

The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases
 and
and
Abstract
:
1. Introduction
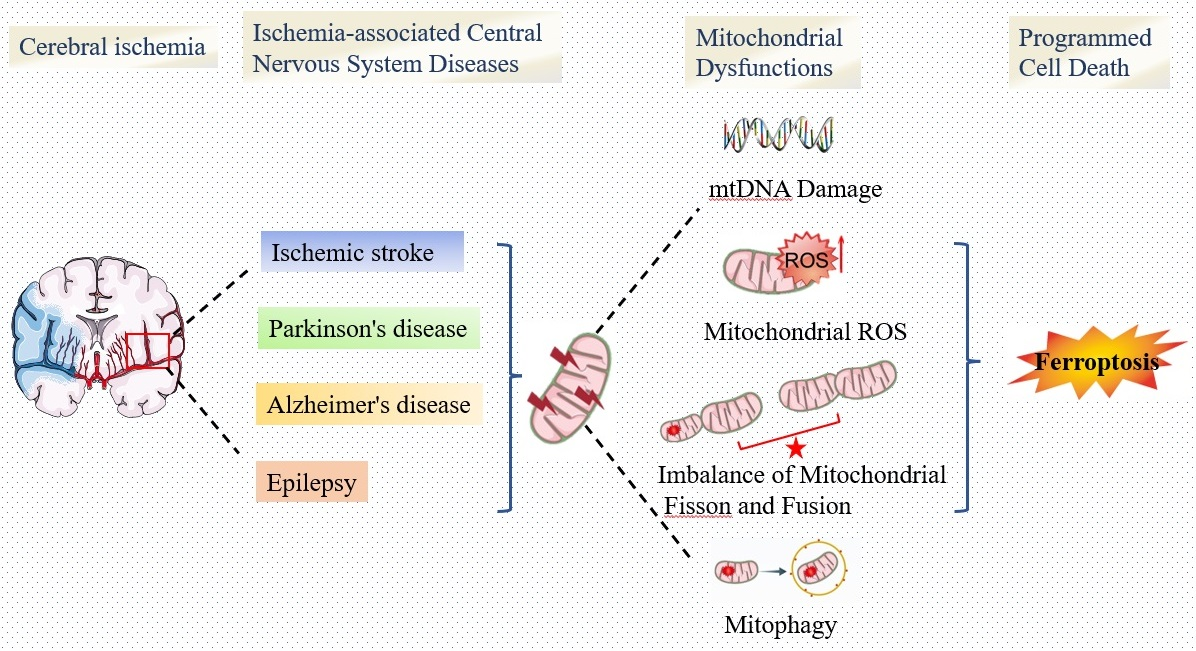
2. Mitochondrial Dysfunction and Ischemia-Associated CNS Diseases
2.1. Mitochondrial Membrane Permeability Transition Pore
2.2. ROS Production and Mitochondrial Dysfunction
2.3. Disturbed Mitochondrial Quality Control
3. Ferroptosis and Ischemia-Associated CNS Diseases
3.1. Lipid Peroxidation
3.2. GSH Depletion and GPX4 Inactivation
3.3. Iron Overload
4. The Interplay between Mitochondrial Dysfunction and Ferroptosis in Ischemia-Associated CNS Diseases
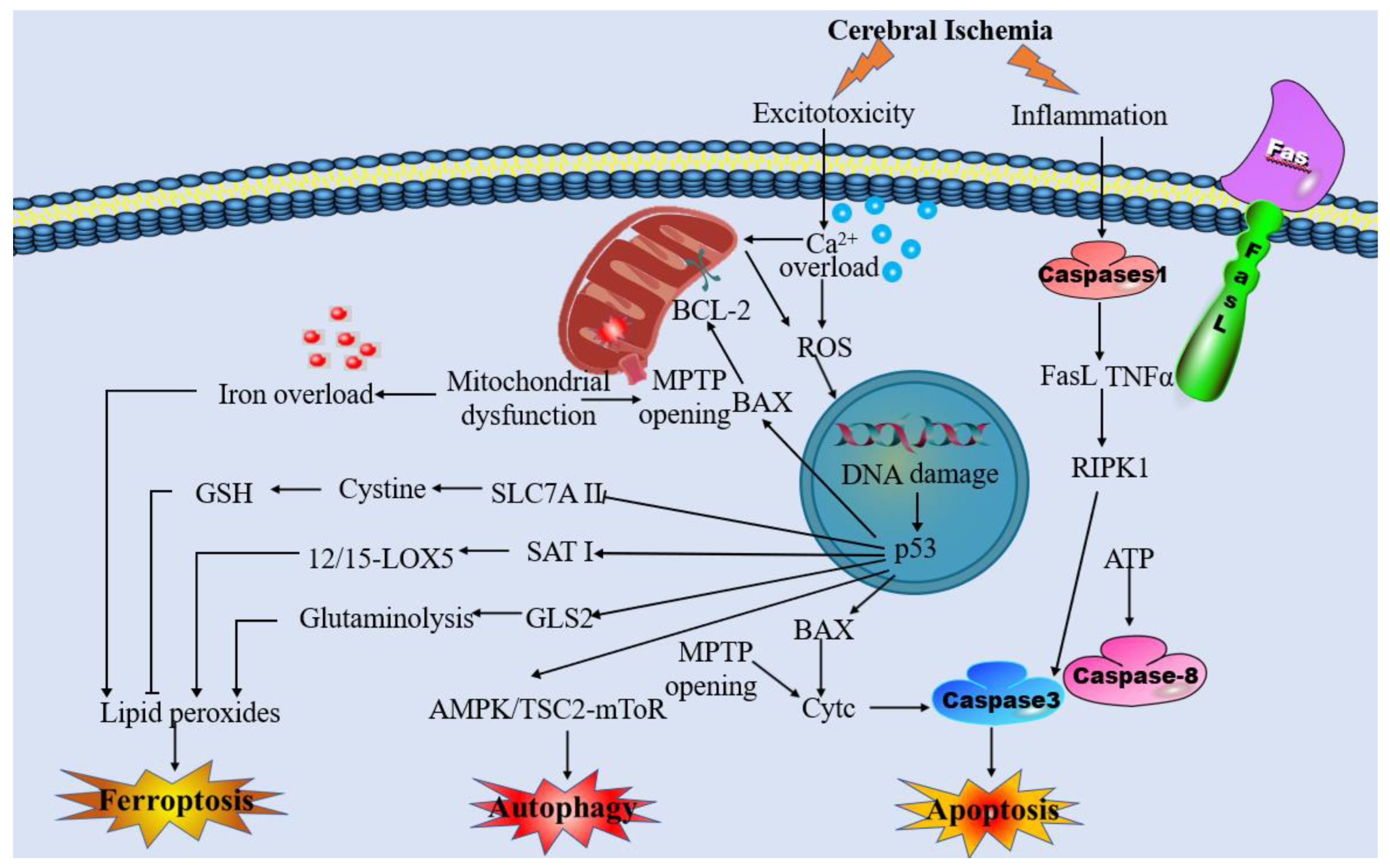
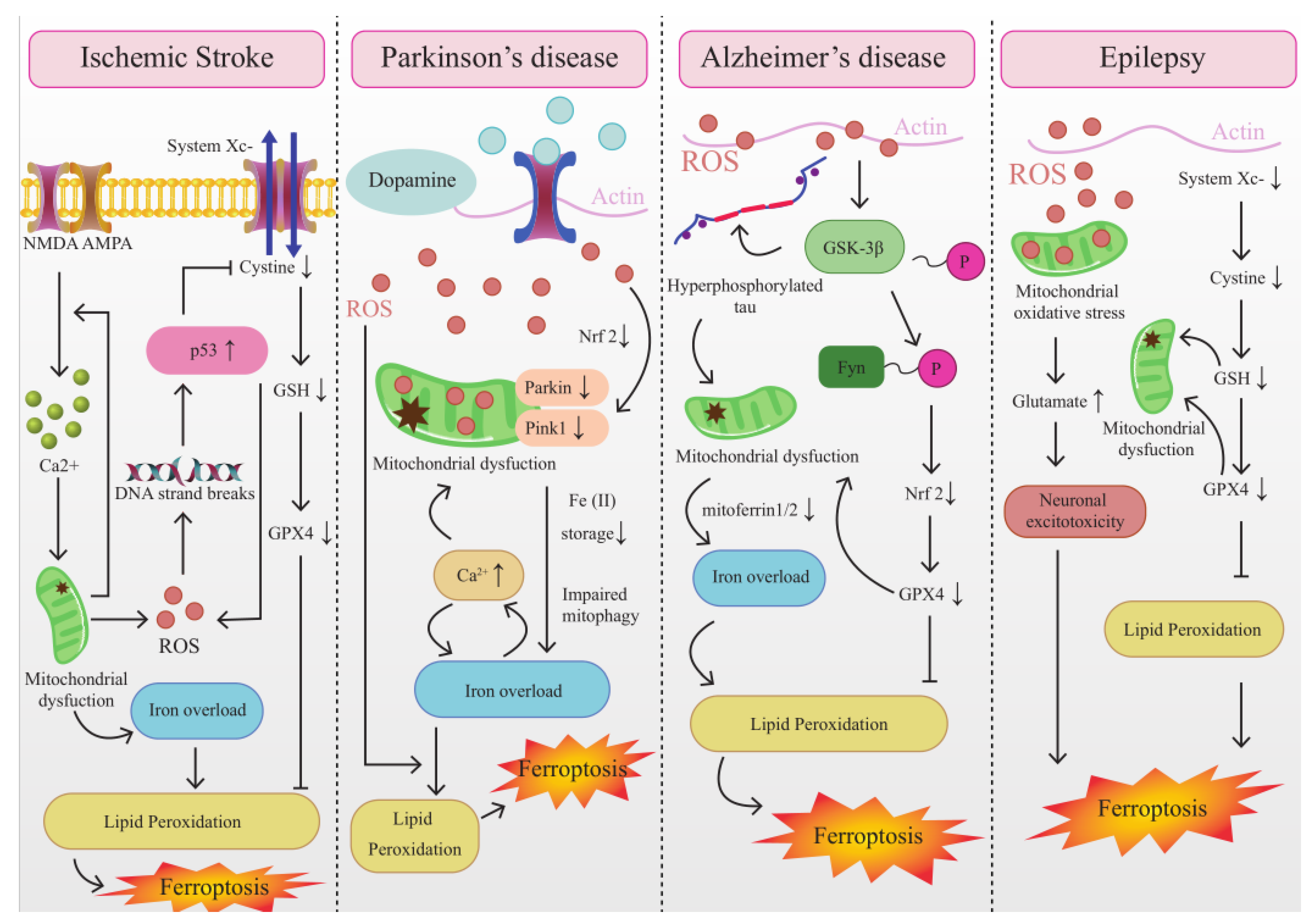
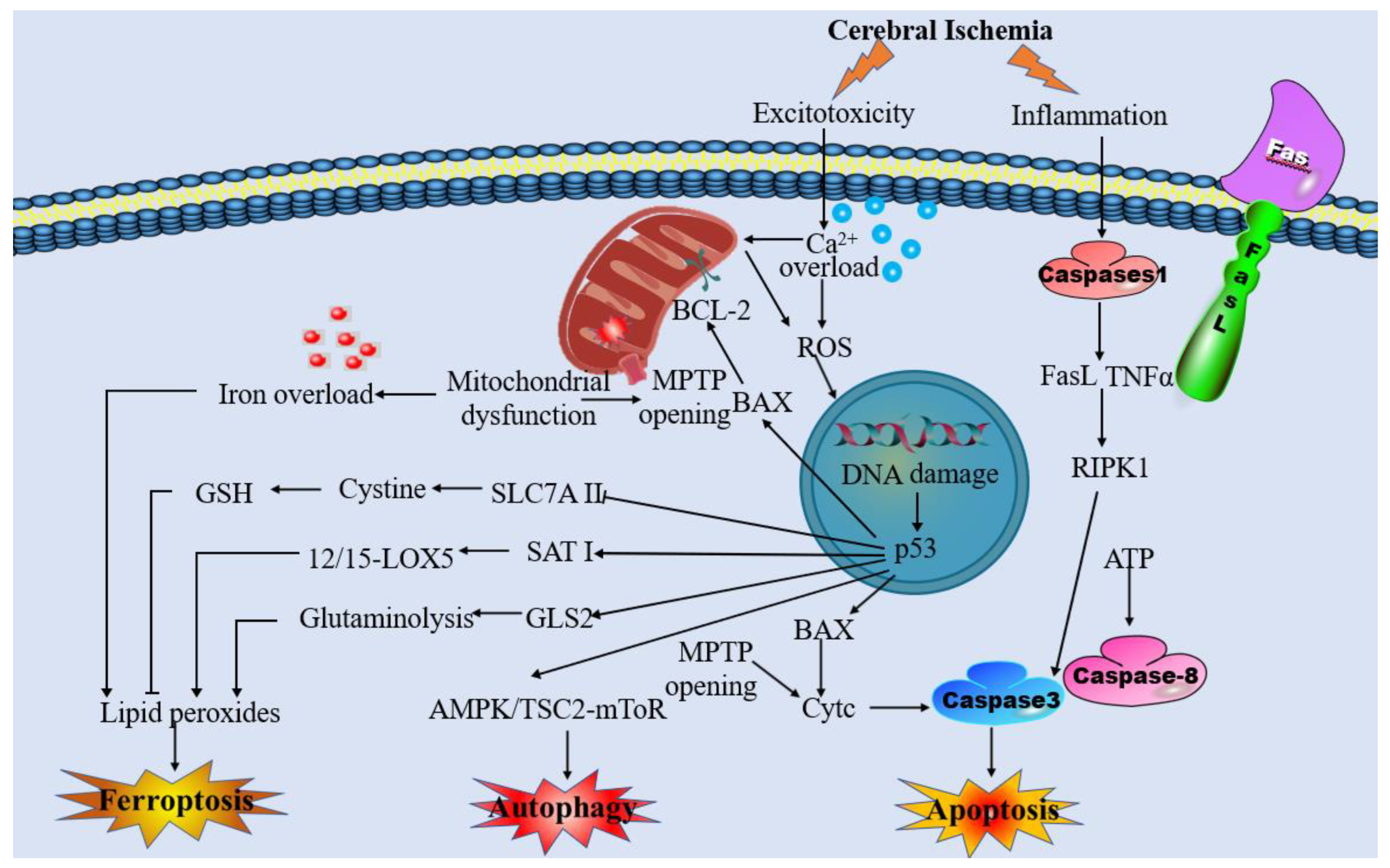
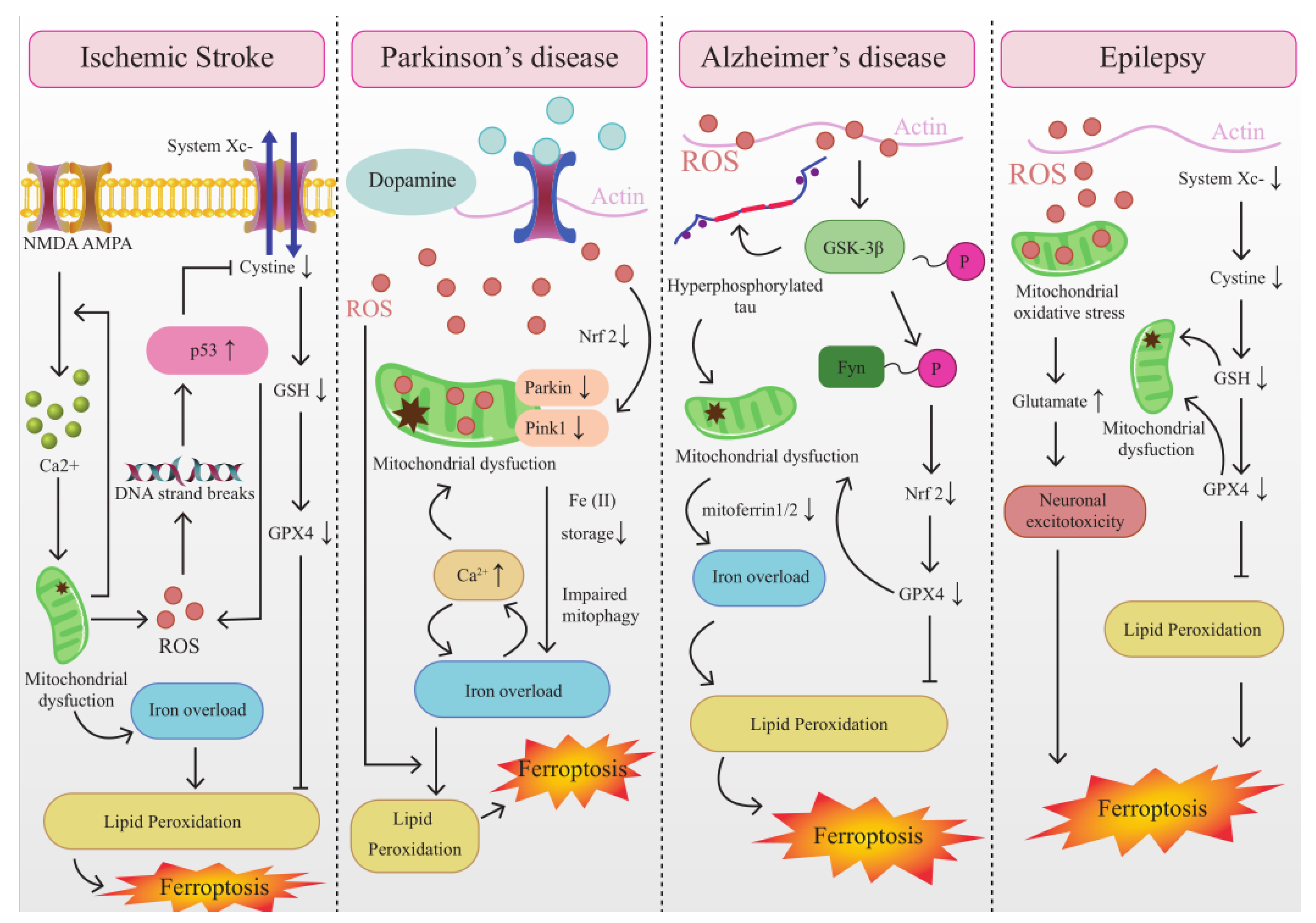
4.1. The Interplay between Mitochondrial Dysfunction and Ferroptosis in IS
4.2. The Interplay between Mitochondrial Dysfunction and Ferroptosis in AD
4.3. The Interplay between Mitochondrial Dysfunction and Ferroptosis in PD
4.4. The Interplay between Mitochondrial Dysfunction and Ferroptosis in Epilepsy
5. Targeting Intervention of Mitochondrial Dysfunction and Ferroptosis for the Treatment against Ischemia-Associated CNS Diseases
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Arachidonoyl |
| ACSL4 | Acyl-CoA synthetase long-chain family member 4 |
| AdA | Adrenoyl |
| AD | Alzheimer’s disease |
| ADP | Adenosine diphosphate |
| AIF | Apoptosis-inducing factor |
| ALOX15 | Arachidonate 15-lipoxygenase |
| AMPA | Amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid |
| ANT | Adenine nucleotide translocator |
| APP | Amyloid β precursor protein |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid β |
| BACE1 | Beta-site amyloid precursor protein cleaving enzyme 1 |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| CAC | Citric acid cycle |
| CISD1 | CDGSH iron sulfur domain 1 |
| Cp | Ceruloplasmin |
| CoQ10 | Coenzyme Q10 |
| CypD | Cyclophilin D |
| DAMPs | Damage-associated molecular patterns |
| DMT1 | Divalent metal transporter 1 |
| DNA | Deoxyribonucleic acid |
| Endo G | Endonuclease G |
| FPN | Ferroportin |
| FTH1 | Ferritin heavy chain 1 |
| FTL | Ferritin light chain |
| FtMt | Mitochondrial ferritin |
| FXN | Frataxin |
| GSH | Glutathione |
| GSSG | Oxidized glutathione |
| GPX4 | Glutathione peroxidase 4 |
| IRP1 | Iron regulatory protein 1 |
| IS | Ischemic stroke |
| LA | α-Lipoic acid |
| LAMP1 | Lysosomal-associated membrane protein 1 |
| LPCAT3 | Lysophosphatidylcholine acyltransferase 3 |
| MDA | Malondialdehyde |
| MPTP | Mitochondrial permeability transition pore |
| MSCs | Mesenchymal stem cells |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NCOA4 | Nuclear receptor coactivator 4 |
| NLRP3 | The NOD LRR pyrin domain-containing protein 3 |
| NO | Nitric oxide |
| Nrf2 | Nuclear factor E2-related factor 2 |
| PD | Parkinson’s disease |
| PEs | Phosphatidylethanolamines |
| PGC-1 | Peroxisome proliferator-activated receptor-coactivator |
| Pi | Inorganic phosphate |
| PKC | Protein kinase C |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PUFAs | Polyunsaturated fatty acids |
| RCD | Regulated cell death |
| RET | Reverse electron transport |
| ROS | Reactive oxygen species |
| Se | Selenium |
| SOD | Superoxide dismutase |
| Sp1 | Specificity protein 1 |
| SNpc | Substantia nigra pars compacta |
| TFAP2C | Transcription factor activating protein 2 gamma |
| tPA | Tissue plasminogen activator |
| TF | Transferrin |
| TFR1 | Transferrin receptor 1 |
| TLE | Temporal lobe epilepsy |
| TSG | Tetrahydroxy stilbene glycoside |
| VDACs | Voltage-dependent anion channels |
| 2Fe-2S | Iron–sulfur protein |
| 4-HNE | 4-Hydroxynonenal |
| 8OHdG | 8 Hydroxy 2V deoxyguanosine |
References
- Levchenkova, O.S.; Novikov, V.E.; Korneva, Y.S.; Dorosevich, A.E.; Parfenov, E.A. Combined Preconditioning Reduces the Negative Influence of Cerebral Ischemia on the Morphofunctional Condition of CNS. Bull. Exp. Biol. Med. 2021, 171, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Sveinsson, O.A.; Kjartansson, O.; Valdimarsson, E.M. Cerebral ischemia/infarction—Epidemiology, causes and symptoms. Laeknabladid 2014, 100, 271–279. [Google Scholar] [PubMed]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Brain Ischemia as a Prelude to Alzheimer’s Disease. Front. Aging Neurosci. 2021, 18, 636653. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, S.; Grigoletto, J.; Bernis, M.E.; Pesch, V.; Ma, L.; Reithofer, S.; Tamgüney, G. Ischemic stroke causes Parkinson’s disease-like pathology and symptoms in transgenic mice overexpressing alpha-synuclein. Acta Neuropathol. Commun. 2022, 10, 26. [Google Scholar] [CrossRef]
- Jiang, L.; Hu, X. Positive Effect of α-Asaronol on the Incidence of Post-Stroke Epilepsy for Rat with Cerebral Ischemia-Reperfusion Injury. Molecules 2022, 27, 1984. [Google Scholar] [CrossRef]
- Goulay, R.; Romo, L.M.; Hol, E.M.; Dijkhuizen, R.M. From Stroke to Dementia: A Comprehensive Review Exposing Tight Interactions Between Stroke and Amyloid-β Formation. Transl. Stroke Res. 2020, 11, 601–614. [Google Scholar] [CrossRef]
