
Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype


Abstract
:1. Genetic Background of Leigh Disease
2. Cytochrome c Oxidase Deficiency and LS
3. Human SURF1 Gene and SURF1 Protein
4. Typical LS Course
5. Typical and Atypical LS Classification
6. The Possible Mechanism of the SURF1 Gene and SURF1 Protein
7. MRI Findings in Typical LS and the SURF1 Mutation
8. Peripheral Neuropathy and Myopathy in LS
9. Genotypes from the Literature and Our Patients with LS and SURF1 Mutations
10. Missense and Nonsense Distributions
11. Correlation of Phenotype and Genotype
12. Therapeutic Approaches
13. Management and Medications
13.1. Ketogenic Diet and High-Fat Diet
13.2. Non-Invasive Positive Pressure Ventilation (NIPPV)
13.3. Drugs that Should Be Avoided
13.4. Coenzyme Q (CoQ) and Vitamin Treatment
13.5. Mitochondrial Biogenesis and Gene Replacement Therapy
14. Outcomes
15. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bourgeron, T.; Rustin, P.; Chretien, D.; Birch-Machin, M.; Bourgeois, M.; Viegas-Péquignot, E.; Munnich, A.; Rötig, A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 1995, 11, 144–149. [Google Scholar]
- Parfait, B.; Chretien, D.; Rötig, A.; Marsac, C.; Munnich, A.; Rustin, P. Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum. Genet. 2000, 106, 236–243. [Google Scholar]
- Dahl, H.H.; Brown, G.K.; Brown, R.M.; Hansen, L.L.; Kerr, D.S.; Wexler, I.D.; Patel, M.S.; De Meirleir, L.; Lissens, W.; Chun, K.; et al. Mutations and polymorphisms in the pyruvate dehydrogenase E1 alpha gene. Hum. Mutat. 1992, 1, 97–102. [Google Scholar]
- Rahman, S.; Blok, R.B.; Dahl, H.H.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar]
- Wong, L.J. Pathogenic mitochondrial DNA mutations in protein-coding genes. Muscle Nerve 2007, 36, 279–293. [Google Scholar]
- Zhu, Z.; Yao, J.; Johns, T.; Fu, K.; De Bie, I.; Macmillan, C.; Cuthbert, A.P.; Newbold, R.F.; Wang, J.; Chevrette, M.; et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998, 20, 337–343. [Google Scholar]
- Zeviani, M.; Klopstock, T. Mitochondrial disorders. Curr. Opin. Neurol. 2001, 14, 553–560. [Google Scholar]
- Zeviani, M. The expanding spectrum of nuclear gene mutations in mitochondrial disorders. Semin. Cell Dev. Biol. 2001, 12, 407–416. [Google Scholar]
- Antonicka, H.; Leary, S.C.; Guercin, G.H.; Agar, J.N.; Horvath, R.; Kennaway, N.G.; Harding, C.O.; Jaksch, M.; Shoubridge, E.A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003, 12, 2693–2702. [Google Scholar]
- Shoubridge, E.A. Cytochrome c oxidase deficiency. Am. J. Med. Genet. 2001, 106, 46–52. [Google Scholar]
- Salviati, L.; Freehauf, C.; Sacconi, S.; DiMauro, S.; Thoma, J.; Tsai, A.C. Novel SURF1 mutation in a child with subacute encephalopathy and without the radiological features of Leigh Syndrome. Am. J. Med. Genet. Part A 2004, 128a, 195–198. [Google Scholar]
