Emerging Cellular and Molecular Strategies for Enhancing Central Nervous System (CNS) Remyelination



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. What Is the Role of Myelin in the CNS?
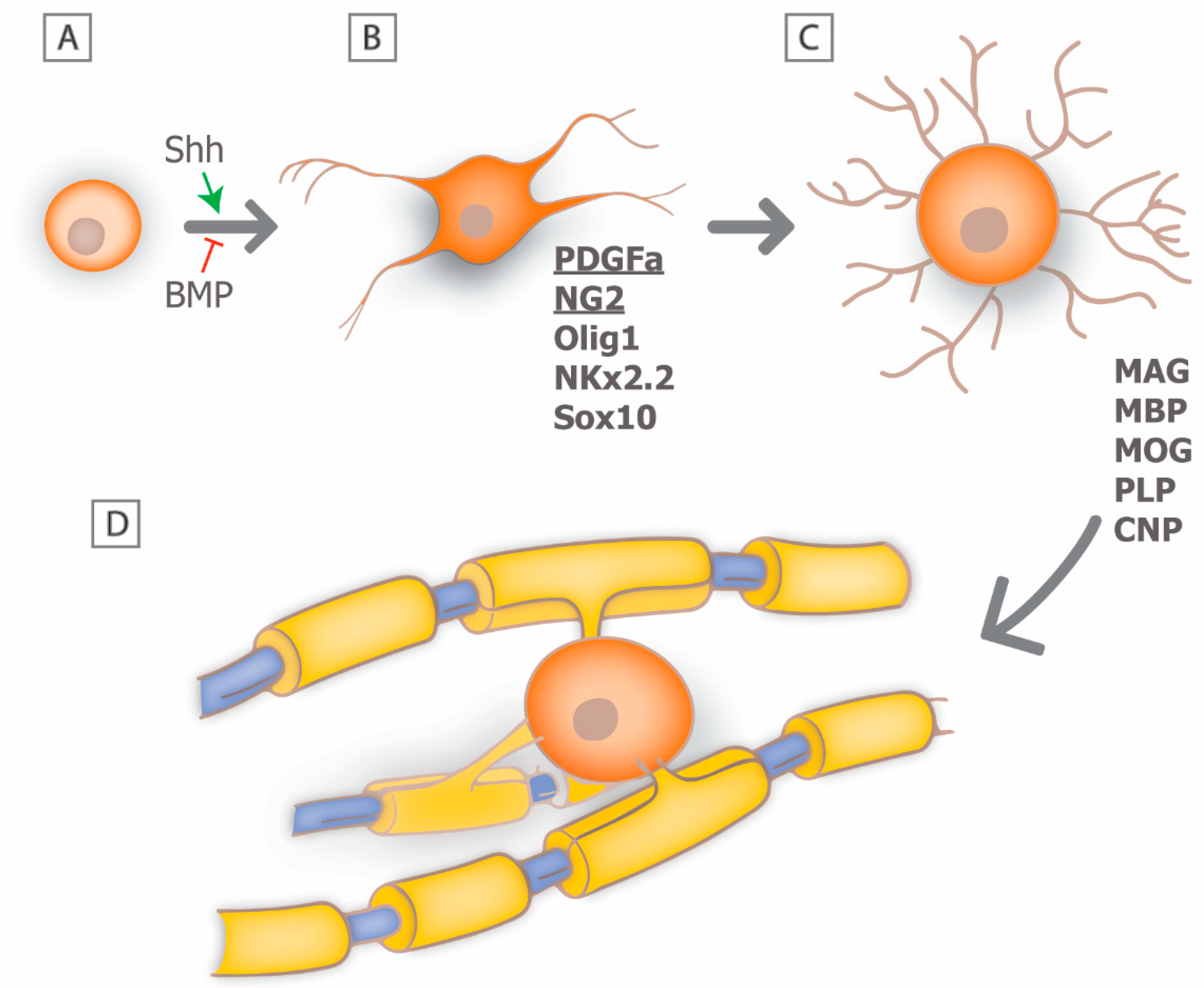
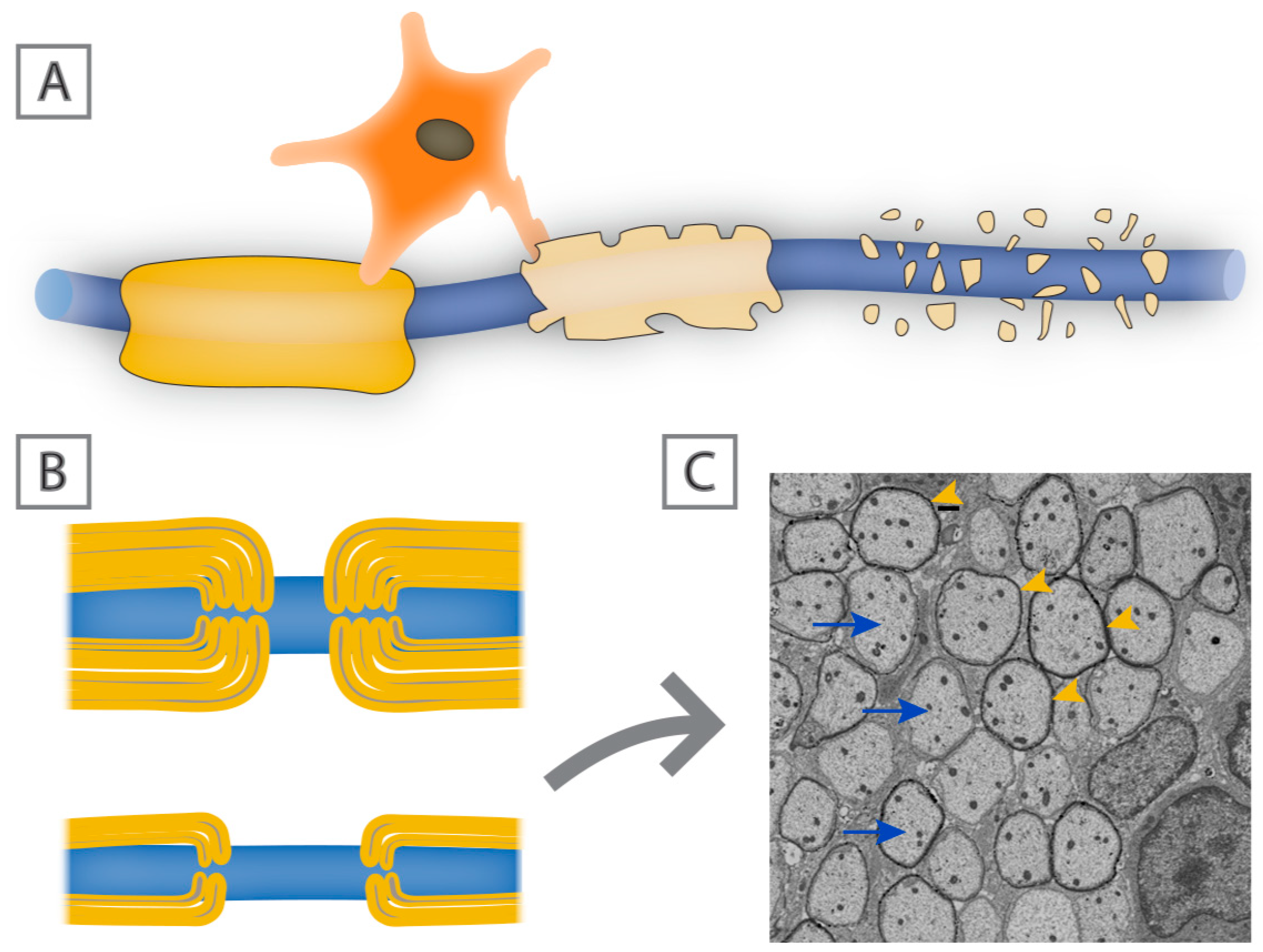
3. Myelin Biology: From Development to Regeneration
4. Mechanisms of Remyelination: Can We Recapitulate Developmental Signals?
5. Insults with Associated Myelin Loss
6. Animal Models of Myelin Loss
7. Therapies Promoting Remyelination
7.1. Oligodendrocyte Progenitor Cells
7.2. MSCs in Myelin Repair
7.3. Bone Marrow Transplants
7.4. Molecular Mediators of Myelin Repair
8. Challenges to Developing Remyelinating Therapies
9. Conclusions
Funding
Conflicts of Interest
References
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Love, S. Demyelinating diseases. J. Clin. Pathol. 2006, 59, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.S. Multiple sclerosis: Remyelination of nascent lesions. Ann. Neurol. 1993, 33, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Balaratnam, M.; Vora, A.; Reynolds, R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol. Appl. Neurobiol. 2007, 33, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple sclerosis: Mechanisms and immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [PubMed]
- Bunge, R.P. Glial cells and the central myelin sheath. Physiol. Rev. 1968, 48, 197–251. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010, 11, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Rasband, M.N.; Peles, E. The nodes of ranvier: Molecular assembly and maintenance. Cold Spring Harb. Perspect. Biol. 2015, 8, a020495. [Google Scholar] [CrossRef] [PubMed]
- McDonald, W.I.; Sears, T.A. Effect of demyelination on conduction in the central nervous system. Nature 1969, 221, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. The pathology of multiple sclerosis and its evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H. Regulation of oligodendrocyte development in the vertebrate CNS. Prog. Neurobiol. 2002, 67, 451–467. [Google Scholar] [CrossRef]
- Tekki-Kessaris, N.; Woodruff, R.; Hall, A.C.; Gaffield, W.; Kimura, S.; Stiles, C.D.; Rowitch, D.H.; Richardson, W.D. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development 2001, 128, 2545–2554. [Google Scholar] [PubMed]
- Crawford, A.H.; Stockley, J.H.; Tripathi, R.B.; Richardson, W.D.; Franklin, R.J. Oligodendrocyte progenitors: Adult stem cells of the central nervous system? Exp. Neurol. 2014, 260, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; Dinsio, K.; Wang, R.; Geertman, R.; Maier, C.E.; Hall, A.K. Patterning of spinal cord oligodendrocyte development by dorsally derived BMP4. J. Neurosci. Res. 2004, 76, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Orentas, D.M.; Hayes, J.E.; Dyer, K.L.; Miller, R.H. Sonic hedgehog signaling is required during the appearance of spinal cord oligodendrocyte precursors. Development 1999, 126, 2419–2429. [Google Scholar] [PubMed]
- Kuhlbrodt, K.; Herbarth, B.; Sock, E.; Hermans-Borgmeyer, I.; Wegner, M. Sox10, a novel transcriptional modulator in glial cells. J. Neurosci. 1998, 18, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; He, X.; Han, X.; Yu, Y.; Ye, F.; Chen, Y.; Hoang, T.; Xu, X.; Mi, Q.S.; Xin, M.; et al. Microrna-mediated control of oligodendrocyte differentiation. Neuron 2010, 65, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Hart, I.K.; Richardson, W.D.; Bolsover, S.R.; Raff, M.C. PDGF and intracellular signaling in the timing of oligodendrocyte differentiation. J. Cell Biol. 1989, 109, 3411–3417. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Nishiyama, A.; Peterson, J.; Prineas, J.; Trapp, B.D. Ng2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J. Neurosci. 2000, 20, 6404–6412. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Tessier-Lavigne, M.; Miller, R.H. Netrin 1 mediates spinal cord oligodendrocyte precursor dispersal. Development 2003, 130, 2095–2105. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Niu, J.; Munji, R.; Davalos, D.; Chang, J.; Zhang, H.; Tien, A.C.; Kuo, C.J.; Chan, J.R.; Daneman, R.; et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science 2016, 351, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Emery, B.; Agalliu, D.; Cahoy, J.D.; Watkins, T.A.; Dugas, J.C.; Mulinyawe, S.B.; Ibrahim, A.; Ligon, K.L.; Rowitch, D.H.; Barres, B.A. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 2009, 138, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Hart, I.K.; Coles, H.S.; Burne, J.F.; Voyvodic, J.T.; Richardson, W.D.; Raff, M.C. Cell death and control of cell survival in the oligodendrocyte lineage. Cell 1992, 70, 31–46. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nishiyama, A.; Cheng, D.; Macklin, W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J. Cell Biol. 1997, 137, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Preston, M.A.; Macklin, W.B. Zebrafish as a model to investigate CNS myelination. Glia 2015, 63, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Baraban, M.; Koudelka, S.; Lyons, D.A. Ca2+ activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 2018, 21, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Krasnow, A.M.; Ford, M.C.; Valdivia, L.E.; Wilson, S.W.; Attwell, D. Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci. 2018, 21, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H. Calcium control of myelin sheath growth. Nat. Neurosci. 2018, 21, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Czopka, T.; Ffrench-Constant, C.; Lyons, D.A. Individual oligodendrocytes have only a few hours in which to generate new myelin sheaths in vivo. Dev. Cell 2013, 25, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.H.; Ravanelli, A.M.; Schwindt, R.; Scott, E.K.; Appel, B. Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci. 2015, 18, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Watkins, T.A.; Emery, B.; Mulinyawe, S.; Barres, B.A. Distinct stages of myelination regulated by gamma-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron 2008, 60, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Demerens, C.; Stankoff, B.; Logak, M.; Anglade, P.; Allinquant, B.; Couraud, F.; Zalc, B.; Lubetzki, C. Induction of myelination in the central nervous system by electrical activity. Proc. Natl. Acad. Sci. USA 1996, 93, 9887–9892. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Lee, P.R.; Fields, R.D. Control of local protein synthesis and initial events in myelination by action potentials. Science 2011, 333, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.M.; Purger, D.; Mount, C.W.; Goldstein, A.K.; Lin, G.L.; Wood, L.S.; Inema, I.; Miller, S.E.; Bieri, G.; Zuchero, J.B.; et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014, 344, 1252304. [Google Scholar] [CrossRef] [PubMed]
- Gautier, H.O.; Evans, K.A.; Volbracht, K.; James, R.; Sitnikov, S.; Lundgaard, I.; James, F.; Lao-Peregrin, C.; Reynolds, R.; Franklin, R.J.; et al. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat. Commun. 2015, 6, 8518. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.A.; Li, A.M.; Grutzendler, J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 2018, 21, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Orthmann-Murphy, J.L.; Langseth, A.J.; Bergles, D.E. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 2018, 21, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Readhead, C.; Popko, B.; Takahashi, N.; Shine, H.D.; Saavedra, R.A.; Sidman, R.L.; Hood, L. Expression of a myelin basic protein gene in transgenic shiverer mice: Correction of the dysmyelinating phenotype. Cell 1987, 48, 703–712. [Google Scholar] [CrossRef]
