dr. Andrea De Biase), as described previously [28]. The total lung RNAs from the Nfkb1+/+ and Nfkb1-/- mice were isolated using RNeasy Mini Kit (Qiagen Inc., Valencia, CA, USA) and cDNA microarray was performed on mouse 430 2.0 arrays (Affymetrix) in the NIEHS Microarray Core Facility, as indicated previously [29]. Array raw data were filtered by a lower expression percentile (at least 1 sample had values within the 20% cut-off rage) and the expression levels normalized to the mean value of the experimental control (wild-type mice/air) for each gene by the quantile algorithm were analyzed statistically using GeneSpring GX14 software (Agilent Technologies, Inc., Santa Clara, CA, USA). O3 exposure time effects in Tnfr+/+ lungs (t-test, p < 0.01) and genotype effects in air exposure (t-test, p < 0.05) or O3 exposure (two-way ANOVA, p < 0.05; Benjamin and Hochberg False Discovery Rate test for the multiple comparisons) were tested to identify the differentially expressed genes. Venn diagram analyses determined common genes varied by O3 between the genotypes. Ingenuity pathway analysis (IPA, Qiagen Inc., Valencia, CA, USA) was used to identify the potential molecular interactions and functions, as well as the downstream and upstream pathways. Microarray data were deposited in the Gene Expression Omnibus (accession numbers: GSE166399 for Tnfr+/+ and Tnfr-/- mice and GSE166398 for Nfkb1+/+ and Nfkb1-/- mice).
2.4. Quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
An aliquot of the total lung RNA was reverse transcribed into cDNAs using GeneAmp PCR System 9700 (Applied Biosystems), and cDNA (40 ng) was subjected to PCR in a 25 μL reaction containing 12.5 μL 2X Power SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and 240 nM of custom-designed 18s rRNA (324F 5′-tacctggttgatcctgccag-3′ and 507R 5′-ccgtcggcatgtattagctc-3′), major urinary protein 1 (Mup1; 228F 5′-tattatcctggcctctgacaa-3′ and 369R 5′-agataattccgagcactcttc-3′), immunoglobulin joining chain (Jchanin; 313F 5′-gaacaacagggagaatatct-3′ and 520R 5′-agtggtatagcacttgtttc-3′), and serum amyloid A3 (Saa3; 191F 5′-tacttccatgctcgggggaacta-3′ and 322R 5′-agctcttgagtcctctgctccat-3′) primers or commercially available ones (Real Time Primers, LLC, Elkins Park, PA, USA) for mouse IL-6, IL-33, tissue inhibitor of metalloproteinase 1 (Timp1), D site albumin promoter binding protein (Dbp), and pituitary tumor-transforming gene 1 (Pttg1), for 10 min at 95 °C, and for up to 45 cycles of 95 °C (15 s)–60 °C (1 min) using an ABI Prism 7700 Sequence Detection System (Applied Biosystems) or CFX Connect Realtime System (Bio-Rad Laboratories, Hercules, CA, USA). The relative quantification of the target gene expression was calculated using the comparative threshold cycle (CT) method by subtracting the fluorescence detected CT of 18s rRNA from that of target gene in the same sample (ΔCT).
2.5. Protein Isolation and Western Blot Analysis
Lung cytosolic and nuclear proteins were isolated from pulverized lungs (2 pooled sample/group and 2 lungs/sample) using a kit following the manufacturer’s direction (Active Motif, Carlsbad, CA, USA). Lung total proteins were isolated from mouse lung homogenates in a radioimmunoprecipitation assay buffer (2 pooled sample/group, 2 lungs/sample). The proteins were quantified and stored in aliquots at −80 °C. The lung total or cytosolic fractions (80–100 µg) or nuclear (20 µg) proteins were separated on 10–20% Tris-HCl SDS-PAGE gels (Bio-Rad) and were analyzed by routine Western blotting using mouse specific antibodies against MARCO (Hycult Biotech), transforming growth factor (TGF)-β1 (Abcam, Cambridge, MA, USA.), c-Fos (Santa Cruz Biotechnology, Inc., Dallas, TX, U.S.A.), MUP1 (Santa Cruz), G2/mitotic-specific cyclin-B1 (CCNB1, Santa Cruz), signal transducer and activator of transcription 1 (STAT1, Santa Cruz), Lamin-B1 (Santa Cruz), and β-actin (Santa Cruz). Protein blot images were scanned and quantified using an Amersham Imager 600 (GE Healthcare Bio-Sciences Co., Piscataway, NJ, USA).
2.6. Sandwich Enzyme-Linked Immunosorbent Assay (ELISA) for IL-6
Aliquots of the lung cytosolic proteins (90 μg) were used to determine IL-6 using a mouse-specific ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer instructions. The optical density was measured at 450 nm and the IL-6 concentrations were determined using a standard curve.
2.7. Statistics
BAL, Western blotting, and qRT-PCR data are expressed as the group mean ± standard error of the mean (S.E.M.). Two-way ANOVA was used to evaluate the effects of exposure and genotype. The Student−Newman−Keuls test was used for a posteriori comparisons of the means for all multiple comparisons (p < 0.05). All of the statistical analyses were performed using SigmaPlot 13.0 program (Systat Software, San Jose, CA, USA)
3. Results
3.1. Time-Dependent Changes of Lung Genes by Subacute O3 in C57Bl/6J Mice
O3 caused time-dependent changes in the lung gene expressions with peak perturbation at 48–72 h of exposure (Figure 1A), when most severe lung protein edema, inflammation, and histopathologic changes take places [24,30]. Venn diagram analyses determined that most of the significantly changed genes were unique at each time (Figure 1A, and Table 1 and Table S1), and the number of common O3 responsive genes throughout the exposure (75 upregulated and 45 downregulated) were limited (Figure 1A). Representative canonical pathways of the enriched genes also dissociated between 24 h and 48–72 h (Figure 1B).
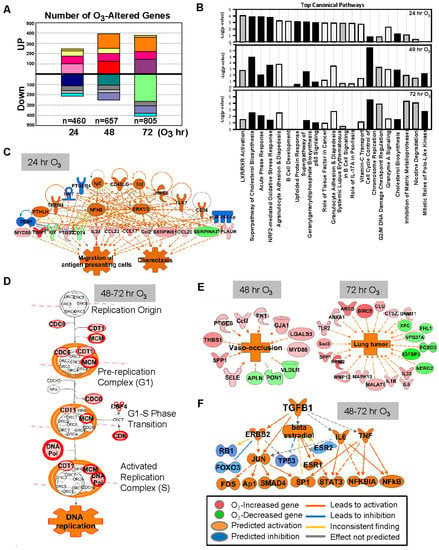
Figure 1.
Effect of ozone (O3) on lung transcriptomics in C57BL/6J mice. (A) The number of lung genes significantly increased or decreased (p < 0.01 with moderated t-test) at 24 (n = 460), 48 (n = 657), and 72 (n = 805) h of 0.3-ppm O3 exposure relative to the air controls. Matching colors of stacks in the graph indicate overlapping genes between different times of exposure. (B) Top-ranked canonical pathway categories of the O3-altered genes at 24, 48, and 72 h are depicted against -log2(P). Black bars = positive z-score (activation); gray bars = negative z-score (inhibition); white bars = no activity pattern available. (C) Pathway analysis determined that tumor necrosis factor (TNF) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) were potential key upstream regulators for the O3-responsive lung genes, which contribute to acute inflammatory and immune responses (e.g., migration of antigen presenting cells) at 24 h. (D) Cell cycle control of chromosome replication was the top canonical pathway of lung genes significantly upregulated by O3 (in red) at 48–72 h. (E) Top disease and biological functions of the genes altered by O3 included blood vessel lesion and vaso-occlusion at 48 h and lung tumor development at 72 h. (F) Transforming growth factor (TGF)-β1 and P53 are predicted to be one of the key upstream regulators orchestrating the lung transcriptome changes at later times (48–72 h) of O3 exposure. Gene or molecule colors indicate upregulation/activation (red/orange) or downregulation/inhibition (green/blue) after O3 exposure compared with air exposure. Analyses were done using ingenuity pathway analysis and GeneSpring software.

Table 1.
Representative lung genes time-dependently changed by ozone (O3) in C57BL/6J mice.
