Abstract
7q11.23 Microduplication (dup7q11.23) syndrome is a rare autosomal dominant disorder due to a recurring 1.5 to 1.8 Mb duplication of the Williams–Beuren Syndrome critical region. Dup7q11.23 has been associated with several neuro-behavioral characteristics such as low cognitive and adaptive functioning, expressive language impairment, anxiety problems and autistic features. In the present study, we analyze the clinical features of ten individuals in which array-CGH detected dup7q11.23, spanning from 1.4 to 2.1 Mb. The clinical characteristics associated with dup7q11.23 are discussed with respect to its reciprocal deletion. Consistent with previous studies, we confirm that individuals with dup7q11.23 syndrome do not have a homogeneous clinical profile, although some recurring dysmorphic features were found, including macrocephaly, prominent forehead, elongated palpebral fissures, thin lip vermilion and microstomia. Minor congenital malformations include patent ductus arteriosus, cryptorchidism and pes planus. A common finding is hypotonia and joint laxity, resulting in mild motor delay. Neuropsychological and psychodiagnostic assessment confirm that mild cognitive impairment, expressive language deficits and anxiety are recurring neurobehavioral features. New insights into adaptive, psychopathological and neurodevelopmental profiles are discussed.
1. Introduction
Interstitial deletions of 7q11.23 cause Williams–Beuren Syndrome (WBS) (MIM 194050). Most deletions are de novo, while inherited deletions are very rare [1,2]. WBS is a distinct neurodevelopmental disorder easily recognized on the basis of a few key features, such as a characteristic facial gestalt, supravalvar aortic stenosis (SAS), prenatal growth deficiency, failure to thrive in infancy and intellectual disability (ID) [3,4]. Sleep disorders, including increased sleep latency and decreased sleep efficiency, occur in 65% of these patients [5]. Reduced grey matter volume in the intraparietal sulcus, occipito-parietal sulcus, brain stem and occipital lobe regions have been detected consistently by brain magnetic resonance imaging (MRI) [6]. Of note, seizures rarely occur [7].
More recently, the reciprocal microduplication of the same chromosomal region implicated in WBS has been identified in several patients who underwent array comparative genomic hybridization analysis (array-CGH) because of ID, language impairment, mild facial dimorphisms and, less frequently, congenital malformations [8,9,10]. Parental transmission of dup7q11.23 is quite common [11]. Unlike its reciprocal deletion, dup7q11.23 is not associated with distinct clinical features. However, some recurring dysmorphic features have been reported, including broad forehead (46%), straight brow line (43%), deep set eyes (33%), high/broad tip (56%), abnormal columella (78%), thin upper lip (64%), and short philtrum (59%) [10]. Macrocephaly occurs in 30–50% of affected individuals [9,10], while 17% of cases display short stature [12]. Dilation of the ascending aorta (DAA) and patent ductus arteriosus (PDA), occurring in approximately 45% and 15% of individuals, respectively, are the most common cardiovascular defects [12]. Seizures are found in 17% of cases and cryptorchidism in 15% of the male population [8,12,13].
To the best of our knowledge, very little information has been collected on the sleep pattern of children with dup7q11.23 syndrome. About 80% of reports have documented abnormal brain MRI, including ventriculomegaly, thin corpus callosum, increased extra-axial spaces, thin white matter, delayed myelination, posterior fossa cysts, and cerebellar vermis hypoplasia [10,12,13,14,15]. However, no recurring distinguishing feature has been reported [10].
Abnormalities in collagen due to an abnormal expression of the ELN gene is found both in WBS and dup7q11.23 syndrome [16,17].
A considerable number of studies have explored the neurobehavioral features of individuals with WBS [3,18,19,20,21,22,23,24]. Several of them have reported mild to severe ID with severely impaired visual spatial abilities, impaired receptive language skills and relatively spared expressive language skills [3,22,23,24,25]. Some investigations have reported that anxiety problems occur in 16.5% to 82.2% of WBS individuals [26,27]. Individual with WBS are well known for their enhanced social motivation (i.e., “hypersociable behaviors”) [3,24]. Nonetheless, some recent studies have highlighted socio-communicative problems in children with WBS, including deficits in shared attention, pragmatic use of language, the understanding of social relationships and emotion comprehension. [24,28]. Thus, enhanced social motivation does not implicate better social skills in individuals with WBS.
Relatively few studies have investigated the neurobehavioral profile of children with dup7q11.23 syndrome [8,9,29,30]. Studies on cognitive and developmental levels reported variable scores ranging from moderate ID/global developmental delay (DD) to average cognitive/developmental level [8,29,30]. The adaptive level of children with dup7q11.2 is usually below the normal range (mild to moderate impairment), and generally lower than their intelligence quotient [9,30,31,32].
Some studies have highlighted the presence of both receptive and/or expressive language deficits, as well as oral motor and vocal sound disorders [13,25,29]. Some of these studies suggest that expressive skills are more severely affected than receptive ones [25,29]. Psychopathological features have also been investigated in past research, reporting that most children with dup7q11.23 syndrome meet the criteria for diagnosis of specific phobias and/or social anxiety [9,29]. More recently, autism spectrum disorder (ASD) [9,30] has been considered a relatively common feature in children with dup7q11.23 syndrome (33%) [9]. However, the use of gold standard diagnostic tools combined with clinical judgments disclosed a lower presence of ASD in this population (around 19%) [30]. The discrepancy between diagnostic tools (parental report vs. clinician report) could be partially explained by the severe social anxiety described in several studies [9,29] which could mime autism-like behaviors [30].
Past studies have contributed to characterizing the phenotype of dup7q11.23 individuals, but only a few of them have comprehensively assessed the neurobehavioral features in representative patients’ samples [9]. Some neuropsychological features, such as visual–motor skills, have not yet been investigated. Therefore, additional studies are warranted in order to extend the available data on neurobehavioral features of dup7q11.23 individuals.
In this study, ten patients with dup7q11.23 identified by array-CGH are described. We provide data on clinical features, cognitive level, adaptive profile, language skills, behavioral problems and psychopathological features. We discuss the present results with published data of patients with duplication and deletion (WBS) 7q11.23 syndromes, in order to outline distinguishing and shared clinical features.
2. Materials and Methods
2.1. Participants
The cohort included ten children with typical dup7q11.23. There were three females and seven males. The mean (M) age was 97.2 months (Standard Deviation (SD): 30.07). All participants were recruited at the Child and Adolescent Psychiatry Unit and Genetics and Rare Diseases Research Division of the Bambino Gesù Children’s Hospital, Rome.