- Snaidero, N.; Velte, C.; Myllykoski, M.; Raasakka, A.; Ignatev, A.; Werner, H.B.; Erwig, M.S.; Möbius, W.; Kursula, P.; Nave, K.A.; et al. Antagonistic functions of MBP and CNP establish cytosolic channels in CNS myelin. Cell Rep. 2017, 18, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.; Sánchez, P.; Schmitt, S.; Snaidero, N.; Mitkovski, M.; Velte, C.; Brückner, B.R.; Alexopoulos, I.; Czopka, T.; Jung, S.Y.; et al. Actin filament turnover drives leading edge growth during myelin sheath formation in the central nervous system. Dev. Cell 2015, 34, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Fu, M.M.; Sloan, S.A.; Ibrahim, A.; Olson, A.; Zaremba, A.; Dugas, J.C.; Wienbar, S.; Caprariello, A.V.; Kantor, C.; et al. CNS myelin wrapping is driven by actin disassembly. Dev. Cell 2015, 34, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.B.; Rivers, L.E.; Young, K.M.; Jamen, F.; Richardson, W.D. Ng2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J. Neurosci. 2010, 30, 16383–16390. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.L.; Röth, P.T.; Stratton, J.A.; Chuang, B.H.; Danne, J.; Ellis, S.L.; Ng, S.W.; Kilpatrick, T.J.; Merson, T.D. Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J. Neurosci. 2014, 34, 14128–14146. [Google Scholar] [CrossRef] [PubMed]
- Fancy, S.P.; Zhao, C.; Franklin, R.J. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol. Cell. Neurosci. 2004, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Duncan, I.D.; Radcliff, A.B. Inherited and acquired disorders of myelin: The underlying myelin pathology. Exp. Neurol. 2016, 283, 452–475. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W.; Stadelmann, C. The spectrum of multiple sclerosis: New lessons from pathology. Curr. Opin. Neurol. 2005, 18, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Garbern, J.; Cambi, F.; Shy, M.; Kamholz, J. The molecular pathogenesis of pelizaeus-merzbacher disease. Arch. Neurol. 1999, 56, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, P.J.; Vanderver, A.; Bernard, G.; Wolf, N.I.; Dreha-Kulczewksi, S.F.; Deoni, S.C.; Bertini, E.; Kohlschütter, A.; Richardson, W.; Ffrench-Constant, C.; et al. Hypomyelinating leukodystrophies: Translational research progress and prospects. Ann. Neurol. 2014, 76, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Van der Knaap, M.S.; Pronk, J.C.; Scheper, G.C. Vanishing white matter disease. Lancet Neurol. 2006, 5, 413–423. [Google Scholar] [CrossRef]
- Bugiani, M.; Boor, I.; Powers, J.M.; Scheper, G.C.; van der Knaap, M.S. Leukoencephalopathy with vanishing white matter: A review. J. Neuropathol. Exp. Neurol. 2010, 69, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Johnson, A.B.; Boespflug-Tanguy, O.; Rodriguez, D.; Goldman, J.E.; Messing, A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with alexander disease. Nat. Genet. 2001, 27, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Kreft, K.L.; Van Nierop, G.P.; Scherbeijn, S.M.J.; Janssen, M.; Verjans, G.; Hintzen, R.Q. Elevated ebna-1 IgG in MS is associated with genetic MS risk variants. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e406. [Google Scholar] [CrossRef] [PubMed]
- Endriz, J.; Ho, P.P.; Steinman, L. Time correlation between mononucleosis and initial symptoms of MS. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e308. [Google Scholar] [CrossRef] [PubMed]
- Koduah, P.; Paul, F.; Dorr, J.M. Vitamin d in the prevention, prediction and treatment of neurodegenerative and neuroinflammatory diseases. EPMA J. 2017, 8, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Belbasis, L.; Bellou, V.; Evangelou, E.; Ioannidis, J.P.; Tzoulaki, I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015, 14, 263–273. [Google Scholar] [CrossRef]