After 24 h of O3 exposure, inflammatory mediators represented by TNF and NF-κB were predicted to be upstream regulators of O3-altered genes (Figure 1C), and acute phase and inflammatory response (e.g., Alb, Saa3, Myd88, Socs3, Ccl17, and Cxcl14) and Nrf2-mediated oxidative stress response (e.g., Nrf2, Maff, Gclc, Gpx2, and Sod2) were predominantlyactivated pathways (Figure 1B,C, and Table 1 and Table S1). At later times of exposure (48 and 72 h), O3 most markedly activated cell cycle and inhibited DNA damage repair responses (e.g., Ccnb1, Cdc6, Cdt1, Cdk1, Plk1, Mcm family, Cks2, and Pcna; Figure 1B,D and Table S1). Distinctively, after 48 h of O3, the enriched genes were predicted to activate cholesterol biosynthesis and affect leucocyte migration (e.g., Ccl17, Retnla, and Timp1) and blood vessel lesion/vaso-occlusion (e.g., Thbs1, Spp1, and Vldlr; Figure 1B,E). Transcriptomics changes at 72 h of O3 may suppress xenobiotic degradation (e.g., Cyp4b1, Fmo3, and Aox3) and tissue repair (e.g., Timp1, Mmp12, and Sdc2) and induce lung tumorigenesis (e.g., Rrm2, Birc5, and Areg; Figure 1B,E). Upstream molecules including TGF-β1 and P53 were indicated to affect O3-induced transcriptome changes at later times (Figure 1F). Among the upstream regulators of O3-responsive transcriptome (24–72 h), chemical drugs including simvastatin, acetaminophen, and sulforaphane (Table S2) were suggested as therapeutic intervention to reverse O3 toxicity, such as reducing inflammation, reactive oxygen species, and lipids. Downregulated lung genes by O3 included clusters of Mup; insulin-like growth factor binding protein 3 (Igfbp3); and xenobiotic metabolizing enzymes including cytochrome P450, family 1, subfamily a, polypeptide 1 (Cyp1a1), and aldehyde oxidase 3 (Table 1 and Table S1). While Hspa1a and Hspa1b encoding TLR4-dependent heat shock protein 70 (HSP70) [28] were upregulated by O3, many other HSP genes (e.g., Hspb1, and Hsph1) were significantly decreased at 48 h of O3 (Table S1).
3.2. TNFR-Dependent Lung Transcriptome Changes
3.2.1. Air-Exposed Lungs
Lung genes basally expressed lower in Tnfr-/- mice than in Tnfr+/+ mice (Table 2 and Table S3) were represented by Mup clusters, Pttg1, HSPs (Hsph1 and Hspa4l), S100 calcium binding proteins (S100a8 and S100a9), chemokines (Ccl5 and Ccl17), cytochrome P450 family (Cyp1a1 and Cyp3a11), and apolipoproteins (Apoa2 and Apoa1). In contrast, basally overexpressed lung genes in Tnfr-/- mice compared with Tnfr+/+ mice (Table 2 and Table S3) included Dbp and nicotineamide nucleotide transhydrogenase (Nnt). Basally Tnf-dependently expressed genes were related to vascular disorders, as well as to the inhibition of inflammatory cell response (Figure 2A,B and Table 3).
Table 2.
Selected tumor necrosis factor receptor (TNFR)-dependent lung genes after air or ozone (O3) exposure.
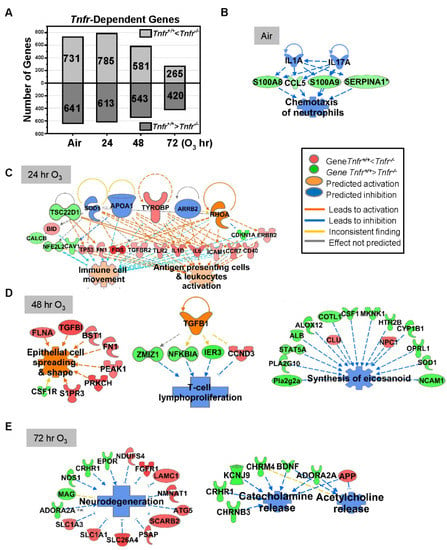
Figure 2.
Tumor necrosis receptor (TNFR)-dependently regulated lung genes. (A) The number of lung transcripts significantly (p < 0.05) upregulated (light gray) or downregulated (dark gray) different Tnfr-deficient (Tnfr-/-) mice relative to wild-type (Tnfr+/+) mice at baseline (air, moderated t-test) or after 24, 48, and 72 h of 0.3-ppm ozone (O3) exposure (two-way ANOVA). (B) In air-exposed basal lungs, the inhibition of interleukins (ILs) 1A and 17A were predicted to suppress TNFR-dependent lung genes (e.g., Ccl5, S100a8, S100a9, and Serpina1), leading to the inhibition of inflammatory cell chemotaxis. (C) After 24 h O3 exposure, a compensatory increase of the genes involved in immune cell activation and movement were manifest in Tnfr-/- lungs compared with Tnfr+/+ mice. (D) After 48 h of O3 exposure, when lung injury and inflammation are greatest, modulation of potential upstream regulators including transforming growth factor (TGF)-β1 may change transcriptomes to suppress lymphocyte proliferation and eicosanoid synthesis and activate epithelial cell spreading/integrity in Tnfr-/- lungs. (E) Tnfr-/- mouse lungs after 72 h of O3 exposure had transcriptome changes to suppress the release of neurotransmitters and inhibit neurodegeneration. Gene or molecule colors indicate upregulation/activation (red or orange) or downregulation/inhibition (green or blue) in Tnfr-/- mice compared with Tnfr+/+ mice after air or O3 exposure. Analyses were done using Ingenuity Pathway Analysis and GeneSpring software.
Table 3.
Representative diseases and biological functions predicted to be affected by tumor necrosis factor receptor (TNFR)-dependent lung transcriptome changes in mice determined by Ingenuity Pathway Analysis (IPA).
3.2.2. O3-Exposed Lungs
Tnfr-dependent variation of lung gene expression was more marked at 24–48 h than at 72 h of O3 exposure (Figure 2A). Transcriptome changes that occurred in Tnfr-/- mice at 24 h were predicted to potentiate immune and inflammatory systems (Figure 2C, and Table 2, Table 3 and Table S4), suggesting a compensatory or adaptive immune response in the absence of TNFR1 and TNFR2 signaling. Upstream regulators of this TNFR-dependent early transcriptome response may include tyrosine kinase-binding protein (TYROBP), APOA1, and arrestin beta-2 (ARRB2) in Tnfr-/- mice (Figure 2C). At 48 h of O3 the pulmonary epithelial proliferation, inflammatory cell influxes, and epithelial injury were significantly more suppressed in Tnfr-/- mice than in Tnfr+/+ mice [24]. TGF-β1 and protein O-GlcNAcase or P53 were potential key upstream regulators, and TNFR-dependently enriched genes in Tnfr-/- mice were predicted to inhibit lymphocyte proliferation/eicosanoid synthesis (e.g., H2-Q4, Il33, Nfkbia, and Tgfbi) and macromolecule oxidation (e.g., multiple cytochrome P450 subfamilies) and activate epithelial cell spreading/integrity (e.g., Fn1 and Flna; Figure 2D, and Table 2, Table 3 and Table S4). Many lipid metabolism (e.g., Fdps, Got2, and Sgms1) and eicosanoid synthesis (e.g., Alb, Alox12, and Ptgs1) genes were also relatively suppressed in Tnfr-/- mice compared with Tnfr+/+ mice at 48 h O3 (Figure 2D, and Table 2, Table 3, and Table S4). At 72 h O3, lack of TNFR signaling inhibited transcriptomes of neurodegeneration (e.g., Slc26a4, Epor, and Nmnat1) and transport for neurotransmitters, acidic amino acids, and anions (e.g., Chrm4, App, and Kcnj9; Figure 2E, Table 2, Table 3 and Table S4).