- Liu, X.; Feng, Z.; Du, L.; Huang, Y.; Ge, J.; Deng, Y.; Mei, Z. The Potential Role of MicroRNA-124 in Cerebral Ischemia Injury. Int. J. Mol. Sci. 2019, 21, 120. [Google Scholar] [CrossRef]
- Koch, R.E.; Josefson, C.C.; Hill, G.E. Mitochondrial function, ornamentation, and immunocompetence. Biol. Rev. Camb. Philos. Soc. 2017, 92, 1459–1474. [Google Scholar] [CrossRef]
- Wang, R.; Liu, Y.-Y.; Liu, X.-Y.; Jia, S.-W.; Zhao, J.; Cui, D.; Wang, L. Resveratrol Protects Neurons and the Myocardium by Reducing Oxidative Stress and Ameliorating Mitochondria Damage in a Cerebral Ischemia Rat Model. Cell. Physiol. Biochem. 2014, 34, 854–864. [Google Scholar] [CrossRef]
- Mehta, S.L.; Kumari, S.; Mendelev, N.; Li, P.A. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci. 2012, 13, 79. [Google Scholar] [CrossRef]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M.; Farbood, Y.; Moghaddam, H.F. Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 2017, 38, 1167–1186. [Google Scholar] [CrossRef] [PubMed]
- Khoshnam, S.E.; Winlow, W.; Farzaneh, M. The Interplay of MicroRNAs in the Inflammatory Mechanisms Following Ischemic Stroke. J. Neuropathol. Exp. Neurol. 2017, 76, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Van der Worp, H.B.; Macleod, M.R.; Kollmar, R.; European Stroke Research Network for Hypothermia. Therapeutic hypothermia for acute ischemic stroke: Ready to start large randomized trials? J. Cereb. Blood Flow Metab. 2010, 30, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-F.; Li, P.-Y.; Wang, X.; Stetler, R.; Chen, J. Anti-inflammatory signaling: The point of convergence for medical gases in neuroprotection against ischemic stroke. Med. Gas Res. 2016, 6, 227–231. [Google Scholar] [CrossRef]
- Luo, T.; Park, Y.; Sun, X.; Liu, C.; Hu, B. Protein Misfolding, Aggregation, and Autophagy After Brain Ischemia. Transl. Stroke Res. 2013, 4, 581–588. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Vuckovic, A.M.; Bosello Travain, V.; Bordin, L.; Cozza, G.; Miotto, G.; Rossetto, M.; Toppo, S.; Venerando, R.; Zaccarin, M.; Maiorino, M. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3epsilon. FEBS Lett. 2020, 594, 611–624. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. PROTEOMICS 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free. Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed]
- Kalani, K.; Yan, S.F.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Drug Discov. Today 2018, 23, 1983–1989. [Google Scholar] [CrossRef]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the Mitochondrial Permeability Transition in Myocardial Disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef]
- Sesso, A.; Belizário, J.; Marques, M.; Higuchi, M.; Schumacher, R.; Colquhoun, A.; Ito, E.; Kawakami, J. Mitochondrial Swelling and Incipient Outer Membrane Rupture in Preapoptotic and Apoptotic Cells. Anat. Rec. 2012, 295, 1647–1659. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. The Mitochondrial Permeability Transition Pore and Cancer: Molecular Mechanisms Involved in Cell Death. Front. Oncol. 2014, 4, 302. [Google Scholar] [CrossRef]
- Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Murozono, M.; Kanazawa, M.; Nara, T.; Ozawa, T.; Watanabe, Y. Edaravone and cyclosporine A as neuroprotective agents for acute ischemic stroke. Acute Med. Surg. 2018, 5, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia–reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial Oxidative Stress: Implications for Cell Death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Boczek, T.; Lisek, M.; Ferenc, B.; Zylinska, L. Plasma membrane Ca2+-ATPase is a novel target for ketamine action. Biochem. Biophys. Res. Commun. 2015, 465, 312–317. [Google Scholar] [CrossRef]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 92–99. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular Mechanisms of Ischemia–Reperfusion Injury in Brain: Pivotal Role of the Mitochondrial Membrane Potential in Reactive Oxygen Species Generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Radak, D.; Resanovic, I.; Isenovic, E.R. Link Between Oxidative Stress and Acute Brain Ischemia. Angiology 2014, 65, 667–676. [Google Scholar] [CrossRef]
- Yang, S.; Li, W. Targeting oxidative stress for the treatment of ischemic stroke: Upstream and downstream therapeutic strategies. Brain Circ. 2016, 2, 153–163. [Google Scholar] [CrossRef]
- Grune, T.; Reinheckel, T.; Joshi, M.; Davies, K.J.A. Proteolysis in Cultured Liver Epithelial Cells during Oxidative Stress. Role of the multicatalytic proteinase complex, proteasome. J. Biol. Chem. 1995, 270, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.A.; Feiring, A.; Cioppi, M.; Hibbard, R.; Gray, B.; Khatib, Y.; Jessup, D.; Bachinsky, W.; Rivera, E.; Tauth, J.; et al. S.M.A.R.T. Self-Expanding Nitinol Stent for the Treatment of Atherosclerotic Lesions in the Superficial Femoral Artery (STROLL): 1-Year Outcomes. J. Vasc. Interv. Radiol. 2015, 26, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Fucci, L.; Oliver, C.N.; Coon, M.J.; Stadtman, E.R. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: Possible implication in protein turnover and ageing. Proc. Natl. Acad. Sci. USA 1983, 80, 1521–1525. [Google Scholar] [CrossRef] [PubMed]
