7q11.23 Microduplication Syndrome: Clinical and Neurobehavioral Profiling

 , , ,
, , , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Cytogenetics and molecular cytogenetics
2.3. Neurobehavioral Assessment
- Expression subtest of the PhonoVocabulary Test (TFL, Test Fono-Lessicale) [37]. The expression subtest contains 45 tables. The examiner indicates a picture on the table and asks the participant to label the targeted picture;
- The Boston Naming Test [38]. In this test, the participant is asked to label the name of each shown picture. Participants have about 20 s to answer each question;
- Expression subtest of “Batteria per la Valutazione del Linguaggio 4-12” (BVL 4–12) [39]. In the expression subtest, participants are required to label 67 images (51 nouns and 16 verbs);
- Expression subtest of “Parole in Gioco” (PinG) [40]. In this subtest, the child must name the pictures shown.
- Receptive subtest of PhonoVocabulary Test (TFL, Test Fono-Lessicale) [37]. The receptive subtest contains 45 tables with four pictures. Participants must choose the picture labeled by examiner;
- Peabody Picture Vocabulary Test–PPVT [41]. In this test the examiner says a word describing one of four drawings and asks the participant to point to the labeled drawing. Raw total score is converted into Lexical Quotient (LQ);
- Receptive subtest of BVL 4-12 [39]. In this test the examiner says a word describing one of four images and asks the participant to point to the labeled images. This subtest contains 42 words (31 nouns, 10 verbs, and 1 adjective);
- Receptive Subtest of Parole in Gioco (PinG) [40]. In this subtest three pictures are shown and the subject must point to the picture corresponding to the word labeled by the examiner.
- Grammar Evaluation Test (PVCL, Prove di Valutazione della Comprensione Linguistica) [42]. In this test, the examiner pronounces a sentence describing one of four pictures. Then, the examiner asks participants to choose the correct picture;