- Prineas, J.W.; Connell, F. Remyelination in multiple sclerosis. Ann. Neurol. 1979, 5, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Kwon, E.E.; Cho, E.S.; Sharer, L.R. Continual breakdown and regeneration of myelin in progressive multiple sclerosis plaques. Ann. N. Y. Acad. Sci. 1984, 436, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Brück, W.; Lucchinetti, C.; Rodriguez, M. Remyelination in multiple sclerosis. Mult. Scler. 1997, 3, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.T.; Collins, D.L.; Atkins, H.L.; Freedman, M.S.; Arnold, D.L.; Canadian, MS/BMT Study Group. Magnetization transfer ratio evolution with demyelination and remyelination in multiple sclerosis lesions. Ann. Neurol. 2008, 63, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.; Antel, J.; Bruck, W.; Stadelmann, C. Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol. 2007, 17, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Zeisel, A.; Codeluppi, S.; van Bruggen, D.; Mendanha Falcao, A.; Xiao, L.; Li, H.; Haring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, W.F. Pattern of remyelination in the CNS. Nature 1974, 249, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef] [PubMed]
- Stoffels, J.M.; de Jonge, J.C.; Stancic, M.; Nomden, A.; van Strien, M.E.; Ma, D.; Sisková, Z.; Maier, O.; Ffrench-Constant, C.; Franklin, R.J.; et al. Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Brain 2013, 136, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Sobel, R.A.; Chen, M.; Maeda, A.; Hinojoza, J.R. Vitronectin and integrin vitronectin receptor localization in multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 1995, 54, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Cuo, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Kutzelnigg, A.; Lassmann, H. Pathology of multiple sclerosis and related inflammatory demyelinating diseases. Handb. Clin. Neurol. 2014, 122, 15–58. [Google Scholar] [PubMed]
- Granqvist, M.; Boremalm, M.; Poorghobad, A.; Svenningsson, A.; Salzer, J.; Frisell, T.; Piehl, F. Comparative effectiveness of rituximab and other initial treatment choices for multiple sclerosis. JAMA Neurol. 2018, 75, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, A.L.; Hauser, S.L. B-cell therapy for multiple sclerosis: Entering an era. Ann. Neurol. 2018, 83, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Rahmanzadeh, R.; Weber, M.S.; Bruck, W.; Navardi, S.; Sahraian, M.A. B cells in multiple sclerosis therapy-a comprehensive review. Acta Neurol. Scand. 2018, 137, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. The history of neuromyelitis optica. J. Neuroinflamm. 2013, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. Igg marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Zekeridou, A.; Lennon, V.A. Aquaporin-4 autoimmunity. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e110. [Google Scholar] [CrossRef] [PubMed]
- Melamed, E.; Levy, M.; Waters, P.J.; Sato, D.K.; Bennett, J.L.; John, G.R.; Hooper, D.C.; Saiz, A.; Bar-Or, A.; Kim, H.J.; et al. Update on biomarkers in neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kumpfel, T.; et al. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the Neuromyelitis Optica Study Group (NEMOS). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Parratt, J.D.; Prineas, J.W. Neuromyelitis optica: A demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult. Scler. 2010, 16, 1156–1172. [Google Scholar] [CrossRef] [PubMed]
- Roemer, S.F.; Parisi, J.E.; Lennon, V.A.; Benarroch, E.E.; Lassmann, H.; Bruck, W.; Mandler, R.N.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007, 130, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.; Hansen, D.B.; Vella, J.; Bond, P.; Harper, G.; Zammit, C.; Valentino, M.; Fern, R. Vesicular glutamate release from central axons contributes to myelin damage. Nat. Commun. 