
Cellular Redox Homeostasis



{kind=link}
Abstract
:1. Introduction
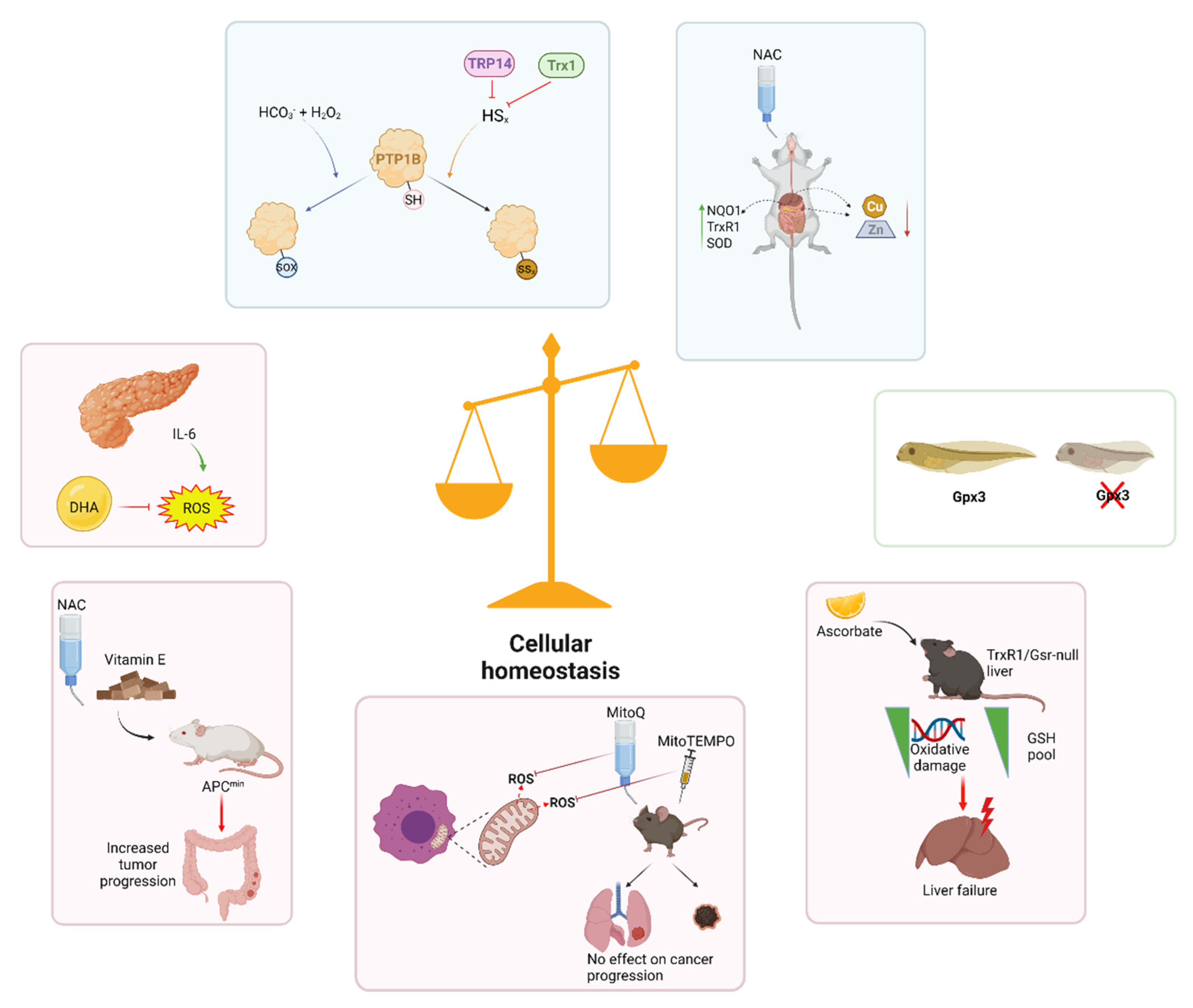
2. Cellular Signaling
3. Development
4. Disease
5. Conclusions
Funding
Conflicts of Interest
References
- Holland, H.D. The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 903–915. [Google Scholar] [CrossRef] [Green Version]
- Sessions, A.L.; Doughty, D.M.; Welander, P.V.; Summons, R.E.; Newman, D.K. The continuing puzzle of the great oxidation event. Curr. Biol. 2009, 19, R567–R574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadenas, E. Basic mechanisms of antioxidant activity. Biofactors 1997, 6, 391–397. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine, 4th ed.; Oxford Biosciences, Oxford University: Oxford, UK, 2007. [Google Scholar]
- Winyard, P.G.; Blake, D.R. Antioxidants, redox-regulated transcription factors, and inflammation. Adv. Pharmacol. 1997, 38, 403–421. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doka, E.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Takata, T.; Espinosa, B.; Nishimura, N.; Cheng, Q.; et al. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.G.; Schmidt, E.E. Sulfur Metabolism Under Stress. Antioxid. Redox Signal. 2020, 33, 1158–1173. [Google Scholar] [CrossRef]
- Dagnell, M.; Cheng, Q.; Arner, E.S.J. Qualitative Differences in Protection of PTP1B Activity by the Reductive Trx1 or TRP14 Enzyme Systems upon Oxidative Challenges with Polysulfides or H2O2 Together with Bicarbonate. Antioxidants 2021, 10, 111. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Watai, Y.; Kobayashi, A.; Nagase, H.; Mizukami, M.; McEvoy, J.; Singer, J.D.; Itoh, K.; Yamamoto, M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 2007, 12, 1163–1178. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M.; Lossow, K.; Kopp, J.F.; Schwerdtle, T.; Kipp, A.P. Crosstalk of Nrf2 with the Trace Elements Selenium, Iron, Zinc, and Copper. Nutrients 2019, 11, 2112. [Google Scholar] [CrossRef] [Green Version]
- Wolfram, T.; Schwarz, M.; Reuss, M.; Lossow, K.; Ost, M.; Klaus, S.; Schwerdtle, T.; Kipp, A.P. N-Acetylcysteine as Modulator of the Essential Trace Elements Copper and Zinc. Antioxidants 2020, 9, 1117. [Google Scholar] [CrossRef]
- Iglesias, M.J.; Terrile, M.C.; Bartoli, C.G.; D’Ippolito, S.; Casalongue, C.A. Auxin signaling participates in the adaptative response against oxidative stress and salinity by interacting with redox metabolism in Arabidopsis. Plant Mol Biol. 2010, 74, 215–222. [Google Scholar] [CrossRef]
- Igamberdiev, A.U.; Hill, R.D. Plant mitochondrial function during anaerobiosis. Ann. Bot. 2009, 103, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Sarath, G.; Hou, G.; Baird, L.M.; Mitchell, R.B. Reactive oxygen species, ABA and nitric oxide interactions on the germination of warm-season C4-grasses. Planta 2007, 226, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernoux, T.; Wilson, R.C.; Seeley, K.A.; Reichheld, J.P.; Muroy, S.; Brown, S.; Maughan, S.C.; Cobbett, C.S.; Van Montagu, M.; Van Montagu, M.; et al. The Root Meristemless1/Cadmium Sensitive2 gene defines a glutathione-dependent pathway involved in initiation and maintenance of cell division during postembryonic root development. Plant. Cell. 2000, 12, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Considine, M.J.; Foyer, C.H. Redox regulation of plant development. Antioxid. Redox Signal. 