- Aizenman, E.; Hartnett, K.A.; Reynoldst, I.J. Oxygen free radicals regulate NMDA receptor function via a redox modulatory site. Neuron 1990, 5, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, C.; Delgado, P.; Vilches, A.; Martín-Gallán, P.; Ribó, M.; Santamarina, E.; Molina, C.; Corbeto, N.; Rodrí;guez-Sureda, V.; Rosell, A.; et al. Oxidative Stress After Thrombolysis-Induced Reperfusion in Human Stroke. Stroke 2010, 41, 653–660. [Google Scholar] [CrossRef]
- Li, S.; Zheng, J.; Carmichael, S.T. Increased oxidative protein and DNA damage but decreased stress response in the aged brain following experimental stroke. Neurobiol. Dis. 2005, 18, 432–440. [Google Scholar] [CrossRef]
- Suárez, I.; Bodega, G.; Fernández, B. Glutamine synthetase in brain: Effect of ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Oliver, C.N.; Starke-Reed, E.P.; Stadtman, E.R.; Liu, G.J.; Carney, J.M.; Floyd, A.R. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5144–5147. [Google Scholar] [CrossRef]
- Jena, N.R. DNA damage by reactive species: Mechanisms, mutation and repair. J. Biosci. 2012, 37, 503–517. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J.; Siggens, L.; Figg, N.; Bennett, M.; Foo, R.; Chastain, P.D.; Nakamura, J.; et al. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Cui, J.; Holmes, E.H.; Greene, T.G.; Liu, P.K. Oxidative DNA damage precedes DNA fragmentation after experimental stroke in rat brain. FASEB J. 2000, 14, 955–967. [Google Scholar] [CrossRef] [PubMed]
- MacManus, J.P.; Fliss, H.; Preston, E.; Rasquinha, I.; Tuor, U. Cerebral Ischemia Produces Laddered DNA Fragments Distinct from Cardiac Ischemia and Archetypal Apoptosis. J. Cereb. Blood Flow Metab. 1999, 19, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Serteser, M.; Ozben, T.; Gumuslu, S.; Balkan, S.; Balkan, E. Lipid Peroxidation in Rat Brain During Focal Cerebral Ischemia: Prevention of Malondialdehyde and Lipid Conjugated Diene Production by a Novel Antiepileptic, Lamotrigine. NeuroToxicology 2002, 23, 111–119. [Google Scholar] [CrossRef]
- Picaud, J.C.; Steghens, J.P.; Auxenfans, C.; Barbieux, A.; Laborie, S.; Claris, O. Lipid peroxidation assessment by malondialdehyde measurement in parenteral nutrition solutions for newborn infants: A pilot study. Acta Paediatr. 2004, 93, 241–245. [Google Scholar] [CrossRef]
- Guéraud, F.; Peiro, G.; Bernard, H.; Alary, J.; Créminon, C.; Debrauwer, L.; Rathahao, E.; Drumare, M.-F.; Canlet, C.; Wal, J.-M.; et al. Enzyme immunoassay for a urinary metabolite of 4-hydroxynonenal as a marker of lipid peroxidation. Free. Radic. Biol. Med. 2006, 40, 54–62. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Edaga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef]
- Slatter, D.A.; Avery, N.C.; Bailey, A.J. Identification of a New Cross-link and Unique Histidine Adduct from Bovine Serum Albumin Incubated with Malondialdehyde. J. Biol. Chem. 2004, 279, 61–69. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, F.; Yu, D.-F.; Wu, P.-F.; Chen, J.-G. The cytotoxic mechanism of malondialdehyde and protective effect of carnosine via protein cross-linking/mitochondrial dysfunction/reactive oxygen species/MAPK pathway in neurons. Eur. J. Pharmacol. 2011, 650, 184–194. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Marnett, L.J.; Tang, M.-S. Malondialdehyde, a major endogenous lipid peroxidation product, sensitizes human cells to UV- and BPDE-induced killing and mutagenesis through inhibition of nucleotide excision repair. Mutat. Res. Mol. Mech. Mutagen. 2006, 601, 125–136. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Daniels, J.S.; Rouzer, C.A.; Greene, R.E.; Marnett, L.J. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J. Biol. Chem. 2003, 278, 31426–31433. [Google Scholar] [CrossRef] [PubMed]
- Bromont, C.; Marie, C.; Bralet, J. Increased lipid peroxidation in vulnerable brain regions after transient forebrain ischemia in rats. Stroke 1989, 20, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Shima, T.; Uozumi, T.; Sogabe, T.; Yamada, K.; Kawasaki, T. A possible role of lipid peroxidation in cellular damages caused by cerebral ischemia and the protective effect of alpha-tocopherol administration. Stroke 1983, 14, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Wei, Y.; Song, Q.; Du, B.; Wang, H.; Chu, Y.; Hu, Y. The Role of Myocardial Mitochondrial Quality Control in Heart Failure. Front. Pharmacol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Simmons, E.C.; Scholpa, N.E.; Schnellmann, R.G. Mitochondrial biogenesis as a therapeutic target for traumatic and neurodegenerative CNS diseases. Exp. Neurol. 2020, 329, 113309. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Chen, S.D.; Lin, T.K.; Lin, J.W.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.-W.; Chuang, Y.-C. Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res. 2010, 88, 3144–3154. [Google Scholar]
- Su, J.; Liu, J.; Yan, X.-Y.; Zhang, Y.; Zhang, J.-J.; Zhang, L.-C.; Sun, L.-K. Cytoprotective Effect of the UCP2-SIRT3 Signaling Pathway by Decreasing Mitochondrial Oxidative Stress on Cerebral Ischemia–Reperfusion Injury. Int. J. Mol. Sci. 2017, 18, 1599. [Google Scholar] [CrossRef]