2018, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Micu, I.; Jiang, Q.; Coderre, E.; Ridsdale, A.; Zhang, L.; Woulfe, J.; Yin, X.; Trapp, B.D.; McRory, J.E.; Rehak, R.; et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 2006, 439, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Kolodziejczyk, K.; Kougioumtzidou, E.; Attwell, D. Proton-gated Ca(2+)-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 2016, 529, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Plemel, J.R.; Keough, M.B.; Duncan, G.J.; Sparling, J.S.; Yong, V.W.; Stys, P.K.; Tetzlaff, W. Remyelination after spinal cord injury: Is it a target for repair? Prog. Neurobiol. 2014, 117, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Myelin damage and repair in pathologic CNS: Challenges and prospects. Front. Mol. Neurosci. 2015, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; Karl, M.; Tognatta, R.; Abu-Rub, M. Model systems to define remyelination therapies. In Neuroplasticity—Insights of Neural Reorganization; InTechOpen: London, UK, 2018. [Google Scholar]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef] [PubMed]
- University of California, San Francisco MS-EPIC Team; Cree, B.A.; Gourraud, P.A.; Oksenberg, J.R.; Bevan, C.; Crabtree-Hartman, E.; Gelfand, J.M.; Goodin, D.S.; Graves, J.; Green, A.J.; et al. Long-term evolution of multiple sclerosis disability in the treatment era. Ann. Neurol. 2016, 80, 499–510. [Google Scholar] [PubMed]
- Hall, S.M. The effect of injections of lysophosphatidyl choline into white matter of the adult mouse spinal cord. J. Cell Sci. 1972, 10, 535–546. [Google Scholar] [PubMed]
- Blakemore, W.F. Ethidium bromide induced demyelination in the spinal cord of the cat. Neuropathol. Appl. Neurobiol. 1982, 8, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Belkadi, A.; Darnall, L.; Hu, T.; Drescher, C.; Cotleur, A.C.; Padovani-Claudio, D.; He, T.; Choi, K.; Lane, T.E.; et al. Cxcr2-positive neutrophils are essential for cuprizone-induced demyelination: Relevance to multiple sclerosis. Nat. Neurosci. 2010, 13, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Caprariello, A.V.; Mangla, S.; Miller, R.H.; Selkirk, S.M. Apoptosis of oligodendrocytes in the central nervous system results in rapid focal demyelination. Ann. Neurol. 2012, 72, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Arasi, K.; Avila, R.L.; Podojil, J.R.; Christakos, A.; Miller, S.D.; Soliven, B.; Popko, B. A genetic mouse model of adult-onset, pervasive central nervous system demyelination with robust remyelination. Brain 2010, 133, 3017–3029. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—From mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.A.; Franklin, R.J.M. Recruitment of endogenous CNS stem cells for regeneration in demyelinating disease. Prog. Brain Res. 2017, 231, 135–163. [Google Scholar] [PubMed]
- Gaesser, J.M.; Fyffe-Maricich, S.L. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Exp. Neurol. 2016, 283, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Wang, S.; Harrison-Restelli, C.; Benraiss, A.; Fraser, R.A.; Gravel, M.; Braun, P.E.; Goldman, S.A. Identification, isolation, and promoter-defined separation of mitotic oligodendrocyte progenitor cells from the adult human subcortical white matter. J. Neurosci. 1999, 19, 9986–9995. [Google Scholar] [CrossRef] [PubMed]
- Windrem, M.S.; Nunes, M.C.; Rashbaum, W.K.; Schwartz, T.H.; Goodman, R.A.; McKhann, G.; Roy, N.S.; Goldman, S.A. Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat. Med. 2004, 10, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.Y.; Du, Z.W.; Zhang, S.C. Differentiation of human oligodendrocytes from pluripotent stem cells. Nat. Protoc. 2009, 4, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Izrael, M.; Zhang, P.; Kaufman, R.; Shinder, V.; Ella, R.; Amit, M.; Itskovitz-Eldor, J.; Chebath, J.; Revel, M. Human oligodendrocytes derived from embryonic stem cells: Effect of noggin on phenotypic differentiation in vitro and on myelination in vivo. Mol. Cell. Neurosci. 2007, 34, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Mozafari, S.; Glatza, M.; Starost, L.; Velychko, S.; Hallmann, A.L.; Cui, Q.L.; Schambach, A.; Kim, K.P.; Bachelin, C.; et al. Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. USA 2017, 114, E2243–E2252. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; McClain, C.R.; Schanz, S.J.; Protack, T.L.; Windrem, M.S.; Goldman, S.A. CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nat. Biotechnol. 2011, 29, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G.; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Windrem, M.S.; Schanz, S.J.; Guo, M.; Tian, G.F.; Washco, V.; Stanwood, N.; Rasband, M.; Roy, N.S.; Nedergaard, M.; Havton, L.A.; et al. Neonatal chimerization with human glial progenitor cells can both remyelinate and rescue the otherwise lethally hypomyelinated shiverer mouse. Cell Stem Cell 2008, 2, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Major, T.; Auyeung, G.; Policarpio, E.; Menon, J.; Droms, L.; Gutin, P.; Uryu, K.; Tchieu, J.; Soulet, D.; et al. Human embryonic stem cell-derived oligodendrocyte progenitors remyelinate the brain and rescue behavioral deficits following radiation. Cell Stem Cell 2015, 16, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; Bai, L.; Lennon, D.P.; Caplan, A.I. The potential of mesenchymal stem cells for neural repair. Discov. Med. 2010, 9, 236–242. [Google Scholar] [PubMed]
- Cohen, J.A. Mesenchymal stem cell transplantation in multiple sclerosis. J. Neurol. Sci. 2013, 333, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Cagliani, J.; Grande, D.; Molmenti, E.P.; Miller, E.J.; Rilo, H.L.R. Immunomodulation by mesenchymal stromal cells and their clinical applications. J. Stem Cell Regen. Biol. 2017, 3. [Google Scholar] [CrossRef]
- Zappia, E.; Casazza, S.; Pedemonte, E.; Benvenuto, F.; Bonanni, I.; Gerdoni, E.; Giunti, D.; Ceravolo, A.; Cazzanti, F.; Frassoni, F.; et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing t-cell anergy. Blood 2005, 106, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Salinas Tejedor, L.; Berner, G.; Jacobsen, K.; Gudi, V.; Jungwirth, N.; Hansmann, F.; Gingele, S.; Prajeeth, C.K.; Baumgartner, W.; Hoffmann, A.; et al. Mesenchymal stem cells do not exert direct beneficial effects on CNS remyelination in the absence of the peripheral immune system. Brain Behav. Immun. 2015, 50, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Lennon, D.P.; Eaton, V.; Maier, K.; Caplan, A.I.; Miller, S.D.; Miller, R.H. Human bone marrow-derived mesenchymal stem cells induce th2-polarized immune response and promote endogenous repair in animal models of multiple sclerosis. Glia 2009, 57, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Kassis, I.; Petrou, P.; Halimi, M.; Karussis, D. Mesenchymal stem cells (MSC) derived from mice with experimental autoimmune encephalomyelitis (EAE) suppress EAE and have similar biological properties with MSC from healthy donors. Immunol. Lett. 2013, 154, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Chen, J.; Cui, Y.; Lu, M.; Elias, S.B.; Mitchell, J.B.; Hammill, L.; Vanguri, P.; Chopp, M. Human bone marrow stromal cell treatment improves neurological functional recovery in EAE mice. Exp. Neurol. 2005, 195, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Jadasz, J.J.; Tepe, L.; Beyer, F.; Samper Agrelo, I.; Akkermann, R.; Spitzhorn, L.S.; Silva, M.E.; Oreffo, R.O.C.; Hartung, H.P.; Prigione, A.; et al. Human mesenchymal factors induce rat hippocampal- and human neural stem cell dependent oligodendrogenesis. Glia 2018, 66, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Zhang, J.F.; Sun, B.; Peng, H.S.; Kong, Q.F.; Bai, S.S.; Liu, Y.M.; Wang, G.Y.; Wang, J.H.; Li, H.L. Reciprocal effect of mesenchymal stem cell on experimental autoimmune encephalomyelitis is mediated by transforming growth factor-beta and interleukin-6. Clin. Exp. Immunol. 2009, 158, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Al Jumah, M.A.; Abumaree, M.H. The immunomodulatory and neuroprotective effects of mesenchymal stem cells (MSCs) in experimental autoimmune encephalomyelitis (EAE): A model of multiple sclerosis (MS). Int. J. Mol. Sci. 2012, 13, 9298–9331. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.W.; Kim, S.S.; Lee, S.Y.; Lee, H.S.; Kim, H.S.; Lee, Y.D.; Suh-Kim, H. Mesenchymal stem cells promote proliferation of endogenous neural stem cells and survival of newborn cells in a rat stroke model. Exp. Mol. Med. 2008, 40, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav. Immun. 2010, 24, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Meamar, R.; Nematollahi, S.; Dehghani, L.; Mirmosayyeb, O.; Shayegannejad, V.; Basiri, K.; Tanhaei, A.P. The role of stem cell therapy in multiple sclerosis: An overview of the current status of the clinical studies. Adv. Biomed. Res. 2016, 5, 46. [Google Scholar] [PubMed]
- Scolding, N.J.; Pasquini, M.; Reingold, S.C.; Cohen, J.A.; International Conference on Cell-Based Therapies for Multiple Sclerosis. Cell-based therapeutic strategies for multiple sclerosis. Brain 2017, 140, 2776–2796. [Google Scholar] [CrossRef] [PubMed]
- Connick, P.; Kolappan, M.; Crawley, C.; Webber, D.J.; Patani, R.; Michell, A.W.; Du, M.Q.; Luan, S.L.; Altmann, D.R.; Thompson, A.J.; et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: An open-label phase 2a proof-of-concept study. Lancet Neurol. 2012, 11, 150–156. [Google Scholar] [CrossRef]
- Lublin, F.D.; Bowen, J.D.; Huddlestone, J.; Kremenchutzky, M.; Carpenter, A.; Corboy, J.R.; Freedman, M.S.; Krupp, L.; Paulo, C.; Hariri, R.J.; et al. Human placenta-derived cells (pda-001) for the treatment of adults with multiple sclerosis: A randomized, placebo-controlled, multiple-dose study. Mult. Scler. Relat. Disord. 2014, 3, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Imrey, P.B.; Planchon, S.M.; Bermel, R.A.; Fisher, E.; Fox, R.J.; Bar-Or, A.; Sharp, S.L.; Skaramagas, T.T.; Jagodnik, P.; et al. Pilot trial of intravenous autologous culture-expanded mesenchymal stem cell transplantation in multiple sclerosis. Mult. Scler. 2018, 24, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Karussis, D.; Karageorgiou, C.; Vaknin-Dembinsky, A.; Gowda-Kurkalli, B.; Gomori, J.M.; Kassis, I.; Bulte, J.W.; Petrou, P.; Ben-Hur, T.; Abramsky, O.; et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Sargent, A.; Bai, L.; Shano, G.; Karl, M.; Garrison, E.; Ranasinghe, L.; Planchon, S.M.; Cohen, J.; Miller, R.H. CNS disease diminishes the therapeutic functionality of bone marrow mesenchymal stem cells. Exp. Neurol. 2017, 295, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Sargent, A.; Shano, G.; Karl, M.; Garrison, E.; Miller, C.; Miller, R.H. Transcriptional profiling of mesenchymal stem cells identifies distinct neuroimmune pathways altered by CNS disease. Int. J. Stem Cells 2018, 11, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Muraro, P.A.; Pasquini, M.; Atkins, H.L.; Bowen, J.D.; Farge, D.; Fassas, A.; Freedman, M.S.; Georges, G.E.; Gualandi, F.; Hamerschlak, N.; et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. 2017, 74, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Saccardi, R.; Freedman, M.S.; Sormani, M.P.; Atkins, H.; Farge, D.; Griffith, L.M.; Kraft, G.; Mancardi, G.L.; Nash, R.; Pasquini, M.; et al. A prospective, randomized, controlled trial of autologous haematopoietic stem cell transplantation for aggressive multiple sclerosis: A position paper. Mult. Scler. 2012, 18, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.K.; Balabanov, R.; Voltarelli, J.; Barreira, A.; Burman, J. Autologous hematopoietic stem cell transplantation for multiple sclerosis—If confused or hesitant, remember: ‘Treat with standard immune suppressive drugs and if no inflammation, no response’. Mult. Scler. 2012, 18, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Wolswijk, G. Oligodendrocyte regeneration in the adult rodent CNS and the failure of this process in multiple sclerosis. Prog. Brain Res. 1998, 117, 233–247. [Google Scholar] [PubMed]
- Mi, S.; Lee, X.; Shao, Z.; Thill, G.; Ji, B.; Relton, J.; Levesque, M.; Allaire, N.; Perrin, S.; Sands, B.; et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat. Neurosci. 2004, 7, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Miller, R.H.; Lee, X.; Scott, M.L.; Shulag-Morskaya, S.; Shao, Z.; Chang, J.; Thill, G.; Levesque, M.; Zhang, M.; et al. Lingo-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 2005, 8, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Hu, B.; Hahm, K.; Luo, Y.; Kam Hui, E.S.; Yuan, Q.; Wong, W.M.; Wang, L.; Su, H.; Chu, T.H.; et al. Lingo-1 antagonist promotes spinal cord remyelination and axonal integrity in mog-induced experimental autoimmune encephalomyelitis. Nat. Med. 2007, 13, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, D.; Balcer, L.; Galetta, S.; Aktas, O.; Ziemssen, T.; Vanopdenbosch, L.; Frederiksen, J.; Skeen, M.; Jaffe, G.J.; Butzkueven, H.; et al. Safety and efficacy of opicinumab in acute optic neuritis (renew): A randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 189–199. [Google Scholar] [CrossRef]
- Mellion, M.; Edwards, K.R.; Hupperts, R.; Drulović, J.; Montalban, X.; Hartung, H.-P.; Brochet, B.; Calabresi, P.A.; Rudick, R.; Ibrahim, A.; et al. Efficacy results from the phase 2b SYNERGY study: Treatment of disabling multiple sclerosis with the anti-LINGO-1 monoclonal antibody opicinumab (s33.004). Neurology 2017, 88. [Google Scholar]
- CenterWatch. Efficacy and Safety of biib033 (Opicinumab) as an Add-On Therapy to Disease-Modifying Therapies (DMTs) in Relapsing Multiple Sclerosis (MS). Available online: https://clinicaltrials.gov/ct2/show/NCT03222973 (accessed on 15 June 2018).
- De Angelis, F.; Bernardo, A.; Magnaghi, V.; Minghetti, L.; Tata, A.M. Muscarinic receptor subtypes as potential targets to modulate oligodendrocyte progenitor survival, proliferation, and differentiation. Dev. Neurobiol. 2012, 72, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (rebuild): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Fancy, S.P.; Baranzini, S.E.; Zhao, C.; Yuk, D.I.; Irvine, K.A.; Kaing, S.; Sanai, N.; Franklin, R.J.; Rowitch, D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009, 23, 1571–1585. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Lang, J.; Sohn, J.; Hammond, E.; Chang, M.; Pleasure, D. Canonical Wnt signaling in the oligodendroglial lineage—Puzzles remain. Glia 2015, 63, 1671–1693. [Google Scholar] [CrossRef] [PubMed]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Tourbah, A.; Lebrun-Frenay, C.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; De Seze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. Md1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult. Scler. 2016, 22, 1719–1731. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu-Rub, M.; Miller, R.H. Emerging Cellular and Molecular Strategies for Enhancing Central Nervous System (CNS) Remyelination. Brain Sci. 2018, 8, 111. https://doi.org/10.3390/brainsci8060111
Abu-Rub M, Miller RH. Emerging Cellular and Molecular Strategies for Enhancing Central Nervous System (CNS) Remyelination. Brain Sciences. 2018; 8(6):111. https://doi.org/10.3390/brainsci8060111
Chicago/Turabian StyleAbu-Rub, Mohammad, and Robert H. Miller. 2018. "Emerging Cellular and Molecular Strategies for Enhancing Central Nervous System (CNS) Remyelination" Brain Sciences 8, no. 6: 111. https://doi.org/10.3390/brainsci8060111
APA StyleAbu-Rub, M., & Miller, R. H. (2018). Emerging Cellular and Molecular Strategies for Enhancing Central Nervous System (CNS) Remyelination. Brain Sciences, 8(6), 111. https://doi.org/10.3390/brainsci8060111