2014, 21, 1305–1326. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A.J.; Kind, K.L.; Thompson, J.G. REDOX regulation of early embryo development. Reproduction 2002, 123, 479–486. [Google Scholar] [CrossRef]
- de Matos, D.G.; Furnus, C.C. The importance of having high glutathione (GSH) level after bovine in vitro maturation on embryo development effect of beta-mercaptoethanol, cysteine and cystine. Theriogenology 2000, 53, 761–771. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Nilton, A.; Ibrahim, M.X.; Agren, P.; Larsson, E.; Petit, M.M.; Hulten, L.M.; Stahlman, M.; Johansson, B.R.; Bergo, M.O.; et al. Zfp148 deficiency causes lung maturation defects and lethality in newborn mice that are rescued by deletion of p53 or antioxidant treatment. PLoS ONE 2013, 8, e55720. [Google Scholar]
- Lee, H.; Ismail, T.; Kim, Y.; Chae, S.; Ryu, H.Y.; Lee, D.S.; Kwon, T.K.; Park, T.J.; Kwon, T.; Lee, H.S. Xenopus gpx3 Mediates Posterior Development by Regulating Cell Death during Embryogenesis. Antioxidants 2020, 9, 1265. [Google Scholar] [CrossRef]
- Fairburn, K.; Grootveld, M.; Ward, R.J.; Abiuka, C.; Kus, M.; Williams, R.B.; Winyard, P.G.; Blake, D.R. Alpha-tocopherol, lipids and lipoproteins in knee-joint synovial fluid and serum from patients with inflammatory joint disease. Clin. Sci. 1992, 83, 657–664. [Google Scholar] [CrossRef]
- Lunec, J.; Blake, D.R. The determination of dehydroascorbic acid and ascorbic acid in the serum and synovial fluid of patients with rheumatoid arthritis (RA). Free Radic. Res. Commun. 1985, 1, 31–39. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, Y.J.; Lim, J.W.; Kim, H. Docosahexaenoic Acid Induces Expression of NAD(P)H: Quinone Oxidoreductase and Heme Oxygenase-1 through Activation of Nrf2 in Cerulein-Stimulated Pancreatic Acinar Cells. Antioxidants 2020, 9, 1084. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Kundert, J.A.; Prigge, J.R.; Amato, J.A.; Perez, A.E.; Coppo, L.; Rizzo, G.N.; Kavanaugh, M.P.; Orlicky, D.J.; Shearn, C.T.; et al. Supplemental Ascorbate Diminishes DNA Damage Yet Depletes Glutathione and Increases Acute Liver Failure in a Mouse Model of Hepatic Antioxidant System Disruption. Antioxidants 2021, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, P.; Holmberg, J.; Tordsson, J.; Lu, S.; Akerstrom, B.; Holmdahl, R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat. Genet. 2003, 33, 25–32. [Google Scholar] [CrossRef]
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. The Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Chandel, N.S.; Tuveson, D.A. The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 2014, 371, 177–178. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228. [Google Scholar] [CrossRef]