- Chung, K.; Chen, Y.; Juan, Y.; Hsu, C.; Nakahira, K.; Huang, Y.; Lin, M.; Wu, S.; Shih, J.; Chang, Y.; et al. Multi-kinase framework promotes proliferation and invasion of lung adenocarcinoma through activation of dynamin-related protein 1. Mol. Oncol. 2021, 15, 560–578. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Alavi, M.V.; Kim, K.-Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.K.Y.; Zhai, D.; Su, P.; Jiang, A.; Boychuk, J.; Liu, F. The receptor-receptor interaction between mGluR1 receptor and NMDA receptor: A potential therapeutic target for protection against ischemic stroke. FASEB J. 2019, 33, 14423–14439. [Google Scholar] [CrossRef] [PubMed]
- Babaei, P. NMDA and AMPA receptors dysregulation in Alzheimer’s disease. Eur. J. Pharmacol. 2021, 908, 174310. [Google Scholar] [CrossRef]
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef]
- Guo, C.; Ma, Y.-Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 17, 711564. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, R.; Li, X.; Fang, W. Porcine Circovirus 2 Induction of ROS Is Responsible for Mitophagy in PK-15 Cells via Activation of Drp1 Phosphorylation. Viruses 2020, 12, 289. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Bhatia, D.; Choi, M.E. The Emerging Role of Mitophagy in Kidney Diseases. J. Life Sci. 2019, 1, 13–22. [Google Scholar] [CrossRef]
- Khalifa, A.R.M.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol. Rep. 2017, 5, e13125. [Google Scholar] [CrossRef]
- Liu, K.-Y.; Mo, Y.; Sun, Y.-Y. Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 2020, 15, 1388–1396. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, Y.; Liu, M.; Li, Y.; Ma, S.; Tang, W.; Yan, W.; Cao, M.; Zheng, W.; Jiang, L.; et al. BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy 2021, 17, 1934–1946. [Google Scholar] [CrossRef]
- Feng, J.; Chen, X.; Guan, B.; Li, C.; Qiu, J.; Shen, J. Inhibition of Peroxynitrite-Induced Mitophagy Activation Attenuates Cerebral Ischemia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 6369–6386. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 23, 342. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef]
- Tuo, Q.-Z.; Lei, P.; Jackman, A.K.; Li, X.-L.; Xiong, H.; Liuyang, Z.-Y.; Roisman, L.C.; Zhang, S.-T.; Ayton, S.; Wang, Q.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef]
- Zhou, R.-P.; Chen, Y.; Wei, X.; Yu, B.; Xiong, Z.-G.; Lu, C.; Hu, W. Novel insights into ferroptosis: Implications for age-related diseases. Theranostics 2020, 10, 11976–11997. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- D’Herde, K.; Krysko, D.V. Ferroptosis: Oxidized PEs trigger death. Nat. Chem. Biol. 2017, 13, 4–5. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Han, H.; Xu, D.; Liu, C.; Claesson, H.-E.; Björkholm, M.; Sjöberg, J. Interleukin-4-Mediated 15-Lipoxygenase-1 Trans-Activation Requires UTX Recruitment and H3K27me3 Demethylation at the Promoter in A549 Cells. PLoS ONE 2014, 9, e85085. [Google Scholar] [CrossRef]
- Hinman, A.; Holst, C.R.; Latham, J.C.; Bruegger, J.J.; Ulas, G.; McCusker, K.P.; Amagata, A.; Davis, D.; Hoff, K.G.; Kahn-Kirby, A.H.; et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS ONE 2018, 13, e0201369. [Google Scholar] [CrossRef]
- van Leyen, K.; Holman, T.R.; Maloney, D.J. The potential of 12/15-lipoxygenase inhibitors in stroke therapy. Future Med. Chem. 2014, 6, 1853–1855. [Google Scholar] [CrossRef]
- Jin, G.; Arai, K.; Murata, Y.; Wang, S.; Stins, M.F.; Lo, E.H. Protecting against cerebrovascular injury: Contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke 2008, 39, 2538–2543. [Google Scholar] [CrossRef]
- Yigitkanli, K.; Zheng, Y.; Pekcec, A.; Lo, E.H.; van Leyen, K. Increased 12/15-Lipoxygenase Leads to Widespread Brain Injury Following Global Cerebral Ischemia. Transl. Stroke Res. 2017, 8, 194–202. [Google Scholar] [CrossRef]
- Krainz, T.; Gaschler, M.M.; Lim, C.; Sacher, J.R.; Stockwell, B.R.; Wipf, P. A Mitochondrial-Targeted Nitroxide Is a Potent Inhibitor of Ferroptosis. ACS Central Sci. 2016, 2, 653–659. [Google Scholar] [CrossRef]
- Chen, C.T.; Green, J.T.; Orr, S.K.; Bazinet, R.P. Regulation of brain polyunsaturated fatty acid uptake and turnover. Prostaglandins, Leukot. Essent. Fat. Acids 2008, 79, 85–91. [Google Scholar] [CrossRef]
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.-F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc–. Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, X.; Tai, B.; Li, W.; Li, T. Ferroptosis and Its Multifaceted Roles in Cerebral Stroke. Front. Cell. Neurosci. 2021, 3, 615372. [Google Scholar] [CrossRef]
- Ahmad, S.; Elsherbiny, N.M.; Haque, R.; Khan, M.B.; Ishrat, T.; Shah, Z.A.; Ali, M.; Jamal, A.; Katare, D.P.; Liou, G.I.; et al. Sesamin attenuates neurotoxicity in mouse model of ischemic brain stroke. NeuroToxicology 2014, 45, 100–110. [Google Scholar] [CrossRef]
- Liu, J.-H.; Wang, T.-W.; Lin, Y.-Y.; Ho, W.-C.; Tsai, H.-C.; Chen, S.-P.; Lin, A.M.-Y.; Liu, T.-Y.; Wang, H.-T. Acrolein is involved in ischemic stroke-induced neurotoxicity through spermidine/spermine-N1-acetyltransferase activation. Exp. Neurol. 2020, 323, 113066. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I. Glutathione and Related Molecules in Parkinsonism. Int. J. Mol. Sci. 2021, 22, 8689. [Google Scholar] [CrossRef]