3.3. Nfkb1-Dependent Lung Transcriptome Changes
3.3.1. Air-Exposed Lungs
NF-κB1 (p50/p105) forms the most abundant heterodimer with RelA, but it also forms a p50−p50 homodimer. The NF-κB1 homodimer is known to work as a transcriptional activator, similar to other NF-κB heterodimer complexes (e.g., RelA-p50 and c-Rel-p50), as well as a transcriptional repressor by inhibiting the binding of other NF-κB dimers to lead to the suppression of NF-κB target gene expressions during innate immune responses [31,32]. Supporting the transcriptional repressor role of NF-κB1, basally different lung genes in Nfkb1-/- mice compared with Nfkb1+/+ mice (t-test p < 0.05 n = 1395 genes; Table 4 and Table S5) were predominantly enriched to increase leukocyte extravasation/adhesion genes (e.g., CCL and CXCL chemokines, Ccr2, claudins, integrins, Tnfrsf1b, Sell, Cd14, and Lbp). In addition, enriched genes for the antigen presentation to CD8+ T lymphocytes (e.g., B2m, Hla-G, Nlrc5, Psmb8, Psmb9, and Tap1) were overexpressed in Nfkb1-/- lungs compared with Nfkb1+/+ lungs (Table 4 and Table S5). Furthermore, the downregulation of other sets of immune genes (e.g., Jchain, Cxcl13, Pcdhb3, and Marco) were also marked in Nfkb1-/- mice compared with in Nfkb1+/+ mice (Table 4 and Table S5). Activation of interferon (IFN) regulatory factors (IRF3 and IRF7) has been predicted to serve as upstream regulators of NF-κB1-dependent genes (e.g., Ifit3, Stat1, and Oas1), which would cause IFN-mediated decrease in infectivity in basal lungs deficient in Nfkb1 (Table 4 and Table S5). This is consistent with the known Nfkb1-/- mouse phenotypes such as defective responses to infection and specific antibody production [33].
Table 4.
Selected nuclear factor of kappa light polypeptide gene enhancer in B-cells p50 (NF-κB1)-dependent lung genes after air or ozone (O3) exposure.
3.3.2. O3-Exposed Lungs
After 48 h of O3, lack of Nfkb1 predominantly suppressed lung cell cycle progression and enhanced DNA damage checkpoint regulation pathways through downregulation of multiple genes in the families of cyclin, cell division cycle, centromere protein, and centrosomal protein (Figure 3A, and Table 4 and Table S6). This corresponded to the significant decrease in O3-induced centriacinar cell proliferation in Nfkb1-/- mice compared with Nfkb1+/+ mice [14]. Similar to basal lung transcriptomics, pathway analyses indicated heightened IFN signaling genes (e.g., Irf1, Psmb8, Oas1, Tap1, and Stat1) and activated upstream regulators, IRF7 and IFN type I receptor (IFAR), in O3-exposed Nfkb1-/- mice compared with Nfkb1+/+ mice (Figure 3A, and Table 4 and Table S6). The results demonstrated suppressed lung cell proliferation and heightened antimicrobial and immune response transcriptomes noticeable in Nfkb1-/- mice relative to Nfkb1+/+ mice after O3. Certain inflammatory genes bearing potential or confirmed NF-kB binding sites were (e.g., Ccl20, Saa3, Fos, Il6, Ido1, Mmp9, and Psmb9) more heightened in Nfkb1-/- mice than in Nfkb1+/+ mice (Table 4 and Table S6), which is suggestive of p50−p50 homodimer-mediated suppression.
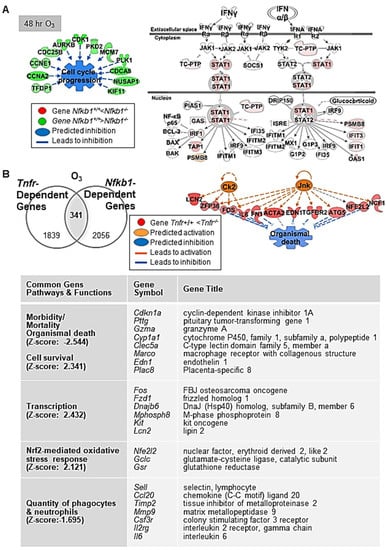
Figure 3.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) dependently regulated lung genes during ozone (O3)-induced lung injury development. (A) At 48 h O3 exposure, downregulation of multiple genes in the family of cyclin (e.g., Ccnb1), cell division cycle (e.g., Cdca8), and centromere protein (e.g., Cenph) in Nfkb1-deficient mice (Nfkb1-/-) compared with wild-type (Nfkb1+/+) mice was marked, suggesting suppressed cell proliferation and enhanced DNA damage checkpoint regulation pathways in Nfkb1-/- mice. In contrast, heightened interferon (IFN) signaling genes (e.g., Irf1, Psmb8, Tap1, and Stat1) and predicted activation of upstream regulators (e.g., IRF7 and IFN type I receptor) indicated an enhanced innate immunity in Nfkb1-/- mice than in Nfkb1+/+ mice during O3-induced lung injury development. (B) Common differentially regulated genes between Tnfr+/+ and Tnfr-/-, and Nfkb1+/+ and Nfkb1-/- (n = 341) were determined by Venn diagram analysis. Transcriptome signatures of TNFR/NF-κB-axis mediated pulmonary O3 toxicity were predicted to modulate cell/organism death and survival, transcriptional regulation, oxidative stress, and inflammation (B). Gene or molecule colors for (A,B) indicate upregulation/activation (red/orange) or downregulation/inhibition (green/blue) in Nfkb1-/- mice compared with Nfkb1+/+ mice after O3 exposure. Analyses were done using Ingenuity Pathway Analysis and GeneSpring software.
3.4. Common Differentially Expressed Genes in Tnfr-/- and Nfkb1-/- Mice Exposed to O3
Venn diagram analysis determined the common genes (n = 341) differentially expressed between Tnfr+/+ and Tnfr-/-, and Nfkb1+/+ and Nfkb1-/- genotypes after O3 exposure (Table S7). These common differentially regulated genes were enriched in host defense functions through activation of cell survival and the inhibition of cell death/mortality and oxidative stress (Figure 3B). A subset of genes suppressed in both Tnfr-/- and Nfkb1-/- mice relative to their wild-type controls may be modulated by the axis of TNFR and p50−p65 NF-κB heterodimers (i.e., transcription activator), while the genes suppressed in Tnfr-/- mice but heightened in Nfkb1-/- mice may include the ones regulated by TNFR and p50−p50 NF-κB homodimer (i.e., transcription suppressor) signaling axis. Among them, mouse Col1a2, Gclc, Il6, Ido1, Junb, Lcn2, Mmp9, Psmb9, and Psme1 are known to possess functional NF-κB binding sites, and the human homology of many other genes (e.g., CCL20, EDN1, HSP90AA1, TNC) are known to be direct NF-κB downstream targets (https://www.bu.edu/nf-kb/gene-resources/target-genes; https://bioinfo.lifl.fr/NF-KB, searched on 30 July 2017). We searched the DECODE database (SAbiosciences.com, searched on 30 July 2017) to find potential NF-κB p50−p65 target genes (bearing NF-κB binding consensus sequences 5′-GGGRNYYYCC-3′ or 5′-GGAATTYCCC-3′; R is A/G, Y is T/C, N is any nucleotide) in the promoter of selected common differentially regulated genes (i.e., genes > 1.5-fold lower in O3-Nfkb1-/- than in O3-Nfkb1+/+ genotypes in Table S7). Genes including endothelin 1 (Edn1), prostate stem cell antigen (Psca), Dbp, and chemokine (C-C motif) ligand 22 (Ccl22) had predicted consensus sequences indicating potential TNFR-NF-κB target genes during O3-induced pulmonary pathogenesis (Table 5).
Table 5.
Potential NF-κB binding motifs on selected Nfkb1-dependently regulated lung genes after ozone (O3) exposure.
3.5. Effects of Tnf and Il6 on O3-Induced Lung Injury
As seen in Tnfr-/- mice [24], the mice deficient in Tnf cluster genes (Tnf, Lta, and Ltb) [34], and the mice treated with the TNF antibody [19], BAL fluids from Tnf-/- mice had significantly reduced numbers of lung neutrophils and epithelial cells and amounts of proteins compared with those from Tnf+/+ mice at 48 h of O3 (Figure 4A). The histopathologic analysis indicated that O3-induced centriacinal proliferation indicated by thickened bronchiolar and terminal bronchiolar epithelium (arrows) were also less marked in Tnf-/- mice compared with Tnf+/+ mice (Figure 4B). The current microarray analysis and a previous study [14] demonstrated that the abundance of Il6 mRNA was higher in both Tnfr-/- and Nfkb1-/- mice compared with their corresponding wild-type mice after O3 (Table 2, Tables S4 and S6). In Il6-/- mice, lung protein hyperpermeability determined by the BAL protein concentration was significantly higher than that in Il6+/+ mice (Figure 4C). However, the numbers of O3-induced neutrophils or epithelial cells in BAL fluids were not significantly different between the two genotypes (data not shown). Consistent with the heightened BAL protein level in Il6-/- mice, H&E-stained lung tissue sections depicted more marked edema and permeability in the perivascular region (arrows), which accompanied protein exudation (pink staining) and congestion (red blood cells) into the alveolar air space in Il6-/- mice compared with Il6+/+ mice after O3 (Figure 4D). Gene expression data and BAL analysis suggested a potential protective role for IL-6 in this model. ELISA determined significantly increased levels of IL-6 in Tnf+/+ (48 h) and Nfkb1+/+ (24 and 48 h) mouse lungs after O3 exposure (Figure 4E). The O3-enhanced IL-6 protein amounts were significantly higher in Tnfr-/- and Nfkb1-/- mice compared with their corresponding wild-type mice (Figure 4E), which supported TNFR- and NF-κB1-dependent Il6 mRNA abundance.