- Tiranti, V.; Hoertnagel, K.; Carrozzo, R.; Galimberti, C.; Munaro, M.; Granatiero, M.; Zelante, L.; Gasparini, P.; Marzella, R.; Rocchi, M.; et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar]
- Tiranti, V.; Jaksch, M.; Hofmann, S.; Galimberti, C.; Hoertnagel, K.; Lulli, L.; Freisinger, P.; Bindoff, L.; Gerbitz, K.D.; Comi, G.P.; et al. Loss-of-function mutations of SURF-1 are specifically associated with Leigh syndrome with cytochrome c oxidase deficiency. Ann. Neurol. 1999, 46, 161–166. [Google Scholar]
- Tiranti, V.; Galimberti, C.; Nijtmans, L.; Bovolenta, S.; Perini, M.P.; Zeviani, M. Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum. Mol. Genet. 1999, 8, 2533–2540. [Google Scholar]
- Tiranti, V.; Lamantea, E.; Uziel, G.; Zeviani, M.; Gasparini, P.; Marzella, R.; Rocchi, M.; Fried, M. Leigh syndrome transmitted by uniparental disomy of chromosome 9. J. Med. Genet. 1999, 36, 927–928. [Google Scholar]
- Tay, S.K.; Sacconi, S.; Akman, H.O.; Morales, J.F.; Morales, A.; De Vivo, D.C.; Shanske, S.; Bonilla, E.; DiMauro, S. Unusual clinical presentations in four cases of Leigh disease, cytochrome C oxidase deficiency, and SURF1 gene mutations. J. Child Neurol. 2005, 20, 670–674. [Google Scholar]
- Mashkevich, G.; Repetto, B.; Glerum, D.M.; Jin, C.; Tzagoloff, A. SHY1, the yeast homolog of the mammalian SURF-1 gene, encodes a mitochondrial protein required for respiration. J. Biol. Chem. 1997, 272, 14356–14364. [Google Scholar]
- Nijtmans, L.G.; Artal Sanz, M.; Bucko, M.; Farhoud, M.H.; Feenstra, M.; Hakkaart, G.A.; Zeviani, M.; Grivell, L.A. Shy1p occurs in a high molecular weight complex and is required for efficient assembly of cytochrome c oxidase in yeast. FEBS Lett. 2001, 498, 46–51. [Google Scholar]
- Rahman, S.; Brown, R.M.; Chong, W.K.; Wilson, C.J.; Brown, G.K. A SURF1 gene mutation presenting as isolated leukodystrophy. Ann. Neurol. 2001, 49, 797–800. [Google Scholar]
- Böhm, M.; Pronicka, E.; Karczmarewicz, E.; Pronicki, M.; Piekutowska-Abramczuk, D.; Sykut-Cegielska, J.; Mierzewska, H.; Hansikova, H.; Vesela, K.; Tesarova, M.; et al. Retrospective, multicentric study of 180 children with cytochrome C oxidase deficiency. Pediatr. Res. 2006, 59, 21–26. [Google Scholar]
- Bruno, C.; Biancheri, R.; Garavaglia, B.; Biedi, C.; Rossi, A.; Lamba, L.D.; Bado, M.; Greco, M.; Zeviani, M.; Minetti, C. A novel mutation in the SURF1 gene in a child with Leigh disease, peripheral neuropathy, and cytochrome-c oxidase deficiency. J. Child Neurol. 2002, 17, 233–236. [Google Scholar]
- Coenen, M.J.; Smeitink, J.A.; Farhoud, M.H.; Nijtmans, L.G.; Rodenburg, R.; Janssen, A.; van Kaauwen, E.P.; Trijbels, F.J.; van den Heuvel, L.P. The first patient diagnosed with cytochrome c oxidase deficient Leigh syndrome. Progress report. J. Inherit. Metab. Dis. 2006, 29, 212–213. [Google Scholar]