- Lan, B.; Ge, J.-W.; Cheng, S.-W.; Zheng, X.-L.; Liao, J.; He, C.; Rao, Z.-Q.; Wang, G.-Z. Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J. Integr. Med. 2020, 18, 344–350. [Google Scholar] [CrossRef]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis (Review). Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Brütsch, S.H.; Wang, C.C.; Li, L.; Stender, H.; Neziroglu, N.; Richter, C.; Kuhn, H.; Borchert, A.; Maiorino, M.; Conrad, M.; et al. Expression of Inactive Glutathione Peroxidase 4 Leads to Embryonic Lethality, and Inactivation of theAlox15Gene Does Not Rescue Such Knock-In Mice. Antioxid. Redox Signal. 2015, 22, 281–293. [Google Scholar] [CrossRef]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, Y.; Yuan, S.; Zhang, P.; Zhang, J.; Li, H.; Li, X.; Shen, H.; Wang, Z.; Chen, G. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 2018, 1701, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Z.; Li, B.; Yao, H.; Zarka, M.; Welch, J.; Sachdev, P.; Bridge, W.; Braidy, N. Supplementation with γ-glutamylcysteine (γ-GC) lessens oxidative stress, brain inflammation and amyloid pathology and improves spatial memory in a murine model of AD. Neurochem. Int. 2021, 144, 104931. [Google Scholar] [CrossRef]
- Mao, X.-Y.; Zhou, H.-H.; Jin, W.-L. Ferroptosis Induction in Pentylenetetrazole Kindling and Pilocarpine-Induced Epileptic Seizures in Mice. Front. Neurosci. 2019, 13, 721. [Google Scholar] [CrossRef]
- Śliwińska, A.; Luty, J.; Aleksandrowicz-Wrona, E.; Małgorzewicz, S. Iron status and dietary iron intake in vegetarians. Adv. Clin. Exp. Med. 2018, 27, 1383–1389. [Google Scholar] [CrossRef]
- Ferris, C.D.; Jaffrey, S.R.; Sawa, A.; Takahashi, M.; Brady, S.D.; Barrow, R.K.; Tysoe, S.A.; Wolosker, H.; Barañano, D.E.; Doré, S.; et al. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat. Cell. Biol. 1999, 1, 152–157. [Google Scholar] [CrossRef]
- Chifman, J.; Laubenbacher, R.; Torti, S.V. A Systems Biology Approach to Iron Metabolism. Adv. Exp. Med. Biol. 2014, 844, 201–225. [Google Scholar] [CrossRef]
- Dong, X.-P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef]
- Hamai, A.; Mehrpour, M. Autophagy and iron homeostasis. Med. Sci. 2017, 33, 260–267. [Google Scholar]
- Garton, T.; Keep, R.F.; Hua, Y.; Xi, G. Brain iron overload following intracranial haemorrhage. Stroke Vasc. Neurol. 2016, 1, 172–184. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Liu, H.; Hua, Y.; Keep, R.F.; Xi, G. Brain Ceruloplasmin Expression After Experimental Intracerebral Hemorrhage and Protection Against Iron-Induced Brain Injury. Transl. Stroke Res. 2019, 10, 112–119. [Google Scholar] [CrossRef]
- Tang, S.; Gao, P.; Chen, H.; Zhou, X.; Ou, Y.; He, Y. The Role of Iron, Its Metabolism and Ferroptosis in Traumatic Brain Injury. Front. Cell. Neurosci. 2020, 14, 590789. [Google Scholar] [CrossRef]
- Qian, Z.M.; Ke, Y. Brain iron transport. Biol. Rev. Camb. Philos. Soc. 2019, 94, 1672–1684. [Google Scholar] [CrossRef]
- DeGregorio-Rocasolano, N.; Martí-Sistac, O.; Gasull, T. Deciphering the Iron Side of Stroke: Neurodegeneration at the Crossroads Between Iron Dyshomeostasis, Excitotoxicity, and Ferroptosis. Front. Neurosci. 2019, 13, 85. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, W.; Jiang, Z.; Tang, X. New progress in the approaches for blood–brain barrier protection in acute ischemic stroke. Brain Res. Bull. 2019, 144, 46–57. [Google Scholar] [CrossRef]
- Lipscomb, D.C.; Gorman, L.G.; Traystman, R.J.; Hurn, P.D. Low Molecular Weight Iron in Cerebral Ischemic Acidosis In Vivo. Stroke 1998, 29, 487–493. [Google Scholar] [CrossRef]
- Palmer, C.; Menzies, S.L.; Roberts, R.L.; Pavlick, G.; Connor, J.R. Changes in iron histochemistry after hypoxic-ischemic brain injury in the neonatal rat. J. Neurosci. Res. 1999, 56, 60–71. [Google Scholar] [CrossRef]
- Selim, M.H.; Ratan, R.R. The role of iron neurotoxicity in ischemic stroke. Ageing Res. Rev. 2004, 3, 345–353. [Google Scholar] [CrossRef]
- Chi, S.I.; Wang, C.K.; Chen, J.J.; Chau, L.Y.; Lin, T.N. Differential regulation of H- and L-ferritin messenger RNA subunits, ferritin protein and iron following focal cerebral ischemia-reperfusion. Neuroscience 2000, 100, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Y.-W.; Zhao, J.-Y.; Liu, Y.-Z.; Holscher, C. Quantitative analysis of iron concentration and expression of ferroportin 1 in the cortex and hippocampus of rats induced by cerebral ischemia. J. Clin. Neurosci. 2009, 16, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, R.B.; Bradley, W.G., Jr. Iron accumulation in the basal ganglia following severe ischemic-anoxic insults in children. Radiology 1988, 168, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Kaye, D.M.; Petrie, M.C.; McKenzie, S.; Hasenfuβ, G.; Malek, F.; Post, M.; Doughty, R.N.; Trochu, J.-N.; Gustafsson, F.; Lang, I.; et al. Impact of an interatrial shunt device on survival and heart failure hospitalization in patients with preserved ejection fraction. ESC Heart Fail. 2019, 6, 62–69. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Hanafy, K.A.; Gomes, J.A.; Selim, M. Rationale and Current Evidence for Testing Iron Chelators for Treating Stroke. Curr. Cardiol. Rep. 2019, 21, 20. [Google Scholar] [CrossRef]