2.2. Cytogenetics and molecular cytogenetics
DNA was extracted from whole peripheral blood by means of Qiagen blood and tissue kit (https://www.qiagen.com), according to the manufacturer’s instructions. CMA (chromosomal microarray analysis) was performed using the Agilent Oligo arrays 8 × 60K (patients 1, 2, 3, 6, 8 and 9), Agilent Oligo arrays 4 × 180K (patients 4, 5, 10) and Illumina CytoSNP 850K (patient 7), using standard procedures. Images were obtained using an Agilent DNA Microarray Scanner and Agilent Scan control Software (v A.8.4.1), and analyses were performed by Agilent CytoGenomics (v 4.0.3.12) or Illumina BlueFuse Multi (v 4.4), depending on the platform. Validation of the genomic rearrangement and segregation analysis was performed by FISH (fluorescence in situ hybridization) on metaphases obtained by lymphocyte cultures from the patients and their parents (when available). Vysis Williams Region Probe–LSI ELN was used for this purpose (https://www.molecular.abbott/int/it/vysis-fish-chromosome-search).
2.3. Neurobehavioral Assessment
Cognitive Level: Cognitive level was assessed by means of appropriate developmental tools—Wechsler Intelligence Scale for Children—Fourth Edition (WISC-IV) [33] and Leiter International Performance Scale-Third Edition (Leiter-3) [34]. WISC-IV is a tool utilized to evaluate cognitive abilities in patients older than 6 years. The test provides four indices: verbal comprehension index (VCI), perceptual reasoning index (PRI), working memory index (WMI), speed processing index (PRI) plus a full scale intelligence quotient (FSIQ). When participants showed severe communicational impairment, Leiter-3 was used to assess the non-verbal cognitive profile. Leiter-3 provides for a non-verbal intelligence quotient (NVIQ).
Adaptive level: The adaptive level was assessed by using Vineland Adaptive Behavior Scales—Second Edition (VABS II) [35], a semi-structured interview with the caregiver. VABS II provides four domains (communication, daily living skills, socialization and motor skills). Adaptive behavior composite (ABC), a global index of adaptive behavior, corresponds to the sum of four domains. Only in one case was Adaptive Behavior Assessment System—Second Edition (ABAS II) [36], a parent-report tool for adaptive behavior measurement, administered. ABAS II provides a composite adaptive behavior score, and four domains: conceptual, social and practical. Although we used different tools for the adaptive functioning, it was possible to combine the scores referring to similar domains given the high correlation between domains of tools [36].
Lexical Production Skills: Lexical production was evaluated by means of the following developmentally appropriate tools:
- Expression subtest of the PhonoVocabulary Test (TFL, Test Fono-Lessicale) [37]. The expression subtest contains 45 tables. The examiner indicates a picture on the table and asks the participant to label the targeted picture;
- The Boston Naming Test [38]. In this test, the participant is asked to label the name of each shown picture. Participants have about 20 s to answer each question;
- Expression subtest of “Batteria per la Valutazione del Linguaggio 4-12” (BVL 4–12) [39]. In the expression subtest, participants are required to label 67 images (51 nouns and 16 verbs);
- Expression subtest of “Parole in Gioco” (PinG) [40]. In this subtest, the child must name the pictures shown.
Lexical Comprehension Skills: Lexical comprehension was evaluated by means of the following developmental appropriate tools:
- Receptive subtest of PhonoVocabulary Test (TFL, Test Fono-Lessicale) [37]. The receptive subtest contains 45 tables with four pictures. Participants must choose the picture labeled by examiner;
- Peabody Picture Vocabulary Test–PPVT [41]. In this test the examiner says a word describing one of four drawings and asks the participant to point to the labeled drawing. Raw total score is converted into Lexical Quotient (LQ);
- Receptive subtest of BVL 4-12 [39]. In this test the examiner says a word describing one of four images and asks the participant to point to the labeled images. This subtest contains 42 words (31 nouns, 10 verbs, and 1 adjective);
- Receptive Subtest of Parole in Gioco (PinG) [40]. In this subtest three pictures are shown and the subject must point to the picture corresponding to the word labeled by the examiner.
Grammar Comprehension Skills: Grammar comprehension skills were evaluated by using the following developmental appropriate tools:
- Grammar Evaluation Test (PVCL, Prove di Valutazione della Comprensione Linguistica) [42]. In this test, the examiner pronounces a sentence describing one of four pictures. Then, the examiner asks participants to choose the correct picture;
- Grammar Evaluation Subtest of BVL 4-12 [39]. In syntactic comprehension subtest, children are asked to recognize each of the 40 examiner’s sentences within one of four pictures.
Visuomotor integration skills: Visuomotor integration skills were investigated by means of the Beery–Buktenica Developmental Test of Visual–Motor Integration (VMI) [43]. In this assessment, the child is required to copy some drawings. The drawings are shown in an increasingly difficulty order. Use of perception (VP) and motor coordination (MC) subtests allow us to assess one skill set, excluding the other.
Behavioral/Psychopathological Problems (Parent Report): The parent-report version of the Child Behavior Checklist (CBCL) [44] was used to measure problem behavior. Both Child Behavior Checklist 1½–5 (CBCL 1½–5) and Child Behavior Checklist 6–18 (CBCL 6–18) were used. Item scoring provides the following subscales: Anxious/ Depressed, Withdrawn/ Depressed, Somatic Complaints, Social Problems, Thought Problems, Attention Problems, Rule-Breaking Behavior, Aggressive Behavior. Furthermore, three composite scales can be computed (Externalizing, Internalizing and a Total Problem). Conners’ Parent Rating Scales-Long Version, Revised (CPRS-R-L) [45], is a parent-report questionnaire that is administered to assess attention deficits and hyperactivity disorders (ADHD) symptoms as well as other symptoms of behavioral and emotional disorders commonly associated based on DSM-IV-TR [46] criteria.
Behavioral/Psychopathological Problems (Clinician Report): Clinical history and actual psychiatric symptomatology were investigated through unstructured interviews performed by a Child Neuropsychiatrist and Licensed Clinical Psychologist. When applicable, the semi-structured interview Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL) [47] was used to evaluate psychiatric comorbidities in our clinical group. Furthermore, the Children’s Global Assessment Scale (C-GAS) was utilized to assess the psychosocial function of the participants [48]. C-GAS scores (ranging from 1 to 100) is a measurement independent by specific diagnosis. Higher scores indicates better functioning. A best-estimate consensus diagnosis was established for participants by a multidisciplinary team (at least one child psychiatrist and one clinical psychologist) based on psychiatric evaluation, psychopathological/behavioral questionnaires and, when administrable, semi-structured interview [47]. Diagnoses were based on Diagnostic and Statistical Manual of mental disorders–Fourth Edition –Text revised (DSM-IV-TR) [46].