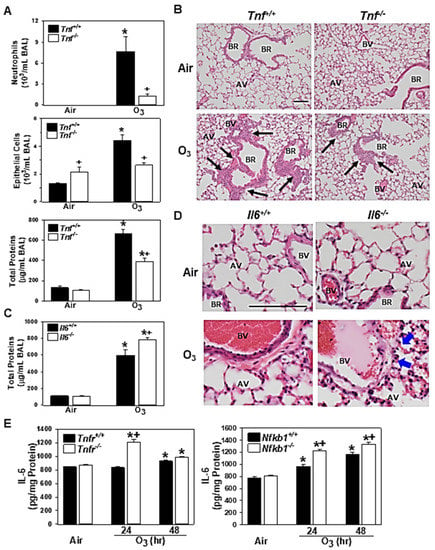
Figure 4.
Functional roles of tumor necrosis factor (TNF) and interleukin 6 (IL-6) in pulmonary O3 pathogenesis. Effect of targeted disruption of Tnf (A) or Il6 (C) was determined by bronchoalveolar lavage (BAL) analyses in gene knockout mice (Tnfr-/-, Il6-/-) and their wild-type mice (Tnf+/+, Il6+/+) after 48 h of exposure to air and 0.3-ppm O3. Data are presented as means ± S.E.M (n = 4–5 mice/group). Representative light photomicrographs of H&E-stained lung sections from Tnf+/+ and Tnfr-/- mice, (B) and Il6+/+ and Il6-/- mice (D) exposed to air or O3 (48 h). Black arrows depict bronchiolar/terminal bronchiolar epithelium under proliferation. Blue arrows depict protein exudation in perivascular regions. AV = alveoli. BV = blood vessel. BR = bronchiole or terminal bronchiole. Bars = 100 μm. TNFR- and NF-κB1-dependent level of lung IL-6 proteins determined by enzyme-linked immunosorbent assay (E). Data are presented as means + S.E.M (n = 3/group). *, significantly different from genotype-matched air control mice (p < 0.05). +, significantly different from O3-exposed corresponding wild-type (Tnf+/+, Il6+/+, Tnfr+/+, or Nfkb1+/+) mice (p < 0.05).
3.6. Validation of Microarray Results
qRT-PCR determined TNFR-dependently increased tissue inhibitor of metalloproteinase (Timp1) and Il33 or decreased Mup1 after air and O3 exposure (Supplemental Figure S1A). Timp1 and Il33 mRNAs were significantly upregulated in O3-resistant Tnfr-/- mice compared with susceptible Tnfr+/+ mice. A significant decline of Mup1 mRNA abundance by O3 was greater in Tnfr-/- mice than in Tnfr+/+ mice. Differential expression of NF-κB1-dependent genes, Jchain, Dbp, and Saa3, were also significantly different between two genotypes at baseline and/or after 48 h O3 (Figure S1B). The mRNA expressions of common differentially regulated genes Pttg1 and Il6 were significantly lower or higher, respectively, in both Tnfr-/- and Nfkb1-/- mice compared with their corresponding wild-type mice (Figure S1C). Western blot analyses found TNFR-dependent variations of the total TGF-β1 and MUP1 proteins and NF-κB1-dependent level of nuclear CCNB1 and STAT1 proteins in the lungs exposed to O3 (Figure 5A). The amount of total MARCO and nuclear c-Fos proteins, common differentially regulated gene products, were also varied similarly in Tnfr-/- and Nfkb1-/- mice compared with their corresponding wild-type mice (Figure 5A). The total lung protein levels of c-Fos were time-dependently increased by O3 in all mice, while the cytoplasmic c-Fos abundances were marginally changed or decreased by O3 (Figure 5A). MARCO was detected in alveolar macrophages and was localized mostly in their plasma membranes and/or cytoplasm (Figure 5B). Consistent with the differential protein levels detected by Western blotting, lower levels of MARCO localization were found in Tnfr-/- and Nfkb1-/- mouse lungs relative to their wild-type mice after 48 h of O3 exposure (Figure 5B).
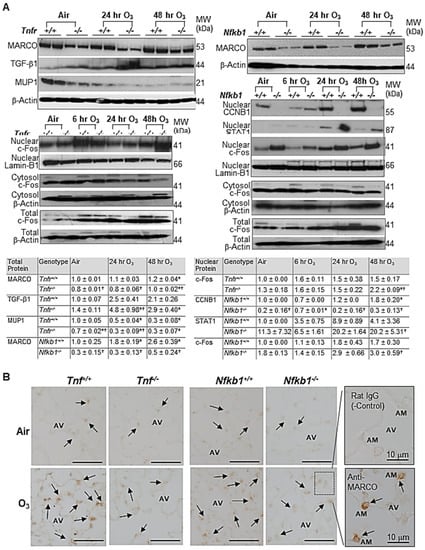
Figure 5.
Microarray analysis validation of tumor necrosis factor receptor (TNFR)- and NF-κB1-dependent transcriptomics in response to ozone (O3). (A) Western blot analyses determined TNFR- and/or NF-κB1-dependent expression of a lung macrophage receptor with a collagenous structure (MARCO), transforming growth factor (TGF)-β1, major urinary protein 1 (MUP1), nuclear c-Fos, nuclear G2/mitotic-specific cyclin-B1 (CCNB1), and nuclear signal transducer and activator of transcription 1 (STAT1). β-Actin (for total and cytosol) and Lamin-B1 (for nuclear) levels detected as the internal controls. Representative digitized lot images are presented. Group mean ± S.E.M. presented for quantified digitized images (n = 4/group for total protein bands, n = 2/group for nuclear protein bands). * Significantly different from genotype-matched air control (p < 0.05). † Significantly different from exposure-matched wild-type mice (p < 0.05). (B) TNFR- and NF-κB1- dependent MARCO protein expression was localized on mouse lungs by an immunohistochemical method after air or O3 (48 h) exposure. Representative light photomicrographs of lung sections (n = 2–3/group) presented. Arrows depict MARCO staining on the plasma membrane and/or cytoplasm of alveolar macrophages (AMs) in the alveoli (AV). A box depicts representative images of magnified AMs in AV stained with rat IgG (negative control) or ant-MARCO antibody. Bars (unlabeled) = 50 μm.
4. Discussion
We elucidated murine lung transcriptional profiles that were time-dependently changed by subacute O3 exposure. Comparative analyses between wild-type and gene knockout mice enriched pulmonary genes modulated via TNFR and/or p50 NF-κB pathways during O3 exposure. Supporting our previous findings that Tnf is a susceptibility gene for subacute O3-induced pulmonary inflammation [19] and that lack of Tnfr and Nfkb1 alleviated pulmonary O3-induced injuries [14,24], the enriched genes may play key roles in pulmonary O3 pathogenesis in mice.