- Grammar Evaluation Subtest of BVL 4-12 [39]. In syntactic comprehension subtest, children are asked to recognize each of the 40 examiner’s sentences within one of four pictures.
2.4. Procedure
3. Results
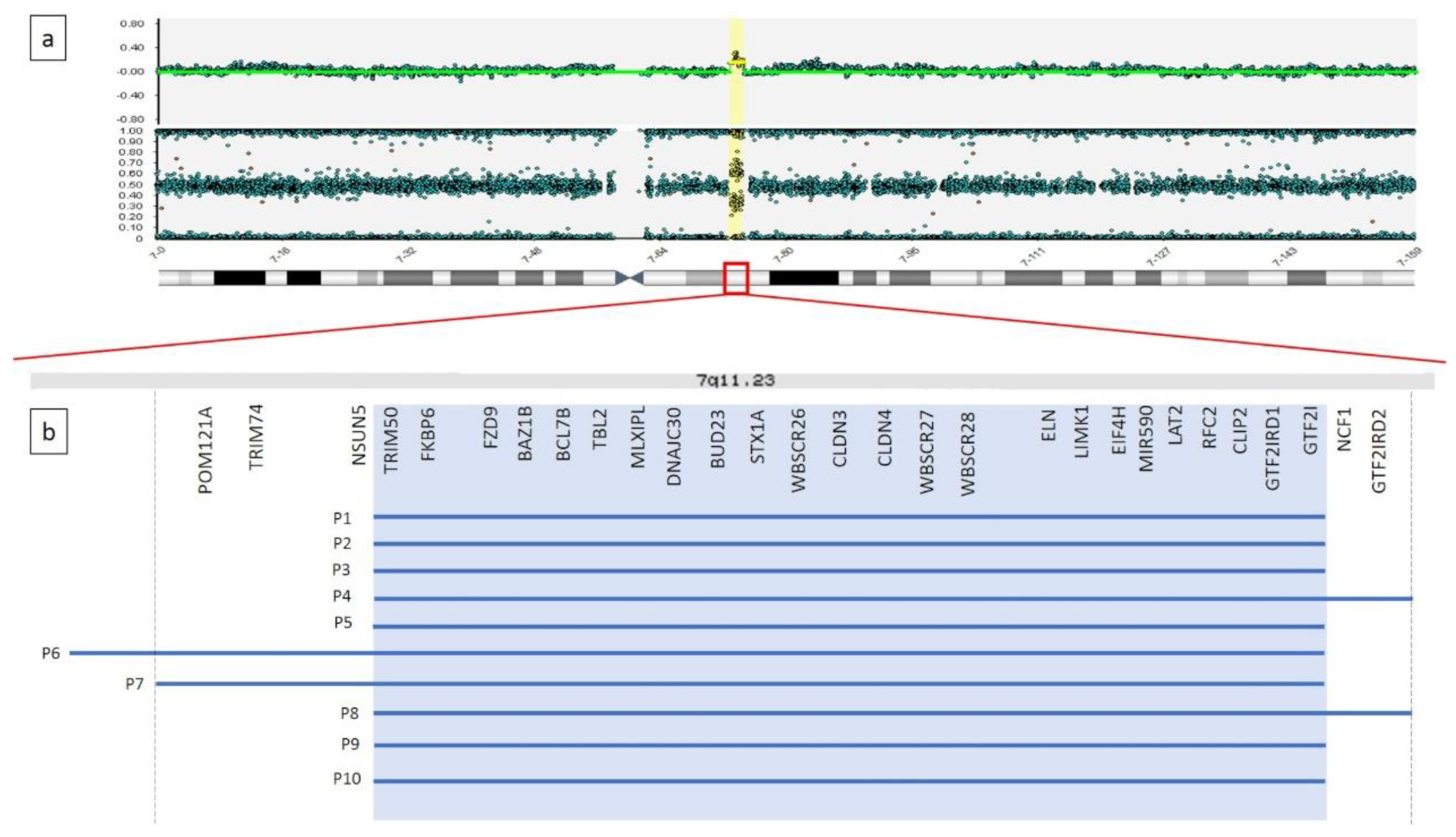
3.1. Cytogenetics and Molecular Cytogenetics Analyses
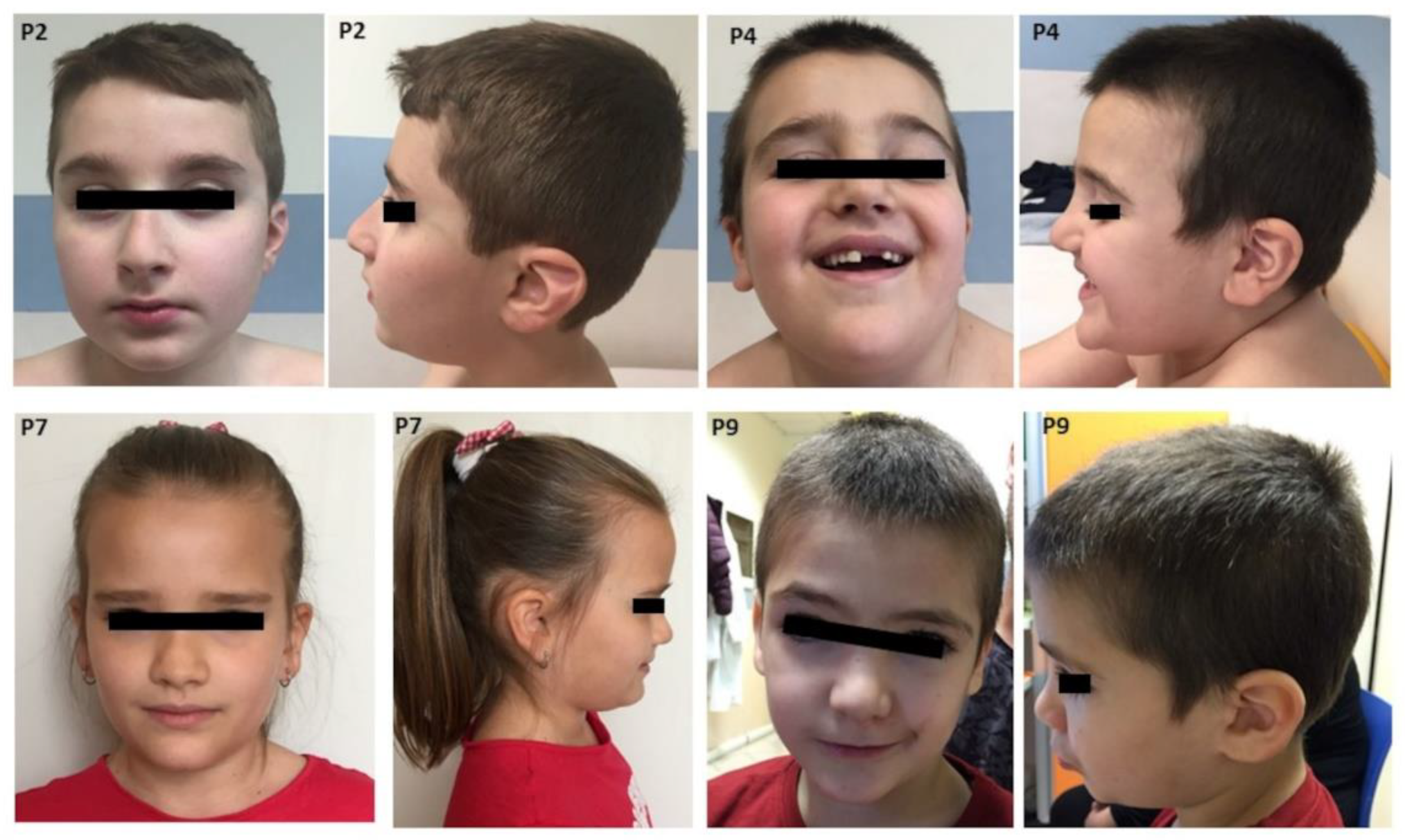
3.2. Clinical Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morris, C.A.; Thomas, I.T.; Greenberg, F. Williams syndrome: Autosomal dominant inheritance. Am. J. Med. Genet. 1993, 47, 478–481. [Google Scholar] [CrossRef]
- Sadler, L.S.; Robinson, L.K.; Verdaasdonk, K.R.; Gingell, R. The Williams syndrome: Evidence for possible autosomal dominant inheritance. Am. J. Med. Genet. 1993, 47, 468–470. [Google Scholar] [CrossRef]
- Pober, B.R. Williams–Beuren Syndrome. N. Engl. J. Med. 2010, 362, 239–252. [Google Scholar] [CrossRef]
- Collins, R.T.; Kaplan, P.; Somes, G.W.; Rome, J.J. Cardiovascular Abnormalities, Interventions, and Long-term Outcomes in Infantile Williams Syndrome. J. Pediatr. 2010, 156, 253–258.e1. [Google Scholar] [CrossRef] [PubMed]
- Mason, T.B.; Arens, R.; Sharman, J.; Bintliff-Janisak, B.; Schultz, B.; Walters, A.S.; Cater, J.R.; Kaplan, P.; Pack, A.I. Sleep in children with Williams Syndrome. Sleep Med. 2011, 12, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Meda, S.A.; Pryweller, J.R.; Thornton-Wells, T.A. Regional brain differences in cortical thickness, surface area and subcortical volume in individuals with Williams syndrome. PLoS ONE 2012, 7, e31913. [Google Scholar] [CrossRef]
- Nicita, F.; Garone, G.; Spalice, A.; Savasta, S.; Striano, P.; Pantaleoni, C.; Spartà, M.V.; Kluger, G.; Capovilla, G.; Pruna, D.; et al. Epilepsy is a possible feature in Williams-Beuren syndrome patients harboring typical deletions of the 7q11.23 critical region. Am. J. Med. Genet. Part A 2016, 170, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.S.; Brunetti-Pierri, N.; Peters, S.U.; Kang, S.-H.L.; Fong, C.-T.; Salamone, J.; Freedenberg, D.; Hannig, V.L.; Prock, L.A.; Miller, D.T.; et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet. Med. 2007, 9, 427–441. [Google Scholar] [CrossRef]