Autism symptoms (Parent report): Autism Symptoms were assessed by means of Social Responsiveness Scale (SRS) [49] a widely used parent report questionnaire that evaluates a child’s social awareness, cognition, communication, motivation, and mannerisms. A total score is also available. Higher scores indicate greater social communication difficulties.
Autism symptoms (Clinician report): a clinician report assessment of autism symptoms was performed by means of Childhood Autism Rating Scale 2 (CARS-2) [50]. CARS-2 has 15 items scoring from 1 (normal behavior) to 4 (severely abnormal behavior). Clinician reports were based on the observation of the child and a parental questionnaire to assign scores. Higher total scores indicate more severe symptoms of autism.
2.4. Procedure
Patients were selected among children referred to our services between 2015 and 2020, because of psychomotor delay, and/or severe language impairment, and/or ASD and/or, more rarely, non-specific facial dysmorphisms. Only participants showing typical dup7q11.23 confirmed by FISH were included in this study. Nine of the ten selected participants were evaluated through neurobehavioral assessment tools. The study was approved by the ethics committee of the Bambino Gesù Children Hospital, Rome (approval number 590). All participating patients/families provided signed informed consent.
3. Results
Results are summarized in Table 1, Table 2, Table 3 and Table 4, and additional information is available in Supplementary Materials.

Table 1.
Clinical features of our cohort of 10 patients affected by 7q11.23 microduplication syndrome.
Table 2.
Cognitive, adaptive, language and visuo-motor assessment.
Table 3.
Autism symptoms, behavioral and psychopathological assessment.
Table 4.
Presence/absence of anxiety/phobias, aggressive behaviors, separation anxiety/social inhibition, and repetitive behaviors/movements, disinhibition.
3.1. Cytogenetics and Molecular Cytogenetics Analyses
Ten patients with dup7q11.23 were grouped out, their features were compared to one another’s and to the clinical features commonly reported in this syndrome (OMIM # 609757, https://omim.org/).
The duplications spanned 1.4 Mb to 2.1 Mb, and all involved the core region reported as critical for the duplication syndrome. Of note, 4 out them presented with atypical breakpoints, spanning proximally or distally the recurrent ones. In particular, the duplication detected in P4 and P8 extended towards the telomere, involving two further OMIM genes NCF1 and GTF2IRD2, while the ones detected in P6 and P7, expanding towards the centromere, involved the extra OMIM genes NSUN5, TRIM74 and POM121A. Details about breakpoints and gene content are summarized in Table 1 and Figure 1.
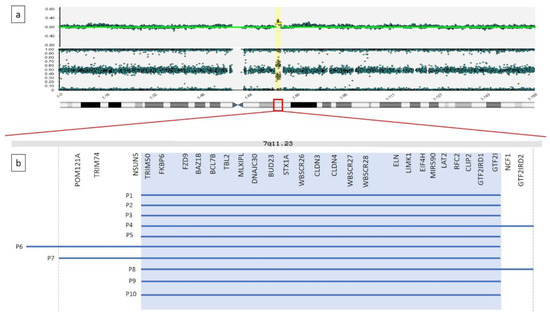
Figure 1.
(a) Graphical representation of SNP-array result for patient 7. A microduplication is detected on the long arm of a chromosome 7, at 7q11.23, 1.9 Mb in size. (b) Enlargement of 7q11.23 genomic region. Gray background shadow highlights the OMIM genes normally included within the recurrent breakpoints reported for the microduplication syndrome. Blue lines represent the microduplications detected in our 10 patients.
3.2. Clinical Features
Table 1 summarizes the cytogenetic, clinical and developmental findings in the present cohort of dup7q11.23 individuals. Figure 2 illustrates the facial features of patients 2, 4, 7 and 9.
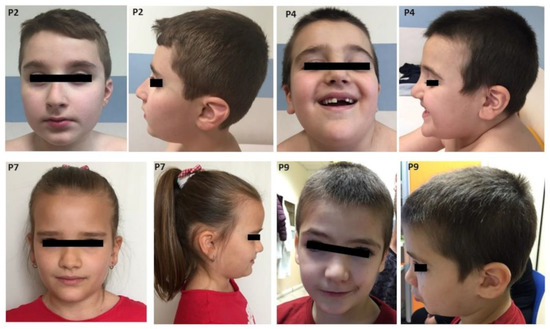
Figure 2.
Photographs of patients with a 7q11.23 microduplication (P2, P4, P7, P9). Note macrocephaly, prominent forehead in some patients, elongated palpebral fissures, deep set eyes, palpebral ptosis, high prominent nasal bridge with bulbous nasal tip, short philtrum and thin vermillion of the lips.
Growth and nutrition: Growth parameters were within normal range in the majority of the individuals and in none of them height was below the 3rd centile.
Facial features: Recurring facial features included macrocephaly (3/8), prominent forehead (6/8), elongated palpebral fissures (7/10). Prominent nose was a common finding, either with a high nasal bridge or a bulbous nasal tip (7/8). Six individuals had thin lip vermilion and three microstomia.
Hearing loss: Hearing loss was detected only in one individual presenting a mild conductive defect due to tubal stenosis.
Congenital heart defects: Congenital heart disease was present in 5/10 patients, including PDA (3/10), pulmonary valve dysplasia (1/10), mild DAA (1/10), mild tricuspid insufficiency (1/10), left ventricular hypertrabeculation and mild aortic insufficiency (1/10).
Musculoskeletal and motor milestone: Both hypotonia and joint laxity were quite common (4/10), resulting in mildly delayed achievement of motor milestones. Four patients presented pes planus (4/10).
Genitourinary system: Features also included cryptorchidism (2/7).
Minor clinical features: Other minor clinical features are listed in Table 1, including periodic migraine (2/10), and single cases with refractive errors (hypermetropia and astigmatism), hepatic steatosis, asthmatic bronchitis and bronchospasm.
Sleep problems: Only one individual reported an irregular sleep pattern resolved after the age of 4.
Brain malformations: A brain MRI was performed on four individuals who did not have any distinct malformation. Thin corpus callosum was recorded in two of them.