There are limited resources of global cDNA expression data in O3-exposed airways. In humans, the microRNA (miRNA) profile on sputum samples exposed to controlled O3 (0.4 ppm for 2 h during exercise) disrupted immune and inflammatory-related miRNAs including neutrophil-specific miR-143 and myeloid cell specific miRNA-223, which supported an increased number of neutrophils in the sputum [35]. The O3-responsive miRNAs were also predicted to post-transcriptionally alter the genes involved in cell cycle (e.g., Ccnd1) and cellular growth and survival (e.g., Arhgdia, Sod2) [35]. In rodents, Gohil et al. [36] first demonstrated microarray profiles after acute exposure to O3 (1 ppm, 8 h/day for 3 days) in adult C57BL/6J mice predominantly upregulated multiple cell cycle progression genes (e.g., Septin5, Nap1, and Cdc2a) and NF-κB-activated genes such as Saa3 and plate-derived growth factor receptor alpha (Pdgfra) in the lungs. In contrast, the suppression of families of transcripts encoding contractile proteins such as troponins, myosins, and actins; cytochrome P450s; and antigen presenting and immune surveillance molecules (e.g., cluster of MHC class and immunoglobulins) were found after O3 exposure [36]. O3-induced repression of the muscle-specific proteins and cytochrome P450 transcription may activate NF-κB [37,38]. Acute O3 (0.8 ppm, 8 h/day for 2 days) also altered the genes involved in oxidative stress and defense, including NRF2 target antioxidants (e.g., Gclc, Gst, and Homx1) in C57BL/6J mouse lungs [39]. The authors did not find significant differences in the lung inflammation and gene expression profiles between mice that lacked Ercc6, the DNA excision repair protein gene, and their heterozygous controls [39]. A recent RNA-seq analysis of acute O3 exposure (1 or 2 ppm for 3 h) was done in two compartments of the lung, dissected conducting airways (no parenchyma) and macrophages collected from BAL, from adult female C57BL/6J mice, in order to segregate transcriptomics in inflammation and tissue injury. Concentration- and compartment-specific profile comparisons indicated more dynamic transcriptional changes in the conducting airways than in the macrophages [40]. Alteration of antioxidant (e.g., Gsta1), immune (e.g., Cx3cl1), cell cycle (e.g., Mcm family and Cdk1), and acute phase (e.g., Saa3 and Lcn2) genes were common in both compartments of the airways, while decreases in the epithelium barrier and marker genes (e.g., Foxj1, Cyp2f2, and Scgb1a1) were distinct in conducting airway compartments, supporting epithelial cell sloughing and metaplasia/hyperplasia in the conducting airways [40]. Neutrophil activation/degranulation and immune signaling genes (e.g., Ccl17 and Retnla) were the most enriched in alveolar macrophage transcriptome after O3 [40]. Another RNA-seq analysis on three compartments of C57BL/6J lungs (i.e., tracheobronchial epithelium, lung parenchyma, and CD11b+ BAL macrophages) exposed to subchronic levels of O3 (0.8 ppm, 4 h/day for total 14 days) determined that lung parenchyma and macrophages were enriched with inflammatory pathway genes (e.g., Ccl2, Ccl17, Timp1, Saa3, Lcn2, and Mmp14) [41]. Transcriptomes of tracheobronchial epithelium and parenchyma, on the other hand, had predominant changes in cell cycle and DNA repair genes (e.g., Cdc20b, Cdk1, and Retnla) in response to subchronic O3 [41]. In this study, most transcriptome changes by subchronic O3 were common in male and female mice, while female mice were more susceptible to inflammatory cell influx, epithelial loss, and compensatory proliferation, which was supported by the more robust changes of the gene expression in females mice [41]. In Fischer rat lungs, acute O3 (5 ppm, 2 h) altered genes with similar functions reported in mouse studies, and upregulation of inflammatory and redox (e.g., Jun, Cxcl2, Nos2, Hsp27, and Nfkb1), cell cycle and DNA repair (Ccne1, Cdc2, and Arrb15b), and lipid metabolism (e.g., Faah and Plaa) genes were evident [42]. In developmental mouse lung at 3 days postnatal (transition from saccular to alveolar stage), transcriptome changes by acute O3 (1 ppm, 3 h) exposure were less robust than those seen in adult mice, and the global suppression of cell cycle-related lung genes (e.g., Cenpf, Cdca8, Cdk1, Cntn14, Cdc45, Mki67, and Pcna) was rather marked until 24 h postexposure [43]. These results indicate that acute O3 exposure disturbs cellular proliferation and differentiation of lungs undergoing development.
Although the current study demonstrated whole lung transcription profiles without dissection of compartment- or cell-specific transcriptomics, our results characterized multiphasic transcriptome changes by O3 exposure time, and only 18% genes were altered commonly in more than two time points. Subacute O3 responsive genes likely have roles in oxidative injury and antioxidant induction, chemotaxis, and immune cell development during early exposure (at 24 h); cell cycle progress, blood vessel lesions, and cholesterol biosynthesis during peak lung injury (at 48 h); and xenobiotic metabolic process, tumorigenesis, and tissue injury/repair at a later time (72 h). After comparison with compartmental transcriptome studies, we predicted the cellular or tissue origin for reproducible O3 responsive genes across multiple transcriptome studies; for example, increased lipocalin 2 (Lcn2) and small proline-rich protein 1A (Sprr1a) and decreased Igfbp3 may be mainly from the parenchyma [41]; increased matrix metalloproteinase 14 (Mmp14) from the macrophages [41]; increased resistin like alpha (Rentnla), leucine-rich alpha-2-glycoprotein 1 (Lrg1), and Timp1 from both the macrophages and parenchyma [40,41]; increased Saa3 from all compartments of airways [40,41]; and increased chromatin licensing and DNA replication factor 1 (Cdk1) and ubiquitin-conjugating enzyme E2C (Ube2c), as well as decreased Cyp1a1, Mup family, and serine (or cysteine) peptidase inhibitor, clade A, member 3K (Serpina3k) from the conducting airways [40,41].
The NF-κB family of proteins has an important role in inflammatory responses initiated by TNF [44]. Despite the identification of a few well-accepted NF-κB target genes in humans and mice (e.g., NFKBIA, TNFAIP3, and MYC), transcriptional outputs through NF-kB are not well understood due to the complexity of NF-κB dynamics and the NF-κB-binding landscape in the gene expression. NF-κB responses in gene transcription are known to vary depending on the cell type as well as the initiating stimulus [45]. In addition, p50 and p52, among five NF-kB family proteins, do not have a transactivation function, and they can activate transcription through heterodimerization with p65 or others [46]. Importantly, the p50−p50 homodimer binding to NF-κB motif inhibits other NF-κB dimer complex binding, and thus it often, but not always, serves as a transcriptional suppressor for NF-κB target genes [47]. The p50−p50 homodimer has thus been shown to have anti-inflammatory functions through repression of proinflammatory genes and enhancement of anti-inflammatory genes [48,49]. TNF I triggered a strong, sustained p65−p50 activation with a relatively lower level of p50−p50 [47]. Therefore, common differentially regulated genes by TNFR and NF-κB1 in the current study may include NF-κB target genes inducible by the TNFR-NF-κB (p50−p65) axis, as well as those suppressible by the p50−p50 homodimer. That is, the genes suppressed in Tnfr-/- and Nfkb1-/- mice (e.g., Pttg1, Mmp3, and Marco) are likely p50−p65-inducible genes. Genes downregulated in Tnfr-/- mice but overexpressed in Nfkb1-/- mice after O3 exposure (e.g., Gzma, Cyp1a1, Nkg7, Il6, Ccl20, and Kit) are possibly p50−p50-repressed genes. Functional NF-κB motifs have been discovered in the murine Il6 promoter [50]. Therefore, together with augmented pulmonary protein hyperpermeability in Il6-deficient mice, IL-6 was predicted as an anti-inflammatory cytokine in the current subacute O3 pathogenesis and p50−p50 homodimer may modulate its transcription. We elucidated the potential NF-κB binding motifs from several common differentially regulated genes (e.g., Psca and Edn1), and these genes are postulated as direct downstream targets of the TNFR-NF-κB signaling pathways.
One of the genes modulated by both TNFR and NF-κB1 is Marco. MARCO expressed in alveolar macrophages recognizes oxidized lipids and provides innate defense against inhaled pathogens [51]. As a downstream effector of TLR4, which is a murine susceptibility gene for subacute O3-induced pulmonary hyperpermeability [17,28], MARCO plays a protective role in subacute O3-exposed mouse lungs through the inhibition of oxidized surfactant lipid production and inflammation [52]. As TLR4 and TNFR are key immune receptors in subacute O3 pathogenesis, and the NF-κB pathway is also known to play an important role in the TLR4-mediated immune responses [53,54], we compared the current transcriptome profiles with TLR4-dependent O3 transcriptomics (GEO accession # GSE20715, [28]). Commonly regulated genes by TLR4 and NF-κB1 were enriched in lipid derangement, including the disruption of membrane phospholipids, reaction with unsaturated fatty acids in airway lining fluids, interruption of fatty acid/steroid metabolism (e.g., Dbp and Cpt1a), as well as in cell-mediated immunity and lymphoid tissue hyperplasia (e.g., Cxcl1, Ccl20, and Ptpn2; Figure S2). Common gene transcripts regulated by TNFR and TLR4 were enriched in the engulfment of phagocytes (e.g., Marco, Icam1, Lcn2, and Il33), protein ubiquitination (e.g., Dnaja1, Hspd1, and Psma3), fatty acid metabolism (e.g., Acot7, Ptges, Elovl1, and Lpin1), and glutathione homeostasis/redox (e.g., Gsr, Gstm1, and Gltx; Figure S3). Overall, the TNFR-NF-κB and TLR4-NF-κB pathways or crosstalk modulated distinct transcriptomes during the development of O3-induced lung injury in mice. Further studies are warranted for these common genes regulated by these three critical immune and inflammatory mediators (Table S7).
Increasing evidence and TNFR-/TLR4-enriched pathways indicate an association of airway O3 responses with extracellular and/or cellular lipid biology. On airway epithelium lining fluids rich in surfactant, inhaled O3 chemically reacts with cholesterol or phospholipids and generates cytotoxic ozonolysis products represented by 5β,6β-epoxycholesterol (β-epoxide) [55,56]. These lipid-ozonized products are proinflammatory and are known to contribute to O3-induced airway inflammation [57,58]. Eicosanoids (e.g., prostaglandins, leukotrienes, and thromboxanes) synthesized by peroxidation of arachidonic acid by lipoxygenases, cyclooxygenases, and cytochrome P450 are also inflammatory mediators increased by O3 leading to airway hyperresponsiveness and extrapulmonary outcomes, including vasoconstriction [59,60]. O3 exposure-released adrenal-derived stress hormones (e.g., epinephrine and corticosterone) disrupted lipid and carbohydrate metabolism, leading to hyperglycemia, glucose intolerance, and lung injury in rats [61,62]. Further rodent studies demonstrated that obesity augmented acute O3-induced airway hyperresponsiveness and inflammation [63,64,65], and diabetes caused early and exacerbated lung inflammation and fibrotic changes in response to subchronic O3 (0.5 ppm, 4 h/day for 13 weeks) [10]. Epidemiological studies also showed a positive association between O3 exposure and adult insulin resistance and preexisting lipid disorders and metabolic conditions (e.g., obesity and diabetes) [66,67,68]. Metabolomic analysis of human serum revealed that acute O3 exposure markedly increased lipid mobilization and catabolism products (e.g., monoacylglycerol and medium- and long-chain free fatty acids) [69]. Interestingly, human population studies indicated an association of gain-of-function TNF −304G/A polymorphism with obesity-related airway hyperresponsiveness in asthmatics [70]. In obese mice, Tnfr2 deficiency reduced body weight and acute O3-induced inflammation and obesity-related airway hyperresponsiveness [64,71]. Overall, these studies suggested a role of lipid derangement in airway and extrapulmonary O3 pathogenesis.
Our transcriptomic and pathway analyses results suggested direct effects of known or potential NF-κB motif-bearing genes in O3-induced pulmonary edema (e.g., Il6), T cell immunity (e.g., Ccl17, Ccl22, and Il27ra), cardiac mortality and vasoconstriction (e.g., Edn1), extracellular matrix degeneration (e.g., Col1a2 and Mmp9), and interruption of lipid metabolism (e.g., Dbp and Tef) via the TNFR-NF-κB signaling axis. However, these TNFR-or NF-κB-dependent genes may be affected by multiple transcription factors or be regulated indirectly by other intracellular signaling pathways during O3 pathogenesis. We previously demonstrated that AP-1 and c-Jun NH2-terminal kinase 1 MAPK are associated with TNFR signaling [14]. The presence of functional AP-1 binding sites in many of the TNFR-or NF-κB1-dependent genes determined in the current study, such as chemokines, cyclins, and E2F transcription factors [72,73,74], supports indirect effects or complex interplays. p38 MAPK and its upstream epidermal growth factor receptor are also known to play key roles in transcriptional activity directly and/or via crosstalk with NF-κB for inflammation and airway hyperreactivity response by O3 [75].
In summary, the time-dependent gene expression and pathway analyses in the current study provided important insight into the downstream molecular events during the development of multi-phasic lung injury by subacute O3. Comparative transcriptome analyses defined common transcriptional profiles and potential cross talk between critical O3-related inflammatory mediators, TNFR and NF-κB, as well as TLR4. Our results increase the understanding of the molecular mechanisms of pulmonary O3 toxicity for further research.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/antiox10091489/s1.
Author Contributions
Conceptualization, methodology, and validation, H.-Y.C. and S.R.K.; software, and formal analysis, H.-Y.C., F.H.C., A.E.J., J.M. and A.K.B.; investigation and resources, H.-Y.C.; data curation, H.-Y.C. and F.H.C.; writing—original draft preparation, H.-Y.C.; writing—review and editing, H.-Y.C., F.H.C., A.E.J., J.M., A.K.B. and S.R.K.; visualization H.-Y.C.; supervision, project administration, and funding acquisition, S.R.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Intramural Research Program of the NIEHS, National Institutes of Health, Department of Health and Human Services.
Institutional Review Board Statement
All animal use in the study was approved by the Animal Care and Use Committee of NIEHS, NIH, U.S.A. (Animal Study Protocol 02-32 approved on 31 May 2017).
Informed Consent Statement
Not applicable.
Data Availability Statement
Microarray data (Gene Expression Omnibus accession numbers: GSE166399, GSE166398) are deposited in a public database repository. Data is contained within the article or supplementary material.
Acknowledgments
The authors thank Don Cook and Kevin Gerrish in NIEHS for their excellent reviews of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Peel, J.L.; Tolbert, P.E.; Klein, M.; Metzger, K.B.; Flanders, W.D.; Todd, K.; Mulholland, J.A.; Ryan, P.B.; Frumkin, H. Ambient air pollution and respiratory emergency department visits. Epidemiology 2005, 16, 164–174. [Google Scholar] [CrossRef]
- Tolbert, P.E.; Mulholland, J.A.; MacIntosh, D.L.; Xu, F.; Daniels, D.; Devine, O.J.; Carlin, B.P.; Klein, M.; Dorley, J.; Butler, A.J.; et al. Air quality and pediatric emergency room visits for asthma in Atlanta, Georgia, USA. Am. J. Epidemiol. 2000, 151, 798–810. [Google Scholar] [CrossRef]
- Bromberg, P.A. Mechanisms of the acute effects of inhaled ozone in humans. Biochim. Biophys. Acta 2016, 1860, 2771–2781. [Google Scholar] [CrossRef]
- Anenberg, S.C.; Henze, D.K.; Tinney, V.; Kinney, P.L.; Raich, W.; Fann, N.; Malley, C.S.; Roman, H.; Lamsal, L.; Duncan, B.; et al. Estimates of the Global Burden of Ambient [Formula: See text], Ozone, and [Formula: See text] on Asthma Incidence and Emergency Room Visits. Environ. Health Perspect. 2018, 126, 107004. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wang, K.; William, W.A.; Zhao, W.; Xia, Z.L. A Systematic Review and Meta-Analysis of Short-Term Ambient Ozone Exposure and COPD Hospitalizations. Int. J. Environ. Res. Public Health 2020, 17, 2130. [Google Scholar] [CrossRef] [PubMed]
- Paulin, L.M.; Gassett, A.J.; Alexis, N.E.; Kirwa, K.; Kanner, R.E.; Peters, S.; Krishnan, J.A.; Paine, R., 3rd; Dransfield, M.; Woodruff, P.G.; et al. Association of Long-term Ambient Ozone Exposure With Respiratory Morbidity in Smokers. JAMA Intern. Med. 2019, 180, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Nuvolone, D.; Petri, D.; Voller, F. The effects of ozone on human health. Environ. Sci. Pollut. Res. Int. 2018, 25, 8074–8088. [Google Scholar] [CrossRef]
- Bennett, W.D.; Hazucha, M.J.; Folinsbee, L.J.; Bromberg, P.A.; Kissling, G.E.; London, S.J. Acute pulmonary function response to ozone in young adults as a function of body mass index. Inhal. Toxicol. 2007, 19, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.A.; Rivera-Sanchez, Y.M.; Schwartzman, I.N.; Johnston, R.A. Responses to ozone are increased in obese mice. J. Appl. Physiol. 2003, 95, 938–945. [Google Scholar] [CrossRef]