- Yang, T.; Sun, Y.; Li, Q.; Li, S.; Shi, Y.; Leak, R.K.; Chen, J.; Zhang, F. Ischemic preconditioning provides long-lasting neuroprotection against ischemic stroke: The role of Nrf2. Exp. Neurol. 2020, 325, 113142. [Google Scholar] [CrossRef]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Wang, S.; Li, F.; Qiao, R.; Hu, X.; Liao, H.; Chen, L.; Wu, J.; Wu, H.; Zhao, M.; Liu, J.; et al. Arginine-Rich Manganese Silicate Nanobubbles as a Ferroptosis-Inducing Agent for Tumor-Targeted Theranostics. ACS Nano 2018, 12, 12380–12392. [Google Scholar] [CrossRef]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Sánchez-Morán, I.; Rodríguez, C. Mitochondrial–nuclear p53 trafficking controls neuronal susceptibility in stroke. IUBMB Life 2021, 73, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gai, C.; Ding, D.; Wang, F.; Li, W. Targeted p53 on Small-Molecules-Induced Ferroptosis in Cancers. Front. Oncol. 2018, 8, 507. [Google Scholar] [CrossRef]
- Zhao, C.; Yu, D.; He, Z.; Bao, L.; Feng, L.; Chen, L.; Liu, Z.; Hu, X.; Zhang, N.; Wang, T.; et al. Endoplasmic reticulum stress-mediated autophagy activation is involved in cadmium-induced ferroptosis of renal tubular epithelial cells. Free. Radic. Biol. Med. 2021, 175, 236–248. [Google Scholar] [CrossRef]
- Feng, J.; Chen, X.; Shen, J. Reactive nitrogen species as therapeutic targets for autophagy: Implication for ischemic stroke. Expert Opin. Ther. Targets 2017, 21, 305–317. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, 242. [Google Scholar] [CrossRef]
- He, Z.; Ning, N.; Zhou, Q.; Khoshnam, S.E.; Farzaneh, M. Mitochondria as a therapeutic target for ischemic stroke. Free. Radic. Biol. Med. 2020, 146, 45–58. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2019, 99, 151058. [Google Scholar] [CrossRef] [PubMed]
- Basit, F.; Van Oppen, L.M.P.E.; Schöckel, L.; Bossenbroek, H.M.; Van Emst-de Vries, S.E.; Hermeling, J.C.W.; Grefte, S.; Kopitz, C.; Heroult, M.; Willems, P.H.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta. 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Hodge, T.; Colombini, M. Regulation of Metabolite Flux through Voltage-Gating of VDAC Channels. J. Membr. Biol. 1997, 157, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Lemasters, J.J. Warburg Revisited: Regulation of Mitochondrial Metabolism by Voltage-Dependent Anion Channels in Cancer Cells. J. Pharmacol. Exp. Ther. 2012, 342, 637–641. [Google Scholar] [CrossRef]
- Yao, G.-Y.; Zhu, Q.; Xia, J.; Chen, F.-J.; Huang, M.; Liu, J.; Zhou, T.-T.; Wei, J.-F.; Cui, G.-Y.; Zheng, K.-Y.; et al. Ischemic postconditioning confers cerebroprotection by stabilizing VDACs after brain ischemia. Cell Death Dis. 2018, 9, 1033. [Google Scholar] [CrossRef]
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 2018, 148, 155–162. [Google Scholar] [CrossRef]
- DeHart, D.N.; Lemasters, J.J.; Maldonado, E.N. Erastin-Like Anti-Warburg Agents Prevent Mitochondrial Depolarization Induced by Free Tubulin and Decrease Lactate Formation in Cancer Cells. SLAS Discov. Adv. Sci. Drug Discov. 2018, 23, 23–33. [Google Scholar] [CrossRef]
- Yin, F.; Zhou, H.; Fang, Y.; Li, C.; He, Y.; Yu, L.; Wan, H.; Yang, J. Astragaloside IV alleviates ischemia reperfusion-induced apoptosis by inhibiting the activation of key factors in death receptor pathway and mitochondrial pathway. J. Ethnopharmacol. 2020, 248, 112319. [Google Scholar] [CrossRef]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Carocci, A.; Catalano, A.; Sinicropi, M.S.; Genchi, G. Oxidative stress and neurodegeneration: The involvement of iron. BioMetals 2018, 31, 715–735. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Dai, Z.; Wu, P.; Shi, H.; Zhao, S. Lack of mitochondrial ferritin aggravated neurological deficits via enhancing oxidative stress in a traumatic brain injury murine model. Biosci. Rep. 2017, 37, BSR20170942. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Aguirre, P.; Tapia, V.; Carrasco, C.M.; Mena, N.P.; Núñez, M.T. Cell death induced by mitochondrial complex I inhibition is mediated by Iron Regulatory Protein 1. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2202–2209. [Google Scholar] [CrossRef]
- Rouault, T.A.; Maio, N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef]
- Calvelage, S.; Tammiranta, N.; Nokireki, T.; Gadd, T.; Eggerbauer, E.; Zaeck, L.M.; Potratz, M.; Wylezich, C.; Höper, D.; Müller, T.; et al. Genetic and Antigenetic Characterization of the Novel Kotalahti Bat Lyssavirus (KBLV). Viruses 2021, 13, 69. [Google Scholar] [CrossRef]
- Huang, J.; Chen, S.; Hu, L.; Niu, H.; Sun, Q.; Li, W.; Tan, G.; Li, J.; Jin, L.; Lyu, J.; et al. Mitoferrin-1 is Involved in the Progression of Alzheimer’s Disease Through Targeting Mitochondrial Iron Metabolism in a Caenorhabditis elegans Model of Alzheimer’s Disease. Neuroscience 2018, 385, 90–101. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Guan, R.; Zou, W.; Dai, X.; Yu, X.; Liu, H.; Chen, Q.; Teng, W. Mitophagy, a potential therapeutic target for stroke. J. Biomed. Sci. 2018, 25, 87. [Google Scholar] [CrossRef]