- Moslemi, A.R.; Tulinius, M.; Darin, N.; Aman, P.; Holme, E.; Oldfors, A. SURF1 gene mutations in three cases with Leigh syndrome and cytochrome c oxidase deficiency. Neurology 2003, 61, 991–993. [Google Scholar]
- Ostergaard, E.; Bradinova, I.; Ravn, S.H.; Hansen, F.J.; Simeonov, E.; Christensen, E.; Wibrand, F.; Schwartz, M. Hypertrichosis in patients with SURF1 mutations. Am. J. Med Genet. Part A 2005, 138, 384–388. [Google Scholar]
- Péquignot, M.O.; Dey, R.; Zeviani, M.; Tiranti, V.; Godinot, C.; Poyau, A.; Sue, C.; Di Mauro, S.; Abitbol, M.; Marsac, C. Mutations in the SURF1 gene associated with Leigh syndrome and cytochrome C oxidase deficiency. Hum. Mutat. 2001, 17, 374–381. [Google Scholar]
- Pecina, P.; Capková, M.; Chowdhury, S.K.; Drahota, Z.; Dubot, A.; Vojtísková, A.; Hansíková, H.; Houst’ková, H.; Zeman, J.; Godinot, C.; et al. Functional alteration of cytochrome c oxidase by SURF1 mutations in Leigh syndrome. Biochim. et Biophys. Acta 2003, 1639, 53–63. [Google Scholar]
- Poyau, A.; Buchet, K.; Bouzidi, M.F.; Zabot, M.T.; Echenne, B.; Yao, J.; Shoubridge, E.A.; Godinot, C. Missense mutations in SURF1 associated with deficient cytochrome c oxidase assembly in Leigh syndrome patients. Hum. Genet. 2000, 106, 194–205. [Google Scholar]
- Pronicka, E.; Piekutowska-Abramczuk, D.H.; Popowska, E.; Pronicki, M.; Karczmarewicz, E.; Sykut-Cegielskâ, Y.; Taybert, J. Compulsory hyperventilation and hypocapnia of patients with Leigh syndrome associated with SURF1 gene mutations as a cause of low serum bicarbonates. J. Inherit. Metab. Dis. 2001, 24, 707–714. [Google Scholar]
- Rossi, A.; Biancheri, R.; Bruno, C.; Di Rocco, M.; Calvi, A.; Pessagno, A.; Tortori-Donati, P. Leigh Syndrome with COX deficiency and SURF1 gene mutations. MR imaging findings. AJNR Am. J. Neuroradiol. 2003, 24, 1188–1191. [Google Scholar]
- Sacconi, S.; Salviati, L.; Sue, C.M.; Shanske, S.; Davidson, M.M.; Bonilla, E.; Naini, A.B.; De Vivo, D.C.; DiMauro, S. Mutation screening in patients with isolated cytochrome c oxidase deficiency. Pediatr. Res. 2003, 53, 224–230. [Google Scholar]
- Santoro, L.; Carrozzo, R.; Malandrini, A.; Piemonte, F.; Patrono, C.; Villanova, M.; Tessa, A.; Palmeri, S.; Bertini, E.; Santorelli, F.M. A novel SURF1 mutation results in Leigh syndrome with peripheral neuropathy caused by cytochrome c oxidase deficiency. Neuromuscul. Disord. NMD 2000, 10, 450–453. [Google Scholar]
- Sue, C.M.; Karadimas, C.; Checcarelli, N.; Tanji, K.; Papadopoulou, L.C.; Pallotti, F.; Guo, F.L.; Shanske, S.; Hirano, M.; De Vivo, D.C.; et al. Differential features of patients with mutations in two COX assembly genes, SURF-1 and SCO2. Ann. Neurol. 2000, 47, 589–595. [Google Scholar]
- Teraoka, M.; Yokoyama, Y.; Ninomiya, S.; Inoue, C.; Yamashita, S.; Seino, Y. Two novel mutations of SURF1 in Leigh syndrome with cytochrome c oxidase deficiency. Hum. Genet. 1999, 105, 560–563. [Google Scholar]
- van Riesen, A.K.; Antonicka, H.; Ohlenbusch, A.; Shoubridge, E.A.; Wilichowski, E.K. Maternal segmental disomy in Leigh syndrome with cytochrome c oxidase deficiency caused by homozygous SURF1 mutation. Neuropediatrics 2006, 37, 88–94. [Google Scholar]
- Von Kleist-Retzow, J.C.; Yao, J.; Taanman, J.W.; Chantrel, K.; Chretien, D.; Cormier-Daire, V.; Rotig, A.; Munnich, A.; Rustin, P.; Shoubridge, E.A. Mutations in SURF1 are not specifically associated with Leigh syndrome. J. Med. Genet. 2001, 38, 109–113. [Google Scholar]
- Williams, S.L.; Taanman, J.W.; Hansíková, H.; Houst’ková, H.; Chowdhury, S.; Zeman, J.; Houstek, J. A novel mutation in SURF1 causes skipping of exon 8 in a patient with cytochrome c oxidase-deficient leigh syndrome and hypertrichosis. Mol. Genet. Metab. 2001, 73, 340–343. [Google Scholar]
- Yüksel, A.; Seven, M.; Cetincelik, U.; Yeşil, G.; Köksal, V. Facial dysmorphism in Leigh syndrome with SURF-1 mutation and COX deficiency. Pediat.Neurol. 2006, 34, 486–489. [Google Scholar]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatry 1951, 14, 216–221. [Google Scholar]
- Willems, J.L.; Monnens, L.A.; Trijbels, J.M.; Veerkamp, J.H.; Meyer, A.E.; van Dam, K.; van Haelst, U. Leigh’s encephalomyelopathy in a patient with cytochrome c oxidase deficiency in muscle tissue. Pediatrics 1977, 60, 850–857. [Google Scholar]
- Mahdieh, N.; Soveizi, M.; Tavasoli, A.R.; Rabbani, A.; Ashrafi, M.R.; Kohlschütter, A.; Rabbani, B. Genetic testing of leukodystrophies unraveling extensive heterogeneity in a large cohort and report of five common diseases and 38 novel variants. Sci. Rep. 2021, 11, 3231. [Google Scholar]
- Lee, I.C.; El-Hattab, A.W.; Wang, J.; Li, F.Y.; Weng, S.W.; Craigen, W.J.; Wong, L.J. SURF1-associated Leigh syndrome: A case series and novel mutations. Hum. Mutat. 2012, 33, 1192–1200. [Google Scholar]
- Baden, K.N.; Murray, J.; Capaldi, R.A.; Guillemin, K. Early developmental pathology due to cytochrome c oxidase deficiency is revealed by a new zebrafish model. J. Biol. Chem. 2007, 282, 34839–34849. [Google Scholar]
- Pronicki, M.; Matyja, E.; Piekutowska-Abramczuk, D.; Szymanska-Debinska, T.; Karkucinska-Wieckowska, A.; Karczmarewicz, E.; Grajkowska, W.; Kmiec, T.; Popowska, E.; Sykut-Cegielska, J. Light and electron microscopy characteristics of the muscle of patients with SURF1 gene mutations associated with Leigh disease. J. Clin. Pathol. 2008, 61, 460–466. [Google Scholar]
- Darin, N.; Moslemi, A.R.; Lebon, S.; Rustin, P.; Holme, E.; Oldfors, A.; Tulinius, M. Genotypes and clinical phenotypes in children with cytochrome-c oxidase deficiency. Neuropediatrics 2003, 34, 311–317. [Google Scholar]
- Ruhoy, I.S.; Saneto, R.P. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 221–234. [Google Scholar]
- Gerards, M.; Sallevelt, S.C.; Smeets, H.J. Leigh syndrome: Resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol. Genet. Metab. 2016, 117, 300–312. [Google Scholar]
- Sofou, K.; de Coo, I.F.M.; Ostergaard, E.; Isohanni, P.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; Lönnqvist, T.; Bindoff, L.A.; et al. Phenotype-genotype correlations in Leigh syndrome: New insights from a multicentre study of 96 patients. J. Med. Genet. 2018, 55, 21–27. [Google Scholar]
- Ogawa, E.; Fushimi, T.; Ogawa-Tominaga, M.; Shimura, M.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; et al. Mortality of Japanese patients with Leigh syndrome: Effects of age at onset and genetic diagnosis. J. Inherit. Metab. Dis. 2020, 43, 819–826. [Google Scholar]
- Yang, Y.L.; Sun, F.; Zhang, Y.; Qian, N.; Yuan, Y.; Wang, Z.X.; Qi, Y.; Xiao, J.X.; Wang, X.Y.; Qi, Z.Y.; et al. Clinical and laboratory survey of 65 Chinese patients with Leigh syndrome. Chin. Med. J. 2006, 119, 373–377. [Google Scholar]
- Zhang, Y.; Yang, Y.L.; Sun, F.; Cai, X.; Qian, N.; Yuan, Y.; Wang, Z.X.; Qi, Y.; Xiao, J.X.; Wang, X.Y.; et al. Clinical and molecular survey in 124 Chinese patients with Leigh or Leigh-like syndrome. J. Inherit. Metab. Dis. 2007, 30, 265. [Google Scholar]
- Wolf, U. Identical mutations and phenotypic variation. Hum. Genet. 1997, 100, 305–321. [Google Scholar]
- Kose, M.; Canda, E.; Kagnici, M.; Aykut, A.; Adebali, O.; Durmaz, A.; Bircan, A.; Diniz, G.; Eraslan, C.; Kose, E.; et al. SURF1 related Leigh syndrome: Clinical and molecular findings of 16 patients from Turkey. Mol. Genet. Metab. Rep. 2020, 25, 100657. [Google Scholar]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar]
- Finsterer, J. Treatment of central nervous system manifestations in mitochondrial disorders. Eur. J. Neurol. 2011, 18, 28–38. [Google Scholar]
- Kretzschmar, H.A.; DeArmond, S.J.; Koch, T.K.; Patel, M.S.; Newth, C.J.; Schmidt, K.A.; Packman, S. Pyruvate dehydrogenase complex deficiency as a cause of subacute necrotizing encephalopathy (Leigh disease). Pediatrics 1987, 79, 370–373. [Google Scholar]
- Horvath, R. Update on clinical aspects and treatment of selected vitamin-responsive disorders II (riboflavin and CoQ 10). J. Inherit. Metab. Dis. 2012, 35, 679–687. [Google Scholar]
- Wexler, I.D.; Hemalatha, S.G.; McConnell, J.; Buist, N.R.; Dahl, H.H.; Berry, S.A.; Cederbaum, S.D.; Patel, M.S.; Kerr, D.S. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology 1997, 49, 1655–1661. [Google Scholar]
- Kang, H.C.; Lee, Y.M.; Kim, H.D.; Lee, J.S.; Slama, A. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia 2007, 48, 82–88. [Google Scholar]
- Panetta, J.; Smith, L.J.; Boneh, A. Effect of high-dose vitamins, coenzyme Q and high-fat diet in paediatric patients with mitochondrial diseases. J. Inherit. Metab. Dis. 2004, 27, 487–498. [Google Scholar]
- Qu, C.; Keijer, J.; Adjobo-Hermans, M.J.; van de Wal, M.; Schirris, T.; van Karnebeek, C.; Pan, Y.; Koopman, W.J. The ketogenic diet as a therapeutic intervention strategy in mitochondrial disease. Int. J. Biochem. Cell Biol. 2021, 138, 106050. [Google Scholar]
- Ortigoza-Escobar, J.D.; Molero-Luis, M.; Arias, A.; Oyarzabal, A.; Darín, N.; Serrano, M.; Garcia-Cazorla, A.; Tondo, M.; Hernández, M.; Garcia-Villoria, J.; et al. Free-thiamine is a potential biomarker of thiamine transporter-2 deficiency. A treatable cause of Leigh syndrome. Brain 2016, 139 Pt 1, 31–38. [Google Scholar]
- Zweers, H.; van Wegberg, A.M.J.; Janssen, M.C.H.; Wortmann, S.B. Ketogenic diet for mitochondrial disease: A systematic review on efficacy and safety. Orphanet J. Rare Dis. 2021, 16, 295. [Google Scholar]
- Saneto, R.P.; Lee, I.C.; Koenig, M.K.; Bao, X.; Weng, S.W.; Naviaux, R.K.; Wong, L.J. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure 2010, 19, 140–146. [Google Scholar]
- Suomalainen, A. Therapy for mitochondrial disorders: Little proof, high research activity, some promise. Semin. Fetal Neonatal Med. 2011, 16, 236–240. [Google Scholar]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V. EPI-743 reverses the progression of the pediatric mitochondrial disease—genetically defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar]
- Tabarki, B.; Al-Shafi, S.; Al-Shahwan, S.; Azmat, Z.; Al-Hashem, A.; Al-Adwani, N.; Biary, N.; Al-Zawahmah, M.; Khan, S.; Zuccoli, G. Biotin-responsive basal ganglia disease revisited: Clinical, radiologic, and genetic findings. Neurology 2013, 80, 261–267. [Google Scholar]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar]
- Kerr, D.S. Treatment of mitochondrial electron transport chain disorders: A review of clinical trials over the past decade. Mol. Genet. Metab. 2010, 99, 246–255. [Google Scholar]
- Mancuso, M.; Orsucci, D.; Calsolaro, V.; Choub, A.; Siciliano, G. Coenzyme Q10 and Neurological Diseases. Pharmaceuticals 2009, 2, 134–149. [Google Scholar]
- Bar-Meir, M.; Elpeleg, O.N.; Saada, A. Effect of various agents on adenosine triphosphate synthesis in mitochondrial complex I deficiency. J. Pediatr. 2001, 139, 868–870. [Google Scholar]
- Mani, S.; Chandak, G.; Singh, K.K.; Singh, R.; Rao, S.N. Novel p. P298L SURF1 mutation in thiamine deficient Leigh syndrome patients compromises cytochrome c oxidase activity. Mitochondrion 2020, 53, 91–98. [Google Scholar]
- Gerards, M.; van den Bosch, B.J.; Danhauser, K.; Serre, V.; van Weeghel, M.; Wanders, R.J.; Nicolaes, G.A.; Sluiter, W.; Schoonderwoerd, K.; Scholte, H.R.; et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: New function for an old gene. Brain 2011, 134 Pt 1, 210–219. [Google Scholar]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar]
- Debs, R.; Depienne, C.; Rastetter, A.; Bellanger, A.; Degos, B.; Galanaud, D.; Keren, B.; Lyon-Caen, O.; Brice, A.; Sedel, F. Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Arch. Neurol. 2010, 67, 126–130. [Google Scholar]
