
Dietary Iron Overload and Hfe−/− Related Hemochromatosis Alter Hepatic Mitochondrial Function

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. High-Resolution Respirometry
2.3. Plasma and Total Tissue Iron
2.4. Prussian Blue Staining
2.5. Isolation of Mitochondria
2.6. RNA Extraction and Quantitative Real-Time PCR
2.7. Protein Extraction and Western Blot
2.8. Aconitase and Citrate Synthase Activity
2.9. Lactate and ATP Concentration
2.10. Mitochondrial DNA Copy Number
2.11. Flow Cytometry and Fluorescence Microscopy
2.12. Statistics
3. Results and Discussion
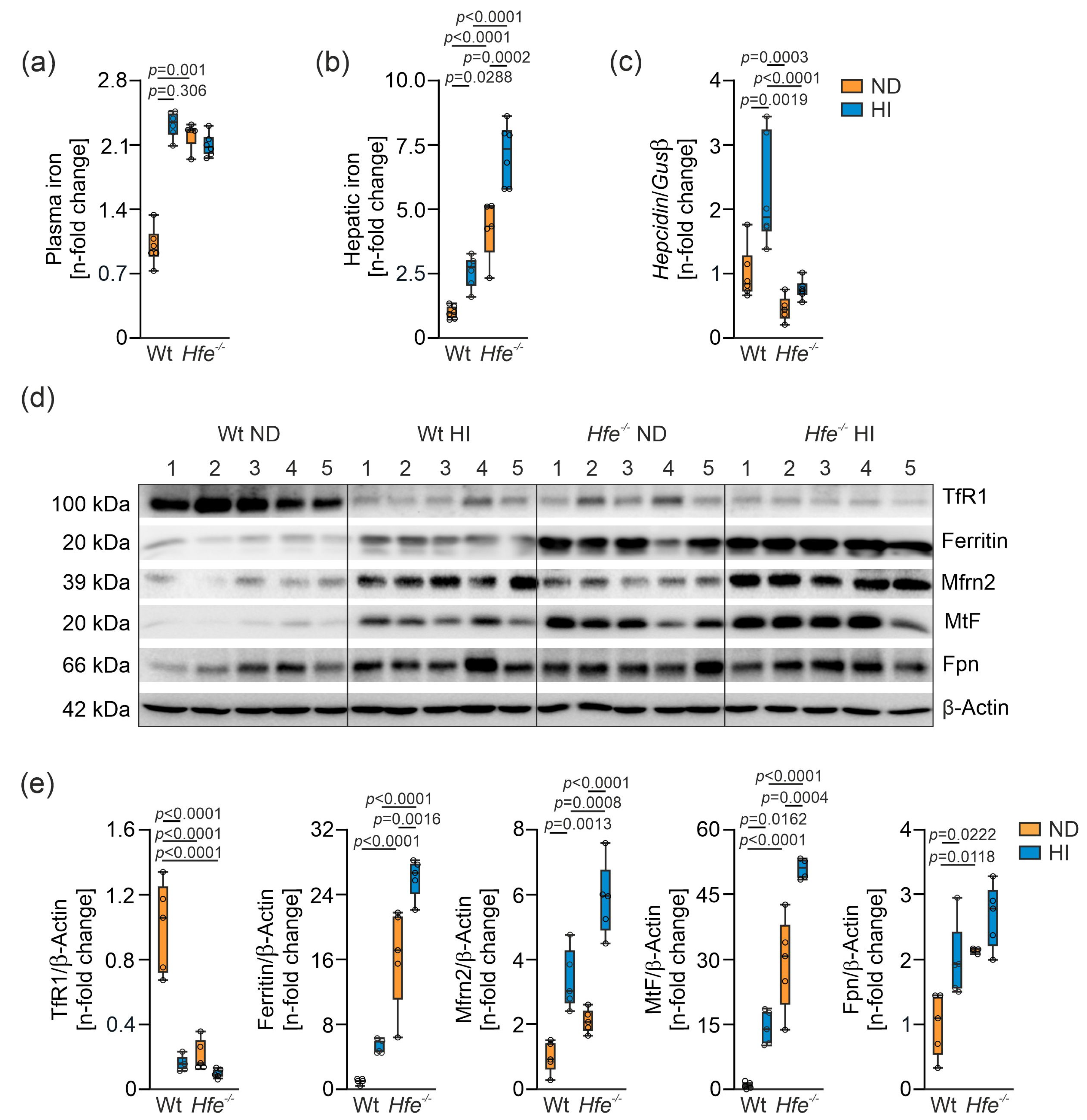
3.1. Alteration of Systemic and Cellular Iron Metabolism by Dietary and Genetic Iron Overload
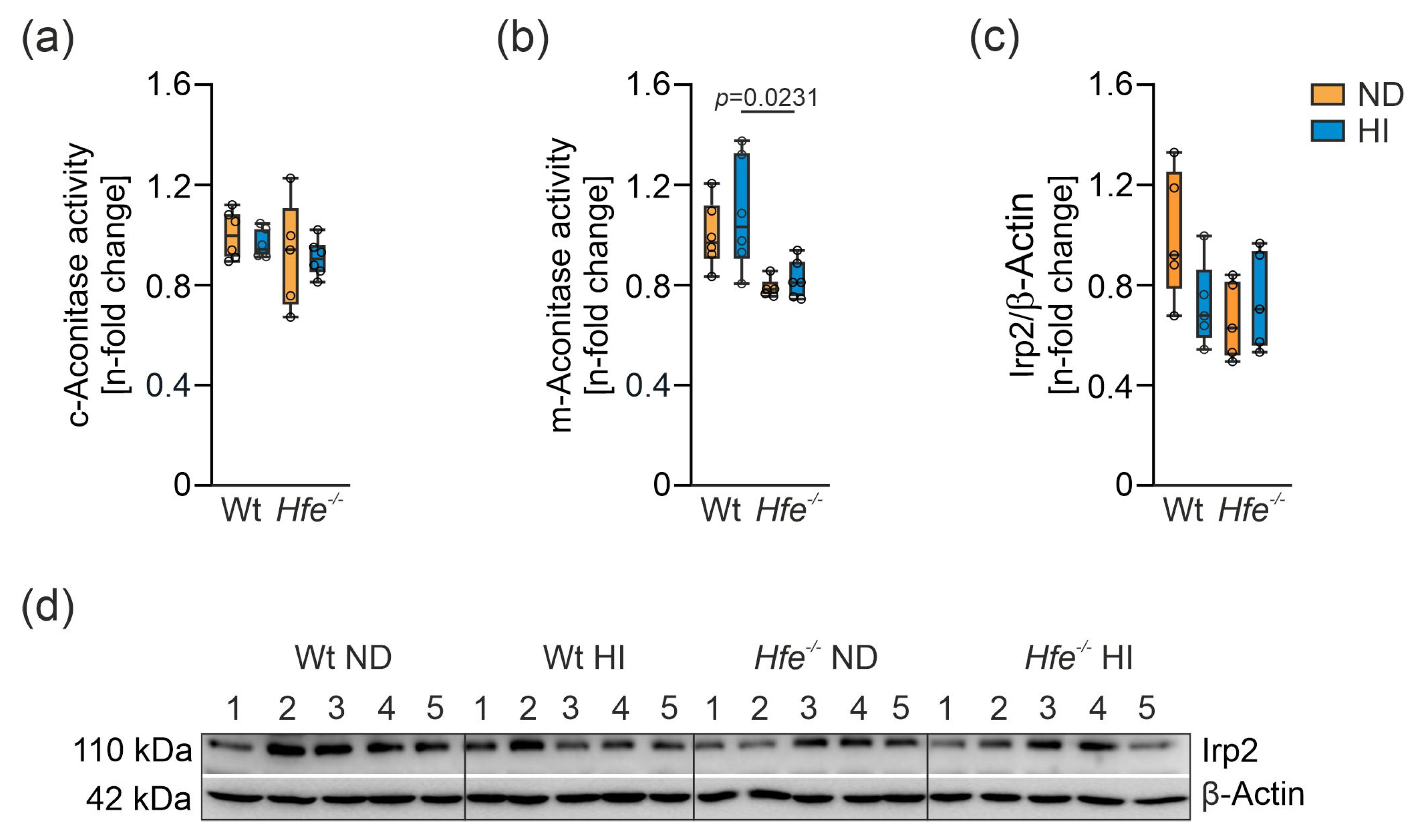
3.2. Iron Content in Liver Mitochondria of Hfe−/− mice was Increased and m-Aconitase Activity Was Decreased
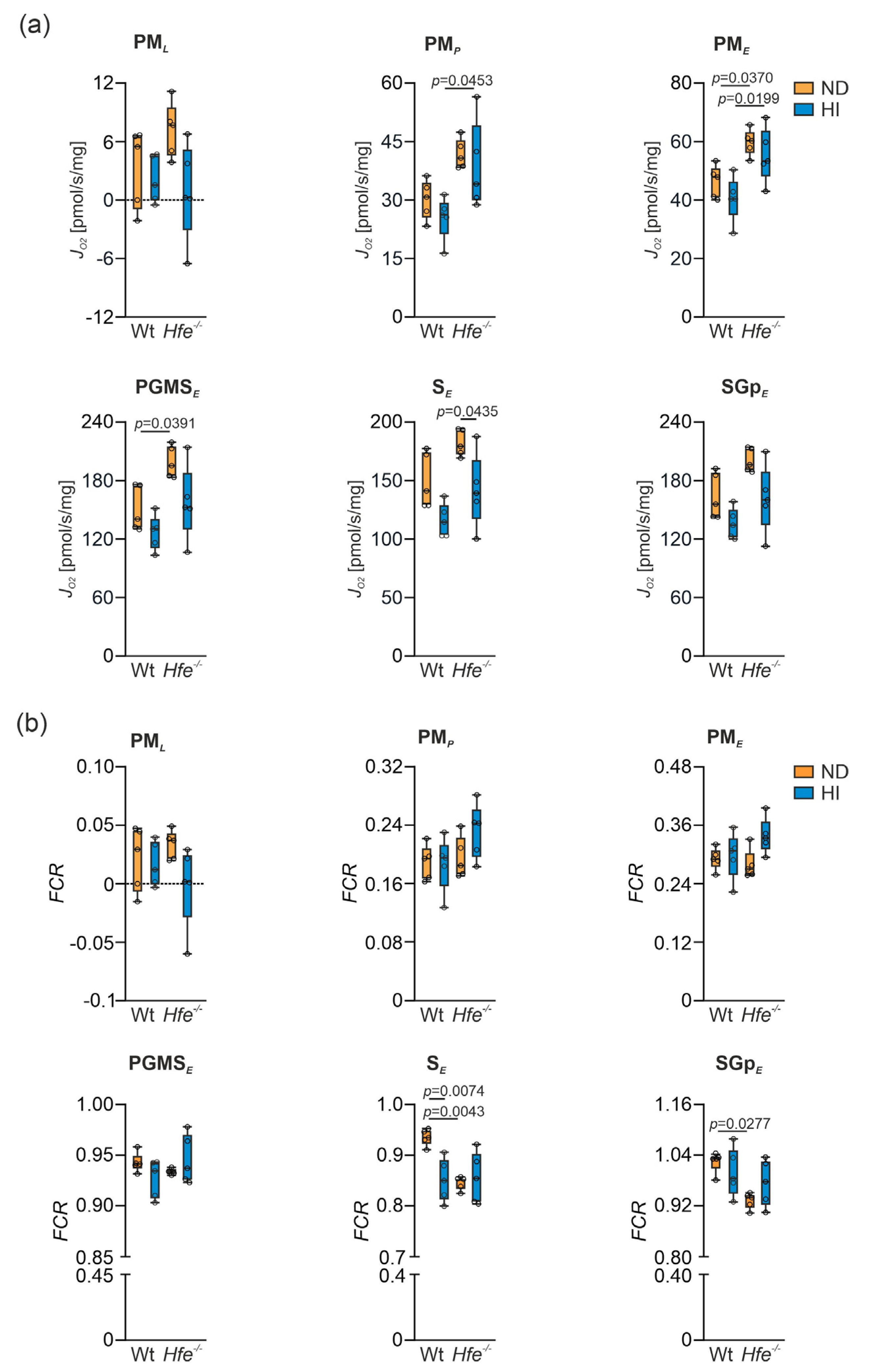
3.3. Mitochondrial Respiration in Wt and Hfe−/− Mice on ND and HI
3.4. Liver Mitochondrial Density Was Unaltered but Protein Expression of Electron Transfer Complexes Was Slightly Upregulated in Genetic Combined with Dietary Iron Overload
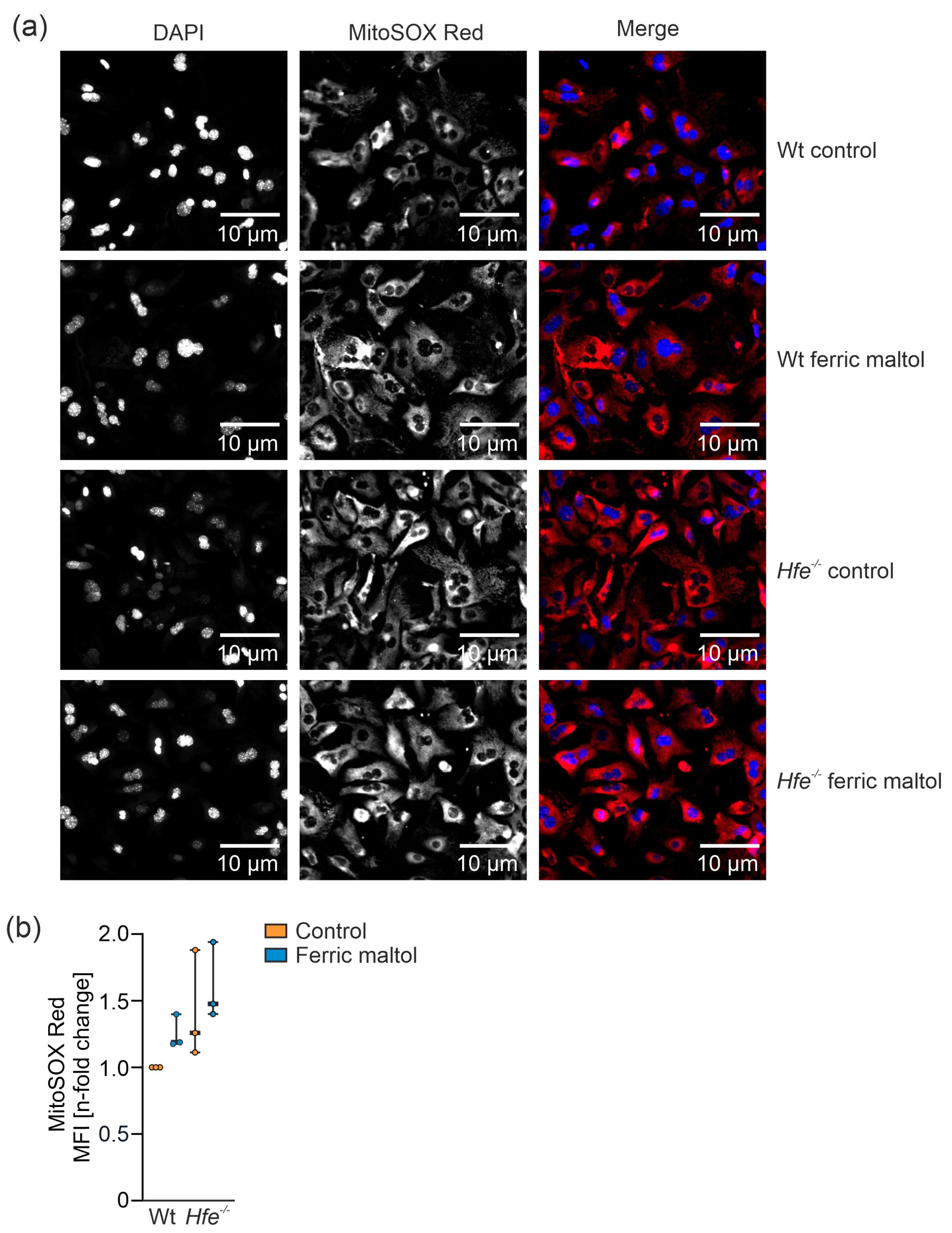
3.5. Dietary and Genetic Iron Overload Increased Liver Mitochondrial Superoxide Formation
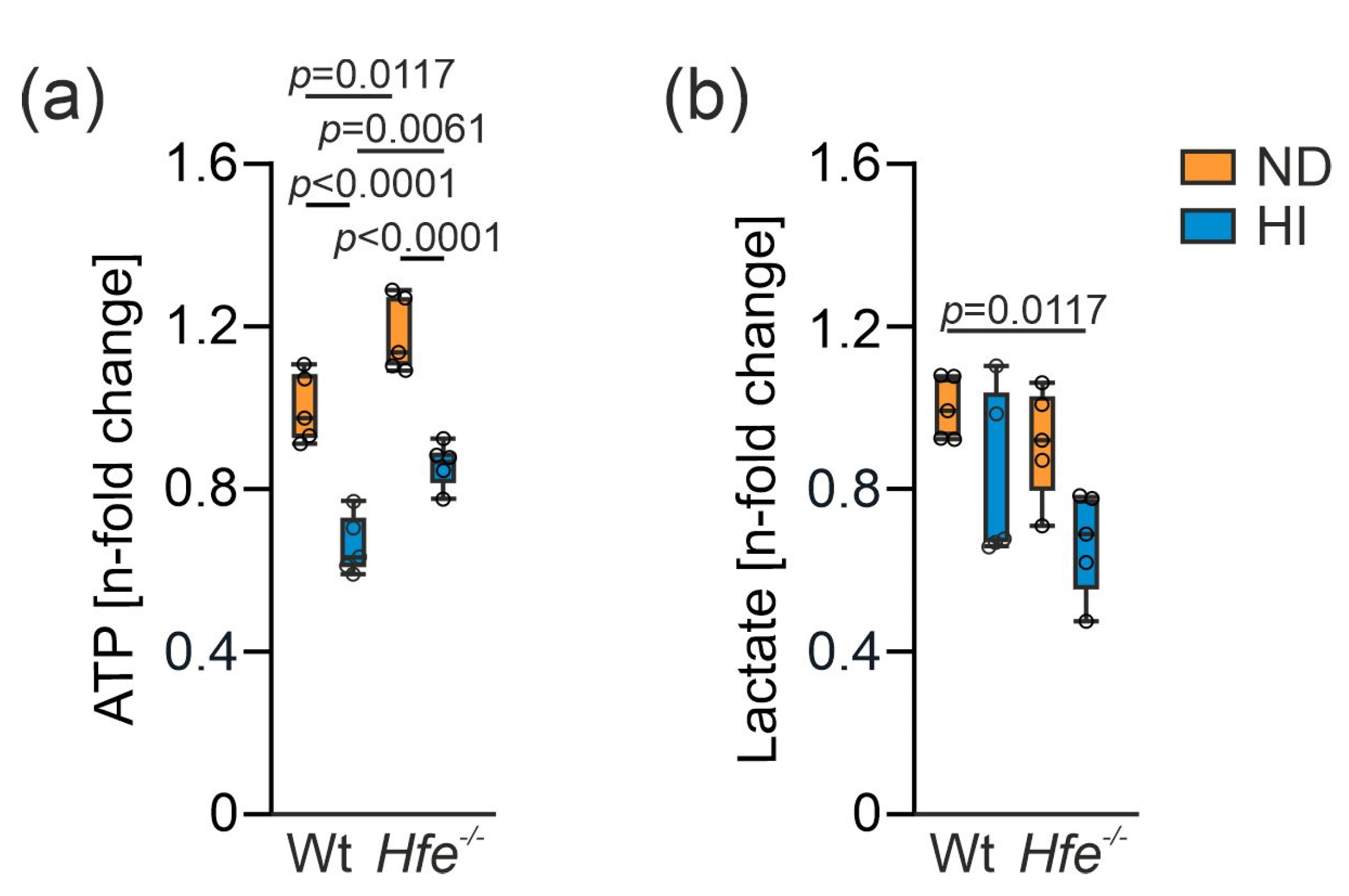
3.6. Summary
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A. Mitochondrial iron overload: Causes and consequences. Curr. Opin. Genet. Dev. 2016, 38, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Lill, R. Function and biogenesis of iron–sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.R.; Lane, D.J.R.; Becker, E.M.; Huang, M.L.H.; Whitnall, M.; Rahmanto, Y.S.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775. [Google Scholar] [CrossRef] [Green Version]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Mészáros, A.T.; Gnaiger, E. High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. Methods Mol. Biol. 2018, 1782, 31–70. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E.; MitoEAGLE TASK Group. Mitochondrial physiology. Bioenerg. Commun. 2020, 2020.1, 1–44. [Google Scholar] [CrossRef]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. An Introduction to OXPHOS Analysis, 5th ed. Bioenerg. Commun. 2020, 2020, 112. [Google Scholar] [CrossRef]
- Cairo, G.; Recalcati, S.; Pietrangelo, A.; Minotti, G. The iron regulatory proteins: Targets and modulators of free radical reactions and oxidative damage. Free Radic. Biol. Med. 2002, 32, 1237–1243. [Google Scholar] [CrossRef]
- Pietrangelo, A. Iron and the liver. Liver Int. 2016, 36 (Suppl. S1), 116–123. [Google Scholar] [CrossRef]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta BBA-Bioenerg. 1999, 1413, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volani, C.; Doerrier, C.; Demetz, E.; Haschka, D.; Paglia, G.; Lavdas, A.A.; Gnaiger, E.; Weiss, G. Dietary iron loading negatively affects liver mitochondrial function. Metallomics 2017, 9, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Primers 2018, 4, 18016. [Google Scholar] [CrossRef]
- Weiss, G. Genetic mechanisms and modifying factors in hereditary hemochromatosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Britton, R.S. Hepatic injury in chronic iron overload. Role of lipid peroxidation. Chem.-Biol. Interact. 1989, 70, 183–226. [Google Scholar] [CrossRef]
- Niemelä, O.; Parkkila, S.; Britton, R.S.; Brunt, E.; Janney, C.; Bacon, B. Hepatic lipid peroxidation in hereditary hemochromatosis and alcoholic liver injury. J. Lab. Clin. Med. 1999, 133, 451–460. [Google Scholar] [CrossRef]
- Sumneang, N.; Siri-Angkul, N.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch. Biochem. Biophys. 2020, 680, 108241. [Google Scholar] [CrossRef]
- Deugnier, Y.; Morcet, J.; Lainé, F.; Hamdi-Roze, H.; Bollard, A.-S.; Guyader, D.; Moirand, R.; Bardou-Jacquet, E. Reduced phenotypic expression in genetic hemochromatosis with time: Role of exposure to non-genetic modifiers. J. Hepatol. 2019, 70, 118–125. [Google Scholar] [CrossRef]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I–like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- Prabhu, A.; Cargill, T.; Roberts, N.; Ryan, J.D. Systematic Review of the Clinical Outcomes of Iron Reduction in Hereditary Hemochromatosis. Hepatology 2020, 72, 1469–1482. [Google Scholar] [CrossRef]
- Zoller, H.; Theurl, I.; Koch, R.O.; McKie, A.T.; Vogel, W.; Weiss, G. Duodenal cytochrome b and hephaestin expression in patients with iron deficiency and hemochromatosis. Gastroenterology 2003, 125, 746–754. [Google Scholar] [CrossRef]
- Theurl, I.; Ludwiczek, S.; Eller, P.; Seifert, M.; Artner, E.; Brunner, P.; Weiss, G. Pathways for the regulation of body iron homeostasis in response to experimental iron overload. J. Hepatol. 2005, 43, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Ludwiczek, S.; Theurl, I.; Bahram, S.; Schümann, K.; Weiss, G. Regulatory networks for the control of body iron homeostasis and their dysregulation in HFE mediated hemochromatosis. J. Cell. Physiol. 2005, 204, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Jouihan, H.A.; Cobine, P.A.; Cooksey, R.C.; Hoagland, E.A.; Boudina, S.; Abel, E.D.; Winge, D.R.; McClain, D.A. Iron-mediated inhibition of mitochondrial manganese uptake mediates mitochondrial dysfunction in a mouse model of hemochromatosis. Mol. Med. 2008, 14, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; O’Neill, R.; Park, C.H. Iron-induced peroxidative injury to isolated rat hepatic mitochondria. J. Free Radic. Biol. Med. 1986, 2, 339–347. [Google Scholar] [CrossRef]
- Saporito-Magriñá, C.; Musacco-Sebio, R.; Acosta, J.M.; Bajicoff, S.; Paredes-Fleitas, P.; Boveris, A.; Repetto, M.G. Rat liver mitochondrial dysfunction by addition of copper(II) or iron(III) ions. J. Inorg. Biochem. 2017, 166, 5–11. [Google Scholar] [CrossRef]
- Bahram, S.; Gilfillan, S.; Kühn, L.C.; Moret, R.; Schulze, J.B.; Lebeau, A.; Schümann, K. Experimental hemochromatosis due to MHC class I HFE deficiency: Immune status and iron metabolism. Proc. Natl. Acad. Sci. USA 1999, 96, 13312–13317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, C.J.; Ostini, L.; Wallace, D.F.; John, A.N.; Watters, D.J.; Subramaniam, V.N. Iron loading and oxidative stress in the Atm−/−mouse liver. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 300, G554–G560. [Google Scholar] [CrossRef] [Green Version]
- Volani, C.; Paglia, G.; Smarason, S.V.; Pramstaller, P.P.; Demetz, E.; Pfeifhofer-Obermair, C.; Weiss, G. Metabolic Signature of Dietary Iron Overload in a Mouse Model. Cells 2018, 7, 264. [Google Scholar] [CrossRef] [Green Version]
- National Research Council. Nutrient Requirements of Laboratory Animals, 4th ed.; The National Academies Press: Washington, DC, USA, 1995; p. 192. [Google Scholar] [CrossRef]
- Gnaiger, E. Bioenergetics at low oxygen: Dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir. Physiol. 2001, 128, 277–297. [Google Scholar] [CrossRef]
- Sonnweber, T.; Ress, C.; Nairz, M.; Theurl, I.; Schroll, A.; Murphy, A.T.; Wroblewski, V.; Witcher, D.R.; Moser, P.; Ebenbichler, C.F.; et al. High-fat diet causes iron deficiency via hepcidin-independent reduction of duodenal iron absorption. J. Nutr. Biochem. 2012, 23, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Demetz, E.; Tymoszuk, P.; Hilbe, R.; Volani, C.; Haschka, D.; Heim, C.; Auer, K.; Lener, D.; Zeiger, L.B.; Pfeifhofer-Obermair, C.; et al. The haemochromatosis gene Hfe and Kupffer cells control LDL cholesterol homeostasis and impact on atherosclerosis development. Eur. Heart J. 2020, 41, 3949–3959. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef]
- Brigo, N.; Pfeifhofer-Obermair, C.; Tymoszuk, P.; Demetz, E.; Engl, S.; Barros-Pinkelnig, M.; Dichtl, S.; Fischer, C.; De Souza, L.V.; Petzer, V.; et al. Cytokine-Mediated Regulation of ARG1 in Macrophages and Its Impact on the Control of Salmonella enterica Serovar Typhimurium Infection. Cells 2021, 10, 1823. [Google Scholar] [CrossRef] [PubMed]
- Ludwiczek, S.; Theurl, I.; Artner-Dworzak, E.; Chorney, M.; Weiss, G. Duodenal HFE expression and hepcidin levels determine body iron homeostasis: Modulation by genetic diversity and dietary iron availability. J. Mol. Med. 2004, 82, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Fazzini, F.; Schöpf, B.; Blatzer, M.; Coassin, S.; Hicks, A.A.; Kronenberg, F.; Fendt, L. Plasmid-normalized quantification of relative mitochondrial DNA copy number. Sci. Rep. 2018, 8, 15347. [Google Scholar] [CrossRef]
- Singh, U.A.; Kumari, M.; Iyengar, S. Method for improving the quality of genomic DNA obtained from minute quantities of tissue and blood samples using Chelex 100 resin. Biol. Proced. Online 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Theurl, M.; Theurl, I.; Hochegger, K.; Obrist, P.; Subramaniam, N.; van Rooijen, N.; Schuemann, K.; Weiss, G. Kupffer cells modulate iron homeostasis in mice via regulation of hepcidin expression. J. Mol. Med. 2008, 86, 825–835. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Tomatsu, S.; Fleming, R.E.; Parkkila, S.; Waheed, A.; Jiang, J.; Fei, Y.; Brunt, E.M.; Ruddy, D.A.; Prass, C.E.; et al. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc. Natl. Acad. Sci. USA 1998, 95, 2492–2497. [Google Scholar] [CrossRef] [Green Version]
- Iwai, K. Regulation of cellular iron metabolism: Iron-dependent degradation of IRP by SCFFBXL5 ubiquitin ligase. Free Radic. Biol. Med. 2019, 133, 64–68. [Google Scholar] [CrossRef]
- Wang, J.; Chen, G.; Muckenthaler, M.; Galy, B.; Hentze, M.W.; Pantopoulos, K. Iron-Mediated Degradation of IRP2, an Unexpected Pathway Involving a 2-Oxoglutarate-Dependent Oxygenase Activity. Mol. Cell. Biol. 2004, 24, 954–965. [Google Scholar] [CrossRef] [Green Version]
- Musacco Sebio, R.; Ferrarotti, N.; Saporito Magriñá, C.; Fuda, J.; Torti, H.; Lairión, F.; Boveris, A.; Repetto, M.G. Redox dyshomeostasis in the experimental chronic hepatic overloads with iron or copper. J. Inorg. Biochem. 2019, 191, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Conte, D.; Piperno, A.; Dongiovanni, P.; Fracanzani, A.L.; Fraquelli, M.; Vergani, A.; Gianni, C.; Carmagnola, L.; Fargion, S. The mitochondrial superoxide dismutase A16V polymorphism in the cardiomyopathy associated with hereditary haemochromatosis. J. Med Genet. 2004, 41, 946. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, H.; Blier, P.U.; Gnaiger, E. Remodeling pathway control of mitochondrial respiratory capacity by temperature in mouse heart: Electron flow through the Q-junction in permeabilized fibers. Sci. Rep. 2017, 7, 2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnaiger, E.; Boushel, R.; Søndergaard, H.; Munch-Andersen, T.; Damsgaard, R.; Hagen, C.; Díez-Sánchez, C.; Ara, I.; Wright-Paradis, C.; Schrauwen, P.; et al. Mitochondrial coupling and capacity of oxidative phosphorylation in skeletal muscle of Inuit and Caucasians in the arctic winter. Scand. J. Med. Sci. Sports 2015, 25, 126–134. [Google Scholar] [CrossRef]
- Huang, J.; Jones, D.; Luo, B.; Sanderson, M.; Soto, J.; Abel, E.D.; Cooksey, R.C.; McClain, D.A. Iron Overload and Diabetes Risk: A Shift From Glucose to Fatty Acid Oxidation and Increased Hepatic Glucose Production in a Mouse Model of Hereditary Hemochromatosis. Diabetes 2011, 60, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Telser, J.; Volani, C.; Hilbe, R.; Seifert, M.; Brigo, N.; Paglia, G.; Weiss, G. Metabolic reprogramming of Salmonella infected macrophages and its modulation by iron availability and the mTOR pathway. Microb. Cell 2019, 6, 531–543. [Google Scholar] [CrossRef]
- Gladden, L.B. Lactate metabolism: A new paradigm for the third millennium. J. Physiol. 2004, 558, 5–30. [Google Scholar] [CrossRef]
- Jeppesen, J.B.; Mortensen, C.; Bendtsen, F.; Møller, S. Lactate metabolism in chronic liver disease. Scand. J. Clin. Lab. Investig. 2013, 73, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O.; Oscai, L.B.; Don, I.J.; Molé, P.A. Mitochondrial citric acid cycle and related enzymes: Adaptive response to exercise. Biochem. Biophys. Res. Commun. 1970, 40, 1368–1373. [Google Scholar] [CrossRef]
- Hood, D.A.; Zak, R.; Pette, D. Chronic stimulation of rat skeletal muscle induces coordinate increases in mitochondrial and nuclear mRNAs of cytochrome-c-oxidase subunits. Eur. J. Biochem. 1989, 179, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Salmons, S.; Newsholme, E.A.; Kaufman, R.E.; Mellor, J. Regulation of nuclear and mitochondrial gene expression by contractile activity in skeletal muscle. J. Biol. Chem. 1986, 261, 376–380. [Google Scholar] [CrossRef]
- Zanninelli, G.; Loréal, O.; Brissot, P.; Konijn, A.M.; Slotki, I.N.; Hider, R.C.; Ioav Cabantchik, Z. The labile iron pool of hepatocytes in chronic and acute iron overload and chelator-induced iron deprivation. J. Hepatol. 2002, 36, 39–46. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhao, Y.; Guo, J.; Zhao, S.; Fei, C.; Xiao, C.; Wu, D.; Wu, L.; Li, X.; Chang, C. Iron overload promotes mitochondrial fragmentation in mesenchymal stromal cells from myelodysplastic syndrome patients through activation of the AMPK/MFF/Drp1 pathway. Cell Death Dis. 2018, 9, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef] [PubMed]
- Abdel Hadi, N.; Reyes-Castellanos, G.; Carrier, A. Targeting Redox Metabolism in Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 1534. [Google Scholar] [CrossRef]
- Campanella, A.; Rovelli, E.; Santambrogio, P.; Cozzi, A.; Taroni, F.; Levi, S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 2008, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scandroglio, F.; Tórtora, V.; Radi, R.; Castro, L. Metabolic control analysis of mitochondrial aconitase: Influence over respiration and mitochondrial superoxide and hydrogen peroxide production. Free Radic. Res. 2014, 48, 684–693. [Google Scholar] [CrossRef]
- Bresgen, N.; Eckl, P.M. Oxidative Stress and the Homeodynamics of Iron Metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Paw, B.H. Cellular and mitochondrial iron homeostasis in vertebrates. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2012, 1823, 1459–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatunic, M.; Finucane, F.M.; Brennan, A.M.; Norris, S.; Pacini, G.; Nolan, J.J. Effect of iron overload on glucose metabolism in patients with hereditary hemochromatosis. Metabolism 2010, 59, 380–384. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, C.; Volani, C.; Komlódi, T.; Seifert, M.; Demetz, E.; Valente de Souza, L.; Auer, K.; Petzer, V.; von Raffay, L.; Moser, P.; et al. Dietary Iron Overload and Hfe−/− Related Hemochromatosis Alter Hepatic Mitochondrial Function. Antioxidants 2021, 10, 1818. https://doi.org/10.3390/antiox10111818
Fischer C, Volani C, Komlódi T, Seifert M, Demetz E, Valente de Souza L, Auer K, Petzer V, von Raffay L, Moser P, et al. Dietary Iron Overload and Hfe−/− Related Hemochromatosis Alter Hepatic Mitochondrial Function. Antioxidants. 2021; 10(11):1818. https://doi.org/10.3390/antiox10111818
Chicago/Turabian StyleFischer, Christine, Chiara Volani, Timea Komlódi, Markus Seifert, Egon Demetz, Lara Valente de Souza, Kristina Auer, Verena Petzer, Laura von Raffay, Patrizia Moser, and et al. 2021. "Dietary Iron Overload and Hfe−/− Related Hemochromatosis Alter Hepatic Mitochondrial Function" Antioxidants 10, no. 11: 1818. https://doi.org/10.3390/antiox10111818
APA StyleFischer, C., Volani, C., Komlódi, T., Seifert, M., Demetz, E., Valente de Souza, L., Auer, K., Petzer, V., von Raffay, L., Moser, P., Gnaiger, E., & Weiss, G. (2021). Dietary Iron Overload and Hfe−/− Related Hemochromatosis Alter Hepatic Mitochondrial Function. Antioxidants, 10(11), 1818. https://doi.org/10.3390/antiox10111818