- Sharma, B.; Pal, D.; Sharma, U.; Kumar, A. Mitophagy: An Emergence of New Player in Alzheimer’s Disease. Front. Mol. Neurosci. 2022, 15, 921908. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L. Quality Control in Neurons: Mitophagy and Other Selective Autophagy Mechanisms. J. Mol. Biol. 2020, 432, 240–260. [Google Scholar] [CrossRef]
- Mumtaz, S.; Rana, J.N.; Choi, E.H.; Han, I. Microwave Radiation and the Brain: Mechanisms, Current Status, and Future Prospects. Int. J. Mol. Sci. 2022, 23, 9288. [Google Scholar] [CrossRef]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links with Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1443. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimer Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef]
- Zalewska, A.; Klimiuk, A.; Zięba, S.; Wnorowska, O.; Rusak, M.; Waszkiewicz, N.; Szarmach, I.; Dzierżanowski, K.; Maciejczyk, M. Salivary gland dysfunction and salivary redox imbalance in patients with Alzheimer’s disease. Sci. Rep. 2021, 11, 23904. [Google Scholar] [CrossRef]
- Matsuo-Tezuka, Y.; Sasaki, Y.; Iwai, T.; Kurasawa, M.; Yorozu, K.; Tashiro, Y.; Hirata, M. T2* Relaxation Time Obtained from Magnetic Resonance Imaging of the Liver Is a Useful Parameter for Use in the Construction of a Murine Model of Iron Overload. Contrast Media Mol. Imaging 2019, 2019, 7463047. [Google Scholar] [CrossRef] [PubMed]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Fengli, W.; Na, T.; Ting, Z.; Weihua, G. The Multifaceted Regulation of Mitochondria in Ferroptosis. Life 2021, 11, 222. [Google Scholar]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Wible, D.J.; Bratton, S.B. Reciprocity in ROS and autophagic signaling. Curr. Opin. Toxicol. 2018, 7, 28–36. [Google Scholar] [CrossRef]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr. Pharm. Des. 2020, 26, 4246–4260. [Google Scholar] [CrossRef]
- Amro, M.S.; Teoh, S.L.; Norzana, A.G.; Srijit, D. The potential role of herbal products in the treatment of Parkinson’s disease. Clin. Ter. 2018, 169, e23–e33. [Google Scholar]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C.; Richmond, R.; Halliwell, B. Inhibition of the iron-catalysed formation of hydroxyl radicals from superoxide and of lipid peroxidation by desferrioxamine. Biochem. J. 1979, 184, 469–472. [Google Scholar] [CrossRef]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Benkovic, S.A.; Lin, L.; Yonutas, H.M.; Crish, S.D.; Sullivan, P.G.; Darvesh, A.S.; Brown, C.M.; Richardson, J.R. MitoNEET (CISD1) Knockout Mice Show Signs of Striatal Mitochondrial Dysfunction and a Parkinson’s Disease Phenotype. ACS Chem. Neurosci. 2017, 8, 2759–2765. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca2+ in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef]
- Brinckmann, R.; Schnurr, K.; Heydeck, D.; Rosenbach, T.; Kolde, G.; Kuhn, H. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 1998, 91, 64–74. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gele, P.; Petrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Yang, N.; Guan, Q.-W.; Chen, F.-H.; Xia, Q.-X.; Yin, X.-X.; Zhou, H.-H.; Mao, X.-Y. Antioxidants Targeting Mitochondrial Oxidative Stress: Promising Neuroprotectants for Epilepsy. Oxidative Med. Cell. Longev. 2020, 2020, 6687185. [Google Scholar] [CrossRef]
- Albrecht, J.; Zielińska, M. Mechanisms of Excessive Extracellular Glutamate Accumulation in Temporal Lobe Epilepsy. Neurochem. Res. 2017, 42, 1724–1734. [Google Scholar] [CrossRef]
- Boison, D.; Steinhäuser, C. Epilepsy and astrocyte energy metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef]
- Fulton, R.E.; Pearson-Smith, J.N.; Huynh, C.Q.; Fabisiak, T.; Liang, L.-P.; Aivazidis, S.; High, B.A.; Buscaglia, G.; Corrigan, T.; Valdez, R.; et al. Neuron-specific mitochondrial oxidative stress results in epilepsy, glucose dysregulation and a striking astrocyte response. Neurobiol. Dis. 2021, 158, 105470. [Google Scholar] [CrossRef]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef]
- Mueller, S.G.; Trabesinger, A.H.; Boesiger, P.; Wieser, H.G. Brain glutathione levels in patients with epilepsy measured by in vivo 1H-MRS. Neurology 2001, 57, 1422–1427. [Google Scholar] [CrossRef]
- Diao, X.; Zhou, Z.; Xiang, W.; Jiang, Y.; Tian, N.; Tang, X.; Chen, S.; Wen, J.; Chen, M.; Liu, K.; et al. Glutathione alleviates acute intracerebral hemorrhage injury via reversing mitochondrial dysfunction. Brain Res. 2020, 1727, 146514. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, Z. Ferroptosis and Its Role in Epilepsy. Front. Cell. Neurosci. 2021, 15, 696889. [Google Scholar] [CrossRef]
- Kurzatkowski, D.M.; Trombetta, L.D. Maneb causes pro-oxidant effects in the hippocampus of Nrf2 knockout mice. Environ. Toxicol. Pharmacol. 2013, 36, 427–436. [Google Scholar] [CrossRef]
- Ye, Q.; Zeng, C.; Dong, L.; Wu, Y.; Huang, Q.; Wu, Y. Inhibition of ferroptosis processes ameliorates cognitive impairment in kainic acid-induced temporal lobe epilepsy in rats. Am. J. Transl. Res. 2019, 11, 875–884. [Google Scholar]