- Goodman, M.; Bostick, R.M.; Kucuk, O.; Jones, D.P. Clinical trials of antioxidants as cancer prevention agents: Past, present, and future. Free Radic Biol. Med. 2011, 51, 1068–1084. [Google Scholar] [CrossRef]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L., Jr.; Valanis, B.; Williams, J.H., Jr.; et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J. Natl. Cancer Inst. 1996, 88, 1550–1559. [Google Scholar] [CrossRef]
- Westerlund, A.; Steineck, G.; Balter, K.; Stattin, P.; Gronberg, H.; Hedelin, M. Dietary supplement use patterns in men with prostate cancer: The Cancer Prostate Sweden study. Ann. Oncol. 2011, 22, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345 e22. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.V.; Le Gal, K.; El Zowalaty, A.E.; Pehlivanoglu, L.E.; Garellick, V.; Gul, N.; Ibrahim, M.X.; Bergh, P.O.; Henricsson, M.; Wiel, C.; et al. Antioxidants Promote Intestinal Tumor Progression in Mice. Antioxidants 2021, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334ra51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.H.; Brooks, A.N.; Wu, X.; Shrestha, Y.; Chouinard, C.; Piccioni, F.; Bagul, M.; Kamburov, A.; Imielinski, M.; Hogstrom, L.; et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell. 2016, 30, 214–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.L.; Papagiannakopoulos, T. The Pleiotropic Role of the KEAP1/NRF2 Pathway in Cancer. Annu. Rev. Cancer Biol. 2020, 4, 413–435. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar] [CrossRef] [PubMed]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [Green Version]
- Supinski, G.S.; Murphy, M.P.; Callahan, L.A. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1095–R1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Feng, D.; Tao, K.; Wang, R.; Shi, Y.; Qin, H.; Murphy, M.P.; Yang, Q.; Zhao, G. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1alpha. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864 Pt B, 2859–2870. [Google Scholar] [CrossRef]
- Le Gal, K.; Wiel, C.; Ibrahim, M.X.; Henricsson, M.; Sayin, V.I.; Bergo, M.O. Mitochondria-Targeted Antioxidants MitoQ and MitoTEMPO Do Not Influence BRAF-Driven Malignant Melanoma and KRAS-Driven Lung Cancer Progression in Mice. Antioxidants 2021, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilip, A.; Cheng, G.; Joseph, J.; Kunnimalaiyaan, S.; Kalyanaraman, B.; Kunnimalaiyaan, M.; Gamblin, T.C. Mitochondria-targeted antioxidant and glycolysis inhibition: Synergistic therapy in hepatocellular carcinoma. Anticancer Drugs 2013, 24, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Gal, K.; Schmidt, E.E.; Sayin, V.I. Cellular Redox Homeostasis. Antioxidants 2021, 10, 1377. https://doi.org/10.3390/antiox10091377
Le Gal K, Schmidt EE, Sayin VI. Cellular Redox Homeostasis. Antioxidants. 2021; 10(9):1377. https://doi.org/10.3390/antiox10091377
Chicago/Turabian StyleLe Gal, Kristell, Edward E. Schmidt, and Volkan I. Sayin. 2021. "Cellular Redox Homeostasis" Antioxidants 10, no. 9: 1377. https://doi.org/10.3390/antiox10091377
APA StyleLe Gal, K., Schmidt, E. E., & Sayin, V. I. (2021). Cellular Redox Homeostasis. Antioxidants, 10(9), 1377. https://doi.org/10.3390/antiox10091377