Cognitive level: Neurobehavioral results are illustrated in Table 2, Table 3 and Table 4. No participant had a normal cognitive level, the results ranging from borderline to mild delay; only one child displayed moderate cognitive impairment.
Adaptive functioning: All participants had an adaptive behavior level below the normal range. The adaptive level tanged from mild to moderate delay, usually being below the cognitive level of the children. An analysis of adaptive relative strengths and weakness showed that the Socialization and Daily Living Skills domains were relatively higher compared to the Communication domain. Language skills: Results on language assessment disclosed global impairment of lexical production and comprehension skills, and grammar comprehension skills in the majority of the children, only one of them having spared lexical production and comprehension skills.
Visuo-motor integration skills: Visuo-motor integration skills were average in 3 of 9 children, while 6 had scores below average. The total sample ranged from borderline to moderate level (M = 75.22; SD = 18.59).
Behavioral problems: Analysis of questionnaires revealed several emotional, behavioral, and attention problems. CBCL (available in 8 participants) [44] showed borderline/clinical results in “Withdrawn/ Depressed” in 50% of children in the study, and “Anxious/ Depressed” and “Social Problems” scales in 37.5% of them. “Internalizing problems” reached the borderline/clinical score in 50% of children. CPRS-R-L (available in 8 participants) [45] revealed that “Cognitive problems/inattention”, “Anxious–shy”, “Perfectionism”, “Social Problems”, “DSM–IV inattentive”, and “DSM–IV total” were in borderline/clinical range in half of the participants.
Psychiatric diagnosis and functioning: The most recurring psychopathological diagnoses assigned by best estimate consensus diagnosis were “Anxiety Disorders” (DSM IV-TR) [46] in 78% of participants. Analysis of C-GAS (available in 5 children) [48] showed a “moderate degree of impairment” in 3 participants, while two subjects had “variable functioning” and “major impairment”.
Autism symptoms: Evaluation of autism symptoms assessed by parent report tool disclosed in 71% of participants mild to severe impairment in at least one scale of SRS [49]. However, only one child met the cut-off for ASD in the clinician report tool CARS-2 [50] even if he does not reach full criteria for ASD by following best estimate consensus diagnosis.
4. Discussion
The present study provides detailed clinical description of ten subjects with dup7q11.23. This rearrangement results in a mild non-characteristic phenotype, thus preventing the possibility to reach the diagnosis based on clinical criteria only [13,51]. However, a few facial features are recurring, including macrocephaly, prominent forehead, deep set eyes, elongated palpebral fissures, short philtrum and thin lips (see Table 1). In general, our results support previous observations [9,10]. The duplications were extended from 1.4 Mb to 2.1 Mb and they all involved the core region reported as critical for the duplication syndrome. Four patients presented atypical breakpoints, spanning proximally or distally the recurrent region. In particular, the duplication detected in P4 and P8 extended towards the telomere, involving NCF1 and GTF2IRD2 genes, while the ones detected in P6 and P7, expanding towards the centromere, involved the NSUN5, TRIM74 and POM121A genes (Table 1 and Figure 1). No other CNV of significant relevance was found. Physical examination of the patients with a larger duplication were compared to the subjects with the duplication spanning the core critical region (1.4 Mb) and no gross phenotypic differences were highlighted (Table 1). Of note, P7 presented only mild facial dimorphism (Figure 1) and no congenital malformation beside PDA, which was the most recurrent coronary heart disease (CHD) reported in P5 and P9 that present the typical 1.4 Mb recurrent microduplication. Some facial features can be regarded as countertypes of periorbital fullness, short nose, long smooth philtrum and thick vermilion of the upper and lower lips found in children with WBS. In WBS, the most common vascular disease is SAS, followed by peripheral pulmonic stenosis that is common in infancy but usually improves over time [4]. The most common cardiovascular disease of dup7q11.23 is DAA, occurring in 46% of the patients [12]. We found DAA in one subject only, while PDA was the most common cardiovascular disease in the present series of patients. The frequency of PDA overlaps previous studies [12]. Hypotonia and joint laxity were common in our patients. This is not surprising considering the abnormal gene dose of ELN in deletion and dup7q11.23 syndromes [16,17].
As far as we know, this is the first study of sleep problems in dup7q11.23 children, which has excluded the presence of major problems, differently from what is generally observed in individuals with WBS [5]. In addition, none of our patients manifested epileptic seizures, a rare complication also in WBS [7].
WBS is characterized by prenatal growth deficiency, followed by a poor linear growth in the first years of life, a normal rate of linear growth in 75% of cases during childhood, and a mean adult height below the third centile. Our dup7q11.23 patients displayed a normal linear growth, differently from published data pointing to short stature in about 17% of cases [12].
We also confirm a rare association with hearing loss, which is reported in 5% of dup7q11.23 individuals [12], a figure much lower compared to WBS in which mild to moderate progressive sensorineural hearing loss occurs in about two thirds of pediatric cases [52]. Our data on cryptorchidism support previous findings [12,13], suggesting a slightly higher association in subjects with duplication compared with those with deletion [53]. Differently from WBS children [54], present results corroborate a grossly normal brain structure in dup7q11.23 subjects, apart for the occasional occurrence of thin corpus callosum [12].
In agreement with previous studies [13], and differently from WBS [1,2], we found parental transmission of the genomic imbalance in 3/8 families. Retrospective evaluation of heterozygous parents disclosed variable developmental delays with learning difficulties and language impairment in infancy.
Neurobehavioral phenotype
All except one of our children displayed mild cognitive impairment. Available data suggests that cognitive impairment in children with dup7q11.23 syndrome is less severe than in WBS. However, comparative studies on larger patients’ cohorts are needed to support this conclusion.
All our subjects had adaptive profiles below their cognitive level. Adaptive behavior is defined as “the collection of conceptual, social, and practical skills that are learned and performed by people in their everyday lives” [55]. The analysis of adaptive relative strengths and weakness provided some interesting insights. In particular, the Communication domain appears to be a severely impaired area differently from previous studies who which showed that “Communication and Socialization” domains were relative strengths [9]. As far as we know, this is the first study reporting separate data on adaptive Communication and Socialization domains, previous studies used combined scales with a common domain for both skills [9]. Our results support that Socialization is a relative strength, even if below the normal range [9]. Weakness in adaptive Communication domain of children with dup7q11.23 enrolled in the present study differs from results obtained in children with WBS, where Communication is usually a relative strength above other domains [23,56,57,58].