- Zhang, P.; Chen, L.; Zhao, Q.; Du, X.; Bi, M.; Li, Y.; Jiao, Q.; Jiang, H. Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Radic. Biol. Med. 2020, 152, 227–234. [Google Scholar] [CrossRef]
- Lu, J.; Xu, F.; Lu, H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020, 260, 118305. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, K.; Zhou, H.; Jiang, L.; Xie, B.; Wang, R.; Xia, W.; Yin, Y.; Gao, Z.; Cui, D.; et al. Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in Alzheimer’s Disease. Aging Cell. 2020, 19, e13125. [Google Scholar] [CrossRef]
- Morrison, R.S.; Kinoshita, Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000, 7, 868–879. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhai, Y.; Chen, J.; Xu, X.; Wang, H. Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules 2021, 11, 923. [Google Scholar] [CrossRef]
- Wu, B.; Luo, H.; Zhou, X.; Cheng, C.-Y.; Lin, L.; Liu, B.-L.; Liu, K.; Li, P.; Yang, H. Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: Therapeutical effects of kaempferol. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2307–2318. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y.; Guo, S.-Q.; Wang, S.; Guo, T.; Wang, Z.-Y.; et al. α-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxid. Med. Cell. Longev. 2019, 2019, 8409329. [Google Scholar] [CrossRef]
- Avcı, B.; Günaydın, C.; Güvenç, T.; Yavuz, C.K.; Kuruca, N.; Bilge, S.S. Idebenone Ameliorates Rotenone-Induced Parkinson’s Disease in Rats Through Decreasing Lipid Peroxidation. Neurochem. Res. 2021, 46, 513–522. [Google Scholar] [CrossRef]
- Yan, J.; Sun, W.; Shen, M.; Zhang, Y.; Jiang, M.; Liu, A.; Ma, H.; Lai, X.; Wu, J. Idebenone improves motor dysfunction, learning and memory by regulating mitophagy in MPTP-treated mice. Cell Death Discov. 2022, 8, 28. [Google Scholar] [CrossRef]
- Shekh-Ahmad, T.; Lieb, A.; Kovac, S.; Gola, L.; Wigley, W.C.; Abramov, A.Y.; Walker, M.C. Combination antioxidant therapy prevents epileptogenesis and modifies chronic epilepsy. Redox Biol. 2019, 26, 101278. [Google Scholar] [CrossRef]
- Kahn-Kirby, A.H.; Amagata, A.; Maeder, C.I.; Mei, J.J.; Sideris, S.; Kosaka, Y.; Hinman, A.; Malone, S.A.; Bruegger, J.J.; Wang, L.; et al. Targeting ferroptosis: A novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy. PLoS ONE 2019, 14, e0214250. [Google Scholar] [CrossRef]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile Response Element-mediated Induction of the Cystine/Glutamate Exchange Transporter Gene Expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef]
- Lee, J.-M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related Factor-2-dependent Genes Conferring Protection against Oxidative Stress in Primary Cortical Astrocytes Using Oligonucleotide Microarray Analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.-J.; Gao, Y.; Bennett, M.V.L.; et al. Peroxisome proliferator-activated receptor gamma (PPARγ): A master gatekeeper in CNS injury and repair. Prog. Neurobiol. 2018, 163–164, 27–58. [Google Scholar] [CrossRef]
- Itoh, T.; Fairall, L.; Amin, K.; Inaba, Y.; Szanto, A.; Balint, B.L.; Nagy, L.; Yamamoto, K.; Schwabe, J.W.R. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct. Mol. Biol. 2008, 15, 924–931. [Google Scholar] [CrossRef]
- Lee, C. Collaborative Power of Nrf2 and PPARγ Activators against Metabolic and Drug-Induced Oxidative Injury. Oxid. Med. Cell. Longev. 2017, 2017, 1378175. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A ΔΨm independent pharmacological regulator of mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingson, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.; Fiskum, G. Brain mitochondria from rats treated with sulforaphane are resistant to redox-regulated permeability transition. J. Bioenerg. Biomembr. 2010, 42, 491–497. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Interventions | Mechanism for Mitochondrial Dysfunction | Mechanism for Ferroptosis | References |
|---|---|---|---|
| Kaempferol |
|
| [206,207] |
| α-Lipoic acid (LA) |
|
| [208,209] |
| Idebenone |
|
| [210,211] |
| RTA 408 |
|
| [212] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, H.-Y.; Huang, B.-Y.; Nie, H.-F.; Chen, X.-Y.; Zhou, Y.; Yang, T.; Cheng, S.-W.; Mei, Z.-G.; Ge, J.-W. The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases. Brain Sci. 2023, 13, 1367. https://doi.org/10.3390/brainsci13101367
Tian H-Y, Huang B-Y, Nie H-F, Chen X-Y, Zhou Y, Yang T, Cheng S-W, Mei Z-G, Ge J-W. The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases. Brain Sciences. 2023; 13(10):1367. https://doi.org/10.3390/brainsci13101367
Chicago/Turabian StyleTian, He-Yan, Bo-Yang Huang, Hui-Fang Nie, Xiang-Yu Chen, Yue Zhou, Tong Yang, Shao-Wu Cheng, Zhi-Gang Mei, and Jin-Wen Ge. 2023. "The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases" Brain Sciences 13, no. 10: 1367. https://doi.org/10.3390/brainsci13101367
APA StyleTian, H.-Y., Huang, B.-Y., Nie, H.-F., Chen, X.-Y., Zhou, Y., Yang, T., Cheng, S.-W., Mei, Z.-G., & Ge, J.-W. (2023). The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases. Brain Sciences, 13(10), 1367. https://doi.org/10.3390/brainsci13101367