- Wagner, J.G.; Barkauskas, C.E.; Vose, A.; Lewandowski, R.P.; Harkema, J.R.; Tighe, R.M. Repetitive Ozone Exposures and Evaluation of Pulmonary Inflammation and Remodeling in Diabetic Mouse Strains. Environ. Health Perspect. 2020, 128, 117009. [Google Scholar] [CrossRef]
- Andersen, Z.J.; Raaschou-Nielsen, O.; Ketzel, M.; Jensen, S.S.; Hvidberg, M.; Loft, S.; Tjonneland, A.; Overvad, K.; Sorensen, M. Diabetes incidence and long-term exposure to air pollution: A cohort study. Diabetes Care 2012, 35, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Allen, K.; Rao, X.; Ying, Z.; Braunstein, Z.; Kankanala, S.R.; Xia, C.; Wang, X.; Bramble, L.A.; Wagner, J.G.; et al. Repeated ozone exposure exacerbates insulin resistance and activates innate immune response in genetically susceptible mice. Inhal. Toxicol. 2016, 28, 383–392. [Google Scholar] [CrossRef]
- Hassett, C.; Mustafa, M.G.; Coulson, W.F.; Elashoff, R.M. Murine lung carcinogenesis following exposure to ambient ozone concentrations. J. Natl. Cancer Inst. 1985, 75, 771–777. [Google Scholar] [PubMed]
- Cho, H.Y.; Morgan, D.L.; Bauer, A.K.; Kleeberger, S.R. Signal transduction pathways of tumor necrosis factor--mediated lung injury induced by ozone in mice. Am. J. Respir. Crit. Care Med. 2007, 175, 829–839. [Google Scholar] [CrossRef]
- McCullough, S.D.; Duncan, K.E.; Swanton, S.M.; Dailey, L.A.; Diaz-Sanchez, D.; Devlin, R.B. Ozone induces a proinflammatory response in primary human bronchial epithelial cells through mitogen-activated protein kinase activation without nuclear factor-kappaB activation. Am. J. Respir. Cell Mol. Biol. 2014, 51, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Jin, Y.; Duan, L.; Yan, Z.; Wang, S.; Li, F.; Liu, Y.; Samet, J.M.; Wu, W. Regulation of ozone-induced lung inflammation by the epidermal growth factor receptor in mice. Environ. Toxicol. 2016, 31, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Kleeberger, S.R.; Reddy, S.; Zhang, L.Y.; Jedlicka, A.E. Genetic susceptibility to ozone-induced lung hyperpermeability: Role of toll-like receptor 4. Am. J. Respir. Cell Mol. Biol. 2000, 22, 620–627. [Google Scholar] [CrossRef]
- Kleeberger, S.R.; Reddy, S.P.; Zhang, L.Y.; Cho, H.Y.; Jedlicka, A.E. Toll-like receptor 4 mediates ozone-induced murine lung hyperpermeability via inducible nitric oxide synthase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L326–L333. [Google Scholar] [CrossRef]
- Kleeberger, S.R.; Levitt, R.C.; Zhang, L.Y.; Longphre, M.; Harkema, J.; Jedlicka, A.; Eleff, S.M.; DiSilvestre, D.; Holroyd, K.J. Linkage analysis of susceptibility to ozone-induced lung inflammation in inbred mice. Nat. Genet. 1997, 17, 475–478. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-kappaB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Cho, H.Y.; Zhang, L.Y.; Kleeberger, S.R. Ozone-induced lung inflammation and hyperreactivity are mediated via tumor necrosis factor-alpha receptors. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L537–L546. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.A.; Schwartzman, I.N.; Le Blanc, B.; Murthy, G.G.; Doerschuk, C.M. Tumor necrosis factor receptor 2 contributes to ozone-induced airway hyperresponsiveness in mice. Am. J. Respir. Crit. Care Med. 2001, 164, 602–607. [Google Scholar] [CrossRef][Green Version]
- Bhalla, D.K.; Reinhart, P.G.; Bai, C.; Gupta, S.K. Amelioration of ozone-induced lung injury by anti-tumor necrosis factor-alpha. Toxicol. Sci. 2002, 69, 400–408. [Google Scholar] [CrossRef]
- Li, Y.F.; Gauderman, W.J.; Avol, E.; Dubeau, L.; Gilliland, F.D. Associations of tumor necrosis factor G-308A with childhood asthma and wheezing. Am. J. Respir. Crit. Care Med. 2006, 173, 970–976. [Google Scholar] [CrossRef]
- Bauer, A.K.; Rondini, E.A.; Hummel, K.A.; Degraff, L.M.; Walker, C.; Jedlicka, A.E.; Kleeberger, S.R. Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: A role for heat-shock protein 70. Environ. Health Perspect. 2011, 119, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; van Houten, B.; Wang, X.; Miller-Degraff, L.; Fostel, J.; Gladwell, W.; Perrow, L.; Panduri, V.; Kobzik, L.; Yamamoto, M.; et al. Targeted Deletion of Nrf2 Impairs Lung Development and Oxidant Injury in Neonatal Mice. Antioxid. Redox Signal. 2012, 17, 1066–1082. [Google Scholar] [CrossRef]
- Backus, G.S.; Howden, R.; Fostel, J.; Bauer, A.K.; Cho, H.Y.; Marzec, J.; Peden, D.B.; Kleeberger, S.R. Protective role of interleukin-10 in ozone-induced pulmonary inflammation. Environ. Health Perspect. 2010, 118, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhang, X.; Edwards, J.P.; Mosser, D.M. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 2006, 281, 26041–26050. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, T.; Perkins, N.D.; Wilson, C.L. NFKB1: A suppressor of inflammation, ageing and cancer. FEBS J. 2016, 283, 1812–1822. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.C.; Liou, H.C.; Tuomanen, E.I.; Baltimore, D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 1995, 80, 321–330. [Google Scholar] [CrossRef]
- Bauer, A.K.; Travis, E.L.; Malhotra, S.S.; Rondini, E.A.; Walker, C.; Cho, H.Y.; Trivedi, S.; Gladwell, W.; Reddy, S.; Kleeberger, S.R. Identification of novel susceptibility genes in ozone-induced inflammation in mice. Eur. Respir. J. 2010, 36, 428–437. [Google Scholar] [CrossRef]
- Fry, R.C.; Rager, J.E.; Bauer, R.; Sebastian, E.; Peden, D.B.; Jaspers, I.; Alexis, N.E. Air toxics and epigenetic effects: Ozone altered microRNAs in the sputum of human subjects. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L1129–L1137. [Google Scholar] [CrossRef]
- Gohil, K.; Cross, C.E.; Last, J.A. Ozone-induced disruptions of lung transcriptomes. Biochem. Biophys. Res. Commun. 2003, 305, 719–728. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef]
- Van Ess, P.J.; Mattson, M.P.; Blouin, R.A. Enhanced induction of cytochrome P450 enzymes and CAR binding in TNF (p55(-/-)/p75(-/-)) double receptor knockout mice following phenobarbital treatment. J. Pharmacol. Exp. Ther. 2002, 300, 824–830. [Google Scholar] [CrossRef]
- Kooter, I.M.; Pennings, J.L.; Fokkens, P.H.; Leseman, D.L.; Boere, A.J.; Gerlofs-Nijland, M.E.; Cassee, F.R.; Schalk, J.A.; Orzechowski, T.J.; Schaap, M.M.; et al. Ozone induces clear cellular and molecular responses in the mouse lung independently of the transcription-coupled repair status. J. Appl. Physiol. (1985) 2007, 102, 1185–1192. [Google Scholar] [CrossRef][Green Version]
- Tovar, A.; Smith, G.J.; Thomas, J.M.; Crouse, W.L.; Harkema, J.R.; Kelada, S.N.P. Transcriptional Profiling of the Murine Airway Response to Acute Ozone Exposure. Toxicol. Sci. 2020, 173, 114–130. [Google Scholar] [CrossRef]
- Choudhary, I.; Vo, T.; Paudel, K.; Patial, S.; Saini, Y. Compartment-specific transcriptomics of ozone-exposed murine lungs reveals sex- and cell type-associated perturbations relevant to mucoinflammatory lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L99–L125. [Google Scholar] [CrossRef] [PubMed]
- Nadadur, S.S.; Costa, D.L.; Slade, R.; Silbjoris, R.; Hatch, G.E. Acute ozone-induced differential gene expression profiles in rat lung. Environ. Health Perspect. 2005, 113, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Gabehart, K.; Correll, K.A.; Yang, J.; Collins, M.L.; Loader, J.E.; Leach, S.; White, C.W.; Dakhama, A. Transcriptome profiling of the newborn mouse lung response to acute ozone exposure. Toxicol. Sci. 2014, 138, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Joy, J.; Zhou, W.; De, S.; Wood, W.H., 3rd; Becker, K.G.; Ji, H.; Sen, R. Transcriptional outcomes and kinetic patterning of gene expression in response to NF-kappaB activation. PLoS Biol. 2018, 16, e2006347. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Regulation of NF-kappaB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Yin, L.; Washington, R.; Rosenberg, D.W.; Giardina, C. The p50-p50 NF-kappaB complex as a stimulus-specific repressor of gene activation. Mol. Cell. Biochem. 2004, 265, 171–183. [Google Scholar] [CrossRef]
- Elsharkawy, A.M.; Oakley, F.; Lin, F.; Packham, G.; Mann, D.A.; Mann, J. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J. Hepatol. 2010, 53, 519–527. [Google Scholar] [CrossRef]
- Wilson, C.L.; Jurk, D.; Fullard, N.; Banks, P.; Page, A.; Luli, S.; Elsharkawy, A.M.; Gieling, R.G.; Chakraborty, J.B.; Fox, C.; et al. NFkappaB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 2015, 6, 6818. [Google Scholar] [CrossRef]
- Wongchana, W.; Palaga, T. Direct regulation of interleukin-6 expression by Notch signaling in macrophages. Cell Mol. Immunol. 2012, 9, 155–162. [Google Scholar] [CrossRef]
- Elomaa, O.; Kangas, M.; Sahlberg, C.; Tuukkanen, J.; Sormunen, R.; Liakka, A.; Thesleff, I.; Kraal, G.; Tryggvason, K. Cloning of a novel bacteria-binding receptor structurally related to scavenger receptors and expressed in a subset of macrophages. Cell 1995, 80, 603–609. [Google Scholar] [CrossRef]
- Dahl, M.; Bauer, A.K.; Arredouani, M.; Soininen, R.; Tryggvason, K.; Kleeberger, S.R.; Kobzik, L. Protection against inhaled oxidants through scavenging of oxidized lipids by macrophage receptors MARCO and SR-AI/II. J. Clin. Investig. 2007, 117, 757–764. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Uhlson, C.; Harrison, K.; Allen, C.B.; Ahmad, S.; White, C.W.; Murphy, R.C. Oxidized phospholipids derived from ozone-treated lung surfactant extract reduce macrophage and epithelial cell viability. Chem. Res. Toxicol. 2002, 15, 896–906. [Google Scholar] [CrossRef]
- Pulfer, M.K.; Murphy, R.C. Formation of biologically active oxysterols during ozonolysis of cholesterol present in lung surfactant. J. Biol. Chem. 2004, 279, 26331–26338. [Google Scholar] [CrossRef]
- Kafoury, R.M.; Pryor, W.A.; Squadrito, G.L.; Salgo, M.G.; Zou, X.; Friedman, M. Induction of inflammatory mediators in human airway epithelial cells by lipid ozonation products. Am. J. Respir. Crit. Care Med. 1999, 160, 1934–1942. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Cui, X.; Li, Z.; Teng, Y.; Barkjohn, K.K.; Norris, C.; Fang, L.; Lin, L.; Wang, Q.; Zhou, X.; et al. Malondialdehyde in Nasal Fluid: A Biomarker for Monitoring Asthma Control in Relation to Air Pollution Exposure. Environ. Sci. Technol. 2020, 54, 11405–11413. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Lin, Y.; Wang, X.; Liu, X.L.; Wang, Y.; Qin, J.; Wang, X.; Day, D.; Xiang, J.; Mo, J.; et al. Associations of ozone exposure with urinary metabolites of arachidonic acid. Environ. Int. 2020, 145, 106154. [Google Scholar] [CrossRef]
- Nakamura, Y.; Kozuka, M.; Naniwa, K.; Takabayashi, S.; Torikai, K.; Hayashi, R.; Sato, T.; Ohigashi, H.; Osawa, T. Arachidonic acid cascade inhibitors modulate phorbol ester-induced oxidative stress in female ICR mouse skin: Differential roles of 5-lipoxygenase and cyclooxygenase-2 in leukocyte infiltration and activation. Free Radic. Biol. Med. 2003, 35, 997–1007. [Google Scholar] [CrossRef]
- Miller, D.B.; Snow, S.J.; Schladweiler, M.C.; Richards, J.E.; Ghio, A.J.; Ledbetter, A.D.; Kodavanti, U.P. Acute Ozone-Induced Pulmonary and Systemic Metabolic Effects Are Diminished in Adrenalectomized Rats. Toxicol. Sci. 2016, 150, 312–322. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, D.B.; Snow, S.J.; Henriquez, A.; Schladweiler, M.C.; Ledbetter, A.D.; Richards, J.E.; Andrews, D.L.; Kodavanti, U.P. Systemic metabolic derangement, pulmonary effects, and insulin insufficiency following subchronic ozone exposure in rats. Toxicol. Appl. Pharmacol. 2016, 306, 47–57. [Google Scholar] [CrossRef]
- Shore, S.A. The Metabolic Response to Ozone. Front. Immunol. 2019, 10, 2890. [Google Scholar] [CrossRef]
- Williams, A.S.; Mathews, J.A.; Kasahara, D.I.; Chen, L.; Wurmbrand, A.P.; Si, H.; Shore, S.A. Augmented pulmonary responses to acute ozone exposure in obese mice: Roles of TNFR2 and IL-13. Environ. Health Perspect. 2013, 121, 551–557. [Google Scholar] [CrossRef]
- Johnston, R.A.; Theman, T.A.; Shore, S.A. Augmented responses to ozone in obese carboxypeptidase E-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R126–R133. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hong, Y.C. GSTM1, GSTT1, and GSTP1 polymorphisms and associations between air pollutants and markers of insulin resistance in elderly Koreans. Environ. Health Perspect. 2012, 120, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Hathout, E.H.; Beeson, W.L.; Ischander, M.; Rao, R.; Mace, J.W. Air pollution and type 1 diabetes in children. Pediatric Diabetes 2006, 7, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Zanobetti, A.; Schwartz, J. Ozone and survival in four cohorts with potentially predisposing diseases. Am. J. Respir. Crit. Care Med. 2011, 184, 836–841. [Google Scholar] [CrossRef]
- Miller, D.B.; Ghio, A.J.; Karoly, E.D.; Bell, L.N.; Snow, S.J.; Madden, M.C.; Soukup, J.; Cascio, W.E.; Gilmour, M.I.; Kodavanti, U.P. Ozone Exposure Increases Circulating Stress Hormones and Lipid Metabolites in Humans. Am. J. Respir. Crit. Care Med. 2016, 193, 1382–1391. [Google Scholar] [CrossRef]
- Castro-Giner, F.; Kogevinas, M.; Imboden, M.; de Cid, R.; Jarvis, D.; Machler, M.; Berger, W.; Burney, P.; Franklin, K.A.; Gonzalez, J.R.; et al. Joint effect of obesity and TNFA variability on asthma: Two international cohort studies. Eur. Respir. J. 2009, 33, 1003–1009. [Google Scholar] [CrossRef]
- Shore, S.A. Mechanistic Basis for Obesity-related Increases in Ozone-induced Airway Hyperresponsiveness in Mice. Ann. Am. Thorac. Soc. 2017, 14, S357–S362. [Google Scholar] [CrossRef]
- Widmer, U.; Manogue, K.R.; Cerami, A.; Sherry, B. Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1 alpha, and MIP-1 beta, members of the chemokine superfamily of proinflammatory cytokines. J. Immunol. 1993, 150, 4996–5012. [Google Scholar] [PubMed]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef]
- Vartanian, R.; Masri, J.; Martin, J.; Cloninger, C.; Holmes, B.; Artinian, N.; Funk, A.; Ruegg, T.; Gera, J. AP-1 regulates cyclin D1 and c-MYC transcription in an AKT-dependent manner in response to mTOR inhibition: Role of AIP4/Itch-mediated JUNB degradation. Mol. Cancer Res. 2011, 9, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Mumby, S.; Chung, K.F.; Adcock, I.M. Transcriptional Effects of Ozone and Impact on Airway Inflammation. Front. Immunol. 2019, 10, 1610. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).