The present data on adaptive communication are not unexpected, given that language impairment is invariably reported in published studies [12,29]. We found that lexical production, lexical comprehension, and grammar comprehension were impaired in the majority of our children. Since we used different assessment tools, we cannot provide any direct comparison of lexical production and lexical comprehension results.
Data on visual–motor integration have shown visual–motor skills at borderline level. As far as we know, this is the first study providing this assessment in dup7q11.23 children, differently from WBS, in which visual motor major impairments have been assessed and documented [59,60].
We found that about half of our children displayed anxiety, social and attentional problems based on parent-report tools. However, best estimate consensus diagnoses have shown that Anxiety Disorders are very common, while Disruptive Behavior Disorders Not Otherwise Specified are less common. In agreement with previous studies, we found that Anxiety Disorders affected more than 75% of dup7q11.23 patients [22]. Furthermore, the prevalence of Anxiety Disorders overlaps with the results obtained in children with WBS [26,27].
Surprisingly, despite several participants showing socially inhibited behaviors, none of our children met the full criteria for Social Phobia diagnosis. This is in contrast with previous results indicating about 50% of dup7q11.23 children having Social Anxiety [22]. In most cases, the qualitative analysis of psychiatric evaluations (Table 4) disclosed a combination of different symptoms that do not meet the criteria for specific disorders—anxiety/phobias, aggressive behaviors, separation anxiety/social inhibition, repetitive behaviors/movements, and social disinhibition. In agreement with previous studies [61], we suggest that some of these symptoms could be related in part to dysregulation of the oxytocin hormone. Past research [61] has identified associations between enhanced social motivation in WBS and elevated oxytocin, suggesting that the typical profile of this syndrome could be related to this neurohormone. Some authors [62] have highlighted that levels of oxytocin are positively associated with social approach and talking with strangers in an experimental environment, while they are negatively associated with social adaptive functioning. Furthermore, these authors found higher oxytocin levels in subjects with WBS when compared to control groups (approximately three times higher). On the contrary, in non-clinical populations, decreased oxytocin is linked to aggressive behaviors [63,64,65] and higher separation anxiety [66]. Thus, authors conclude that oxytocin could play a role in phenotypical expression of deletion vs. duplication syndrome. However, further experimental studies are needed to explore these hypotheses.
We found that 11% of our patients met the cut-off for ASD on the clinicians’ report tool, while a more significant proportion of children showed autistic-like behaviors, such as poor eye contact, repetitive behaviors and motor stereotypies, when evaluated by the self–parent tool. However, none of our children met the full diagnostic criteria for ASD following best estimate consensus diagnoses. Our data differ from previous figures on the prevalence of ASD in dup7q11.23 syndrome (19%) [67] and WBS (30/35%) [67,68]. While the use of different tools (CARS-2 [50] vs. Autism Diagnostic Observation Schedule—Second Edition [69]) could explain part of the difference in the prevalence of children who met instrument cut-offs [70], the present findings suggest a possible overestimation of ASD in dup7q11.23 individuals. The combination of symptoms like anxiety/phobias, separation anxiety/social inhibition, and repetitive behaviors/movements (Table 4) could have been inferred as ASD.
The present parental reports have highlighted that 44% of the children displayed socially disinhibited behaviors, such as hugging unfamiliar people (occasionally in alternation with social inhibited behavior) or showing sexual parts. So far, these behaviors have not been investigated in depth by using specific assessment tools. Future investigations should consider these tools for assessing social motivation.
The heterogeneous psychopathological and neurodevelopmental profiles emerging from the present study cast some doubt on the phenotype of dup7q11.23 syndrome as “opposite” to WBS. One could speculate that the psychopathological and neurodevelopmental heterogeneity could be associated in part with “social dysregulation” features. In fact, our children do not appear to be completely avoidant or disengaged from social relationships. These observations appear to be supported by the relative strength in socialization upon adaptive behavior evaluation. We suggest that children with dup7q11.23 might have difficulty in emotional regulation during social interaction, bringing them alternatively towards aggressive behaviors, social inhibition/separation anxiety or social disinhibited behaviors. However, further experimental studies on social motivation, social orientation and social engagement in these children are warranted to support this hypothesis.
5. Conclusions
In conclusion, the present study extends the available data on genotype–phenotype correlations in individuals with dup7q11.23, and provides new insight into the diagnosis, management and treatment of affected individuals. We confirm that facial dysmorphisms are not characteristic, although some recurring features, including macrocephaly, prominent forehead, elongated palpebral fissures, prominent nose, thin lip vermilion and microstomia, could provide some clues to diagnosis. We found that the adaptive and cognitive impairment of dup7q11.23 children is milder compared to WBS. The analysis of neurobehavioral profile has shown that the communication adaptive level is the most impaired domain compared to other adaptive domains. Differently from WBS, we found that dup7q11.23 individuals display only minor impairment in visual–motor integration, and confirm that language impairment is a major problem in most of the children studied so far. In agreement with previous studies, we confirm that the more common psychopathological issues include a wide spectrum of anxiety disorders, not necessarily social anxiety. The presence of isolated social disinhibited behaviors reported in some of our patients warrants further investigation in future studies. Finally, our data suggest that ASD could affect only a small proportion of dup7q11.23 individuals.
This study has some limitations, given the relatively small sample of evaluated subjects. In addition, participants were tested with different tools, based on their developmental level and compliance. Finally, carrier parents have not been assessed directly, since their retrospective evaluation was based on verbal reports. Future studies on larger number of patients are required to support our hypothesis on cognitive and adaptive profiles in dup7q11.23 children. Furthermore, cross-syndrome studies are warranted in order to compare the neurobehavioral patterns of children with 7q11.23 duplication and deletion syndromes.
Supplementary Materials
The supplementary materials are available online at https://www.mdpi.com/2076-3425/10/11/839/s1.
Author Contributions
Conceptualization, P.A. and M.L.D.; methodology, P.A. and M.L.D.; software, V.A. and A.N.; validation, V.A. and A.N.; formal analysis, P.B., C.C., F.C., V.A., A.N. and P.D.R.; investigation, P.B., V.A., A.N., M.D.L., F.S., F.C., L.S., G.Z., P.A., C.C., R.C.; resources, M.L.D. and P.A.; data curation, M.L.D. and P.B.; writing—original draft preparation, M.D.L., P.A., F.S., F.C., L.S., G.Z.; writing—review and editing, B.D., M.T., M.D.L., F.S., F.C., L.S., G.Z., P.A., C.C., R.C.; visualization, F.S., C.C., F.C.; supervision, G.Z., R.C., M.T., M.C.D., B.D. and S.V.; project administration M.L.D., P.D.R. and P.B.; funding acquisition, M.L.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministero della Salute, grant number RC2020, to M.L.D.
Acknowledgments
We are grateful to the participating family. Clinical investigations and genetic analyses were approved by the institutional Ethics Committee of the Bambino Gesù Children’s Hospital.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Morris, C.A.; Thomas, I.T.; Greenberg, F. Williams syndrome: Autosomal dominant inheritance. Am. J. Med. Genet. 1993, 47, 478–481. [Google Scholar] [CrossRef]
- Sadler, L.S.; Robinson, L.K.; Verdaasdonk, K.R.; Gingell, R. The Williams syndrome: Evidence for possible autosomal dominant inheritance. Am. J. Med. Genet. 1993, 47, 468–470. [Google Scholar] [CrossRef]
- Pober, B.R. Williams–Beuren Syndrome. N. Engl. J. Med. 2010, 362, 239–252. [Google Scholar] [CrossRef]
- Collins, R.T.; Kaplan, P.; Somes, G.W.; Rome, J.J. Cardiovascular Abnormalities, Interventions, and Long-term Outcomes in Infantile Williams Syndrome. J. Pediatr. 2010, 156, 253–258.e1. [Google Scholar] [CrossRef] [PubMed]
- Mason, T.B.; Arens, R.; Sharman, J.; Bintliff-Janisak, B.; Schultz, B.; Walters, A.S.; Cater, J.R.; Kaplan, P.; Pack, A.I. Sleep in children with Williams Syndrome. Sleep Med. 2011, 12, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Meda, S.A.; Pryweller, J.R.; Thornton-Wells, T.A. Regional brain differences in cortical thickness, surface area and subcortical volume in individuals with Williams syndrome. PLoS ONE 2012, 7, e31913. [Google Scholar] [CrossRef]
- Nicita, F.; Garone, G.; Spalice, A.; Savasta, S.; Striano, P.; Pantaleoni, C.; Spartà, M.V.; Kluger, G.; Capovilla, G.; Pruna, D.; et al. Epilepsy is a possible feature in Williams-Beuren syndrome patients harboring typical deletions of the 7q11.23 critical region. Am. J. Med. Genet. Part A 2016, 170, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.S.; Brunetti-Pierri, N.; Peters, S.U.; Kang, S.-H.L.; Fong, C.-T.; Salamone, J.; Freedenberg, D.; Hannig, V.L.; Prock, L.A.; Miller, D.T.; et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet. Med. 2007, 9, 427–441. [Google Scholar] [CrossRef]
- Mervis, C.B.; Klein-Tasman, B.P.; Huffman, M.J.; Velleman, S.L.; Pitts, C.H.; Henderson, D.R.; Woodruff-Borden, J.; Morris, C.A.; Osborne, L.R. Children with 7q11.23 duplication syndrome: Psychological characteristics. Am. J. Med. Genet. Part A 2015, 167, 1436–1450. [Google Scholar] [CrossRef]
- Abbas, E.; Cox, D.M.; Smith, T.; Butler, M.G. The 7q11.23 Microduplication Syndrome: A Clinical Report with Review of Literature. J. Pediatr. Genet. 2016, 5, 129–140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Merla, G.; Brunetti-Pierri, N.; Micale, L.; Fusco, C. Copy number variants at Williams–Beuren syndrome 7q11.23 region. Hum. Genet. 2010, 128, 3–26. [Google Scholar] [CrossRef]
- Morris, C.A.; Mervis, C.B.; Paciorkowski, A.P.; Abdulrahman, O.A.; Dugan, S.L.; Rope, A.F.; Bader, P.; Hendon, L.G.; Velleman, S.L.; Klein-Tasman, B.P.; et al. 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am. J. Med. Genet. Part A 2015, 167, 2916–2935. [Google Scholar] [CrossRef] [PubMed]
- Van Der Aa, N.; Rooms, L.; Vandeweyer, G.; Ende, J.V.D.; Reyniers, E.; Fichera, M.; Romano, C.; Chiaie, B.D.; Mortier, G.; Menten, B.; et al. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Med. Genet. 2009, 52, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; McKee, S.; Mansour, S.; Mehta, S.; Tanteles, G.A.; Anastasiadou, V.; Patsalis, P.; Martin, K.; McCullough, S.; Suri, M.; et al. 7q11.23 Microduplication: A recognizable phenotype. Clin. Genet. 2012, 83, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Prontera, P.; Serino, D.; Caldini, B.; Scarponi, L.; Merla, G.; Testa, G.; Muti, M.; Napolioni, V.; Mazzotta, G.; Piccirilli, M.; et al. Brief Report: Functional MRI of a Patient with 7q11.23 Duplication Syndrome and Autism Spectrum Disorder. J. Autism Dev. Disord. 2014, 44, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Mari, A.; Amati, F.; Mingarelli, R.; Giannotti, A.; Sebastio, G.; Colloridi, V.; Novelli, G.; Dallapiccola, B. Analysis of the elastin gene in 60 patients with clinical diagnosis of Williams syndrome. Hum. Genet. 1995, 96, 444–448. [Google Scholar] [CrossRef]
- Guemann, A.-S.; Andrieux, J.; Petit, F.; Halimi, E.; Bouquillon, S.; Manouvrier-Hanu, S.; Van De Kamp, J.; Boileau, C.; Hanna, N.; Jondeau, G.; et al. ELN gene triplication responsible for familial supravalvular aortic aneurysm. Cardiol. Young 2014, 25, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Karmiloff-Smith, A.; Klima, E.; Bellugi, U.; Grant, J.; Baron-Cohen, S. Is There a Social Module? Language, Face Processing, and Theory of Mind in Individuals with Williams Syndrome. J. Cogn. Neurosci. 1995, 7, 196–208. [Google Scholar] [CrossRef]
- Karmiloff-Smith, A.; Brown, J.H.; Grice, S.; Paterson, S. Dethroning the Myth: Cognitive Dissociations and Innate Modularity in Williams Syndrome. Dev. Neuropsychol. 2003, 23, 227–242. [Google Scholar] [CrossRef]
- Vicari, S.; Caselli, M.C.; Gagliardi, C.; Tonucci, F.; Volterra, V. Language acquisition in special populations: A comparison between Down and Williams syndromes. Neuropsychologia 2002, 40, 2461–2470. [Google Scholar] [CrossRef]
- Volterra, V.; Caselli, M.C.; Capirci, O.; Tonucci, F.; Vicari, S. Early Linguistic Abilities of Italian Children With Williams Syndrome. Dev. Neuropsychol. 2003, 23, 33–58. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A. The behavioral phenotype of Williams syndrome: A recognizable pattern of neurodevelopment. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2010, 154, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, P.; Menghini, D.; Marotta, L.; De Peppo, L.; Ravà, L.; Salvaguardia, F.; Varuzza, C.; Vicari, S. A comparison between linguistic skills and socio-communicative abilities in Williams syndrome. J. Intellect. Disabil. Res. 2017, 61, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Vivanti, G.; Hamner, T.; Lee, N.R. Neurodevelopmental Disorders Affecting Sociability: Recent Research Advances and Future Directions in Autism Spectrum Disorder and Williams Syndrome. Curr. Neurol. Neurosci. Rep. 2018, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Somerville, M.J.; Mervis, C.B.; Young, E.J.; Seo, E.-J.; Del Campo, M.; Bamforth, S.; Peregrine, E.; Loo, W.; Lilley, M.; Pérez-Jurado, L.A.; et al. Severe Expressive-Language Delay Related to Duplication of the Williams–Beuren Locus. N. Engl. J. Med. 2005, 353, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Stinton, C.; Elison, S.; Howlin, P. Mental Health Problems in Adults With Williams Syndrome. Am. J. Intellect. Dev. Disabil. 2010, 115, 3–18. [Google Scholar] [CrossRef]
- Woodruff-Borden, J.; Kistler, D.J.; Henderson, D.R.; Crawford, N.A.; Mervis, C.B. Longitudinal course of anxiety in children and adolescents with Williams syndrome. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2010, 154, 277–290. [Google Scholar] [CrossRef]
- Alfieri, P.; Scibelli, F.; Digilio, M.C.; Novello, R.L.; Caciolo, C.; Valeri, G.; Vicari, S. Comparison of Adaptive Functioning in Children with Williams Beuren Syndrome and Autism Spectrum Disorder: A Cross-Syndrome Study. 2020; submitted. [Google Scholar]
- Velleman, S.L.; Mervis, C.B. Children With 7q11.23 Duplication Syndrome: Speech, Language, Cognitive, and Behavioral Characteristics and Their Implications for Intervention. Perspect. Lang. Learn. Educ. 2011, 18, 108–116. [Google Scholar] [CrossRef]
- Klein-Tasman, B.P.; Mervis, C.B. Autism Spectrum Symptomatology Among Children with Duplication 7q11.23 Syndrome. J. Autism Dev. Disord. 2018, 48, 1982–1994. [Google Scholar] [CrossRef]
- Mervis, C.B.; Morris, C.A.; Klein-Tasman, B.P.; Velleman, S.; Osborne, L.R. 7q11.23 Duplication Syndrome; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews®; University of Washington: Seattle, WA, USA, 1993–2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK327268/ (accessed on 26 June 2020).
- Earhart, B.A.; Williams, M.E.; Zamora, I.; Randolph, L.M.; Votava-Smith, J.K.; Marcy, S.N. Phenotype of 7q11.23 duplication: A family clinical series. Am. J. Med. Genet. Part A 2016, 173, 114–119. [Google Scholar] [CrossRef]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; (WISC-IV); Psychological Corporation: San Antonio, TX, USA, 2003. [Google Scholar]
- Roid, G.H.; Miller, L.J.; Pomplun, M.; Koch, C. Leiter International Performance Scale, 3rd ed.; (Leiter-3); Western Psychological Services: Los Angeles, CA, USA, 2013. [Google Scholar]
- Sparrow, S.S.; Cicchetti, D.; Balla, D.A. Vineland Adaptive Behavior Scales, 2nd ed.; NCS Pearson Inc.: Minneapolis, MN, USA, 2005. [Google Scholar]
- Harrison, P.L.; Oakland, T. ABAS-II Adaptive Behavior Assessment System, 2nd ed.; Harcourt Assessment, Inc.: San Antonio, TX, USA, 2003. [Google Scholar]
- Vicari, S.; Marotta, L.; Luci, A. TFL Test Fono-Lessicale: Valutazione Delle Abilità Lessicali in età Prescolare; Edizioni Erickson: Trento, Italy, 2007. [Google Scholar]
- Kaplan, E.; Goodglass, H.; Weintraub, S. Boston Naming Test; Lea & Febiger: Philadelphia, PA, USA, 1983. [Google Scholar]
- Marini, A.; Marotta, L.; Bulgheroni, S.; Fabbro, F. Batteria per la Valutazione del Linguaggio in Bambini dai 4 ai 12 anni; Giunti OS: Firenze, Italy, 2015. [Google Scholar]
- Bello, A.; Caselli, M.C.; Pettinati, P.; Stefanini, S. PinG: Parole in Gioco; Giunti O.S.: Florence, Italy, 2010. [Google Scholar]
- Dunn, L.M.; Dunn, L.M. Peabody Picture Vocabulary Test, 3rd ed.; American Guidance Services, Inc.: Circle Pines, MN, USA, 1997. [Google Scholar]
- Lancaster, D.R.M. Prove di Valutazione della Comprensione Linguistica PVCL; Giunti Psychometrics: Firenze, Italy, 2007. [Google Scholar]
- Beery, K.E.; Buktenica, N.A. The Beery–Buktenica Developmental Test of Visual–Motor Integration; NCS Pearson: Minneapolis, MN, USA, 1997. [Google Scholar]
- Achenbach, T.M.; Rescorla, L. Manual for the ASEBA School-Age Forms & Profiles; University of Vermont: Burlington, VT, USA, 2001. [Google Scholar]
- Conners, C.K. Conners’ Rating Scales–Revised: User’s Manual; MultiHealth Systems, Incorporated: North Tonawanda, NY, USA, 1997. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar]
- Kaufman, J.; Birmaher, B.; Brent, D.; Rao, U.; Flynn, C.; Moreci, P.; Williamson, D.; Ryan, N. Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL): Initial Reliability and Validity Data. J. Am. Acad. Child. Adolesc. Psychiatry 1997, 36, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, D.; Gould, M.S.; Brasic, J.; Ambrosini, P.; Fisher, P.; Bird, H.; Aluwahlia, S. A Children’s Global Assessment Scale (CGAS). Arch. Gen. Psychiatry 1983, 40, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N.; Davis, S.A.; Todd, R.D.; Schindler, M.K.; Gross, M.M.; Brophy, S.L.; Metzger, L.M.; Shoushtari, C.S.; Splinter, R.; Reich, W. Validation of a Brief Quantitative Measure of Autistic Traits: Comparison of the Social Responsiveness Scale with the Autism Diagnostic Interview-Revised. J. Autism Dev. Disord. 2003, 33, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Schopler, E.; Van Bourgondien, M.E.; Wellman, G.J.; Love, S.R. Childhood Autism Rating Scale, 2nd ed.; (CARS2); Western Psychological Services: Los Angeles, CA, USA, 2010. [Google Scholar]
- Castiglia, L.; Husain, R.A.; Marquardt, I.; Fink, C.; Liehr, T.; Serino, D.; Elia, M.; Coci, E.G. 7q11.23 microduplication syndrome: Neurophysiological and neuroradiological insights into a rare chromosomal disorder. J. Intellect. Disabil. Res. 2018, 62, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Marler, J.A.; Sitcovsky, J.L.; Mervis, C.B.; Kistler, D.J.; Wightman, F.L. Auditory function and hearing loss in children and adults with Williams syndrome: Cochlear impairment in individuals with otherwise normal hearing. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154, 249–265. [Google Scholar] [CrossRef]
- Sammour, Z.M.; Gomes, C.M.; De Bessa, J.; Pinheiro, M.S.; Kim, C.A.; Hisano, M.; Bruschini, H.; Srougi, M. Congenital genitourinary abnormalities in children with Williams–Beuren syndrome. J. Pediatr. Urol. 2014, 10, 804–809. [Google Scholar] [CrossRef]
- Jackowski, A.P.; Rando, K.; De Araújo, C.M.; Del Cole, C.G.; Silva, I.; De Lacerda, A.L.T. Brain abnormalities in Williams syndrome: A review of structural and functional magnetic resonance imaging findings. Eur. J. Paediatr. Neurol. 2009, 13, 305–316. [Google Scholar] [CrossRef]
- Association on Intellectual and Developmental Disabilities. Definition of Intellectual Disability. 2010. Available online: https://www.aaidd.org/intellectual-disability/definition#:~:text=Adaptive%20behavior%20is%20the%20collection,people%20in%20their%20everyday%20lives (accessed on 26 October 2020).
- Greer, M.K.; Brown, F.R.; Pai, G.S.; Choudry, S.H.; Klein, A.J. Cognitive, adaptive, and behavioral characteristics of Williams syndrome. Am. J. Med. Genet. 1997, 74, 521–525. [Google Scholar] [CrossRef]
- Mervis, C.B.; Klein-Tasman, B.P.; Mastin, M.E. Adaptive Behavior of 4- Through 8-Year-Old Children With Williams Syndrome. Am. J. Ment. Retard. 2001, 106. [Google Scholar] [CrossRef]
- Mervis, C.B.; Klein-Tasman, B.P. Williams syndrome: Cognition, personality, and adaptive behavior. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 148–158. [Google Scholar] [CrossRef]
- Vicari, S.; Bates, E.; Caselli, M.C.; Pasqualetti, P.; Gagliardi, C.; Tonucci, F.; Evolterra, V. Neuropsychological profile of Italians with Williams syndrome: An example of a dissociation between language and cognition? J. Int. Neuropsychol. Soc. 2004, 10, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Heiz, J.; Barisnikov, K. Visual-motor integration, visual perception and motor coordination in a population with Williams syndrome and in typically developing children. J. Intellect. Disabil. Res. 2016, 60, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B.; Procyshyn, T.L. Williams syndrome deletions and duplications: Genetic windows to understanding anxiety, sociality, autism, and schizophrenia. Neurosci. Biobehav. Rev. 2017, 79, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Carter, C.S.; Ying, J.; Bellugi, U.; Pournajafi-Nazarloo, H.; Korenberg, J.R. Oxytocin and Vasopressin Are Dysregulated in Williams Syndrome, a Genetic Disorder Affecting Social Behavior. PLoS ONE 2012, 7, e38513. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Ferris, C.; Van De Kar, L.; Coccaro, E.F. Cerebrospinal fluid oxytocin, life history of aggression, and personality disorder. Psychoneuroendocrinology 2009, 34, 1567–1573. [Google Scholar] [CrossRef]
- Beitchman, J.H.; Zai, C.C.; Muir, K.; Berall, L.; Nowrouzi, B.; Choi, E.; Kennedy, J.L. Childhood aggression, callous-unemotional traits and oxytocin genes. Eur. Child. Adolesc. Psychiatry 2012, 21, 125–132. [Google Scholar] [CrossRef]
- Malik, A.I.; Zai, C.C.; Abu, Z.; Nowrouzi-Kia, B.; Beitchman, J.H. The role of oxytocin and oxytocin receptor gene variants in childhood-onset aggression. Genes Brain Behav. 2012, 11, 545–551. [Google Scholar] [CrossRef]
- Thompson, R.J.; Parker, K.J.; Hallmayer, J.F.; Waugh, C.E.; Gotlib, I.H. Oxytocin receptor gene polymorphism (rs2254298) interacts with familial risk for psychopathology to predict symptoms of depression and anxiety in adolescent girls. Psychoneuroendocrinology 2011, 36, 144–147. [Google Scholar] [CrossRef]
- Klein-Tasman, B.P.; Van Der Fluit, F.; Mervis, C.B. Autism Spectrum Symptomatology in Children with Williams Syndrome Who Have Phrase Speech or Fluent Language. J. Autism Dev. Disord. 2018, 48, 3037–3050. [Google Scholar] [CrossRef]
- Klein-Tasman, B.P.; Phillips, K.D.; Lord, C.E.; Mervis, C.B.; Gallo, F.J. Overlap With the Autism Spectrum in Young Children With Williams Syndrome. J. Dev. Behav. Pediatr. 2009, 30, 289–299. [Google Scholar] [CrossRef]
- Lord, C.; Rutter, M.; DiLavore, P.; Risi, S.; Gotham, K.; Bishop, S. Autism Diagnostic Observation Schedule, 2nd ed.; (ADOS-2); Western Psychological Corporation: Los Angeles, CA, USA, 2012. [Google Scholar]
- Randall, M.; Egberts, K.J.; Samtani, A.; Scholten, R.J.; Hooft, L.; Livingstone, N.; Sterling-Levis, K.; Woolfenden, S.; Williams, K. Diagnostic tests for autism spectrum disorder (ASD) in preschool children. Cochrane Database Syst. Rev. 2018, 7, CD009044. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).