- Pinard, J.M.; Marsac, C.; Barkaoui, E.; Desguerre, I.; Birch-Machin, M.; Reinert, P.; Ponsot, G. Leigh syndrome and leukodystrophy due to partial succinate dehydrogenase deficiency: Regression with riboflavin. Arch. Pediatrie Organe Off. Soc. Fr. Pediatrie 1999, 6, 421–426. [Google Scholar]
- Garone, C.; Donati, M.A.; Sacchini, M.; Garcia-Diaz, B.; Bruno, C.; Calvo, S.; Mootha, V.K.; Dimauro, S. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol. 2013, 70, 1177–1179. [Google Scholar]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar]
- Ramaekers, V.; Weis, J.; Sequeira, J.; Quadros, E.; Blau, N. Mitochondrial complex I encephalomyopathy and cerebral 5-methyltetrahydrofolate deficiency. Neuropediatrics 2007, 38, 184–187. [Google Scholar]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar]
- Song, C.; Li, M.; Xu, L.; Shen, Y.; Yang, H.; Ding, M.; Liu, X.; Xie, Z. Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport 2018, 29, 1517–1524. [Google Scholar]
- Day, J.W.; Finkel, R.S.; Chiriboga, C.A.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021, 20, 284–293. [Google Scholar]
- Ling, Q.; Rioux, M.; Hu, Y.; Lee, M.; Gray, S.J. Adeno-associated viral vector serotype 9-based gene replacement therapy for SURF1-related Leigh syndrome. Mol. Ther. Methods Clin. Dev. 2021, 23, 158–168. [Google Scholar]
- Vasta, V.; Merritt, J.L.; 2nd Saneto, R.P.; Hahn, S.H. Next-generation sequencing for mitochondrial diseases: A wide diagnostic spectrum. Pediatr. Int. Off. J. Jpn. Pediat.Soc. 2012, 54, 585–601. [Google Scholar]
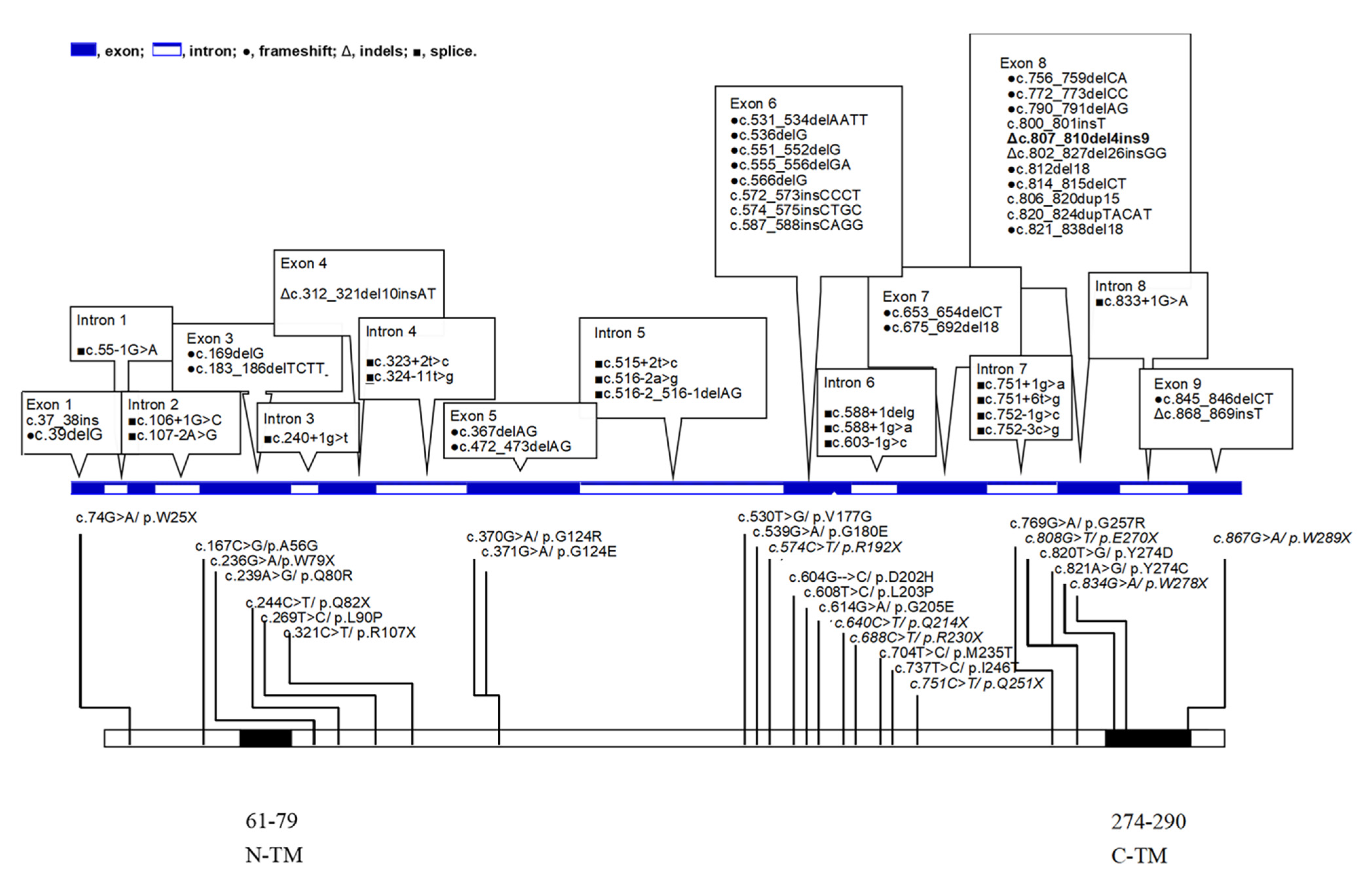

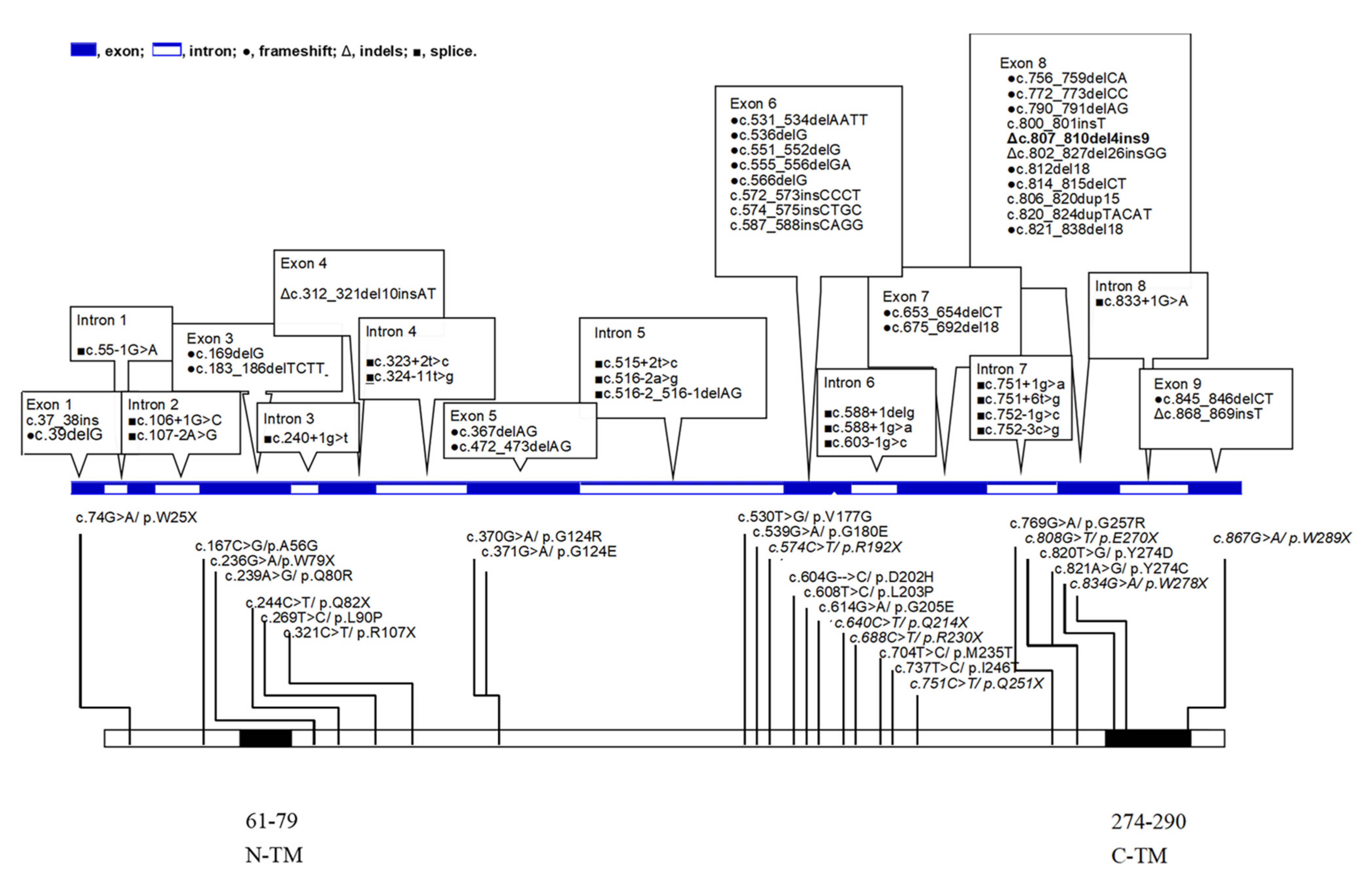{kind=link}
| DNA Change (Allele 1/Allele 2) | Age(y) (Dx/Alive/ Died/Gender) | Phenotype | Matched Criteria | Lactate/ Pyruvate | CT/MRI | Ethnicity | COX Activity | Reference |
|---|---|---|---|---|---|---|---|---|
| c.867G>A/ c.G867G>A | 2/died 5/M | Early motor delay, hypotonia, ataxia, tongue fasciculation, hypertrichosis, apnea with sudden death at 5 years old | 3/4 | +/NA | Caudal part of medulla, cerebellum | German | <30% | [34] |
| c.312del10insAT/ c.312del10insAT | 3/died 9/M | Mental retardation, ataxia, cannot walk, ophthalmoplegia, central apnea, died due to diazepam injection at 9 years old | 2/4 | +/NA | CT: normal | Danish | 5–18% | [24] |
| c.312-321delinsAT/ c.572_573insCCCT | 3.5/alive 10/M | MR, brain stem signs, tube feeding, wheelchair dependency | 2/4 | −/− | mild MRI change | NA | NA | [11] |
| c.790-791delAG/ c.790-791delAG | 2/alive 2/F | Early motor delay, failure to thrive, microcephaly, short stature, regression, hypertrichosis | 2/4 | +/+ | Leukodystrophy, dentate nucleus., medulla | NA | <30% | [19] |
| c.618G>C/ c.751C>T | 2/died 7.8/F | Hypotonia, tremor, ataxia and deafness, brain stem signs, and progressive | 2/4 | −/NA | Leukodystrophy | French | NA | [27] |
| c.688C>T/ c.751+1G>A | 0.4/died 2.1/M | Feeding difficulties, short stature, muscle weakness, nystagmus | 3/4 | + | Autopsy at 5 years old, brain stem | NA | <30% | [23] |
| c.653_54delCT/ c.807_810del4ins9 | 22/alive 22/F | Ataxia, ophthalmoparesis, hearing loss, short stature | 2/4 | − | Basal ganglia | Asian | Decreased | [41] |
| c.169delG/ c.530T>G | 1.8/alive 1.8/M | Developmental delay, muscle weakness, hypotonia, seizures, ptosis, dysmorphism, episodic coma | 2/4 | +/+ | Leukodystrophy | Hispanic | NA | [41] |
| c.324-11T>G/ c.324-11T>G | 2/alive 2/F | Developmental regression, failure to thrive, microcephaly | 2/4 | −/− | Leukodystrophy | Asian | NA | [41] |
| c.167C> G/ c.751+6T>C | 10/alive 10/M | Mental retardation, ataxia, dystonia chorea, intractable seizures | 2/4 | −/+ | Basal ganglia, leukodystrophy | Caucasian | 32% | [41] |
| c.688C>T/ c.806_820dup | 1.3/alive 10/M | Feeding intolerance, short stature, muscle weakness since 1 year, ophthalmoplegia, dystonia, choreoathetosis, ataxia, hirsutism, alive at 10 years old | 3/4 | +/NA | Swedish | <25% | [23] |
| DNA Change (Allele 1/Allele 2) | Age (y) Dx/Alive/Died | Caudate Nulceus (62%) | Subthalamic Nulceus (54%) | Substantia Nigra (62%) | Tecmentum (46%) | Dentate Nulceus (46%) | Cerebellar White Matter (38%) | Medulla/S. Cord (62%) | Cerebral White Matter (8%) | Referrence |
|---|---|---|---|---|---|---|---|---|---|---|
| c.244C>T/ c.244C>T | 2/alive 2 | + | + | + | +/− | + | [37] | |||
| c.530T>G/ c.530T>G | 1.9/alive 3 | + | + | + | [37] | |||||
| c.312del10insAT/ c.688C>T | 1.5/alive 1.5 | + | + | + | + | [17] | ||||
| c.312del10insAT/ c.845delCT | NA/alive 2.6 | + | + | + | + | + | + | [24] | ||
| c.312del10insAT/ c.688C>T | NA/alive 0.8 | + | [24] | |||||||
| c.312del10insAT/ c.820_824dupTACAT | NA/alive 0.8 | + | [16] | |||||||
| c.240 + 1G>T/ c.531_534del AAAT | NA/died 1.8 | + | + | + | + | + | [29] | |||
| c.320del10insAT/ c.320 del10insAt | 1.3/alive 6.7 | + | + | + | + | + | + | [23] | ||
| c.566delG | 1/alive 3 | + | + | + | + | +/+ | [29] | |||
| c.772_773delCC/ c.772_773delCC | 1/alive 4 | + | + | + | + | + | + | + | [29] | |
| c.240 + 1G>T/ c.531_534delAAAT | 1.3/died 1.8 | + | + | [21] | ||||||
| c.320del10insAT/ c.812del18 | 1/died 2.8 | + | [36] | |||||||
| c.790delAG/ c.820T>G | 0.8/alive 1.6 | + | + | + | [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, I.-C.; Chiang, K.-L. Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype. Antioxidants 2021, 10, 1950. https://doi.org/10.3390/antiox10121950
Lee I-C, Chiang K-L. Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype. Antioxidants. 2021; 10(12):1950. https://doi.org/10.3390/antiox10121950
Chicago/Turabian StyleLee, Inn-Chi, and Kuo-Liang Chiang. 2021. "Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype" Antioxidants 10, no. 12: 1950. https://doi.org/10.3390/antiox10121950
APA StyleLee, I.-C., & Chiang, K.-L. (2021). Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype. Antioxidants, 10(12), 1950. https://doi.org/10.3390/antiox10121950