- Mervis, C.B.; Klein-Tasman, B.P.; Huffman, M.J.; Velleman, S.L.; Pitts, C.H.; Henderson, D.R.; Woodruff-Borden, J.; Morris, C.A.; Osborne, L.R. Children with 7q11.23 duplication syndrome: Psychological characteristics. Am. J. Med. Genet. Part A 2015, 167, 1436–1450. [Google Scholar] [CrossRef]
- Abbas, E.; Cox, D.M.; Smith, T.; Butler, M.G. The 7q11.23 Microduplication Syndrome: A Clinical Report with Review of Literature. J. Pediatr. Genet. 2016, 5, 129–140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Merla, G.; Brunetti-Pierri, N.; Micale, L.; Fusco, C. Copy number variants at Williams–Beuren syndrome 7q11.23 region. Hum. Genet. 2010, 128, 3–26. [Google Scholar] [CrossRef]
- Morris, C.A.; Mervis, C.B.; Paciorkowski, A.P.; Abdulrahman, O.A.; Dugan, S.L.; Rope, A.F.; Bader, P.; Hendon, L.G.; Velleman, S.L.; Klein-Tasman, B.P.; et al. 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am. J. Med. Genet. Part A 2015, 167, 2916–2935. [Google Scholar] [CrossRef] [PubMed]
- Van Der Aa, N.; Rooms, L.; Vandeweyer, G.; Ende, J.V.D.; Reyniers, E.; Fichera, M.; Romano, C.; Chiaie, B.D.; Mortier, G.; Menten, B.; et al. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Med. Genet. 2009, 52, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; McKee, S.; Mansour, S.; Mehta, S.; Tanteles, G.A.; Anastasiadou, V.; Patsalis, P.; Martin, K.; McCullough, S.; Suri, M.; et al. 7q11.23 Microduplication: A recognizable phenotype. Clin. Genet. 2012, 83, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Prontera, P.; Serino, D.; Caldini, B.; Scarponi, L.; Merla, G.; Testa, G.; Muti, M.; Napolioni, V.; Mazzotta, G.; Piccirilli, M.; et al. Brief Report: Functional MRI of a Patient with 7q11.23 Duplication Syndrome and Autism Spectrum Disorder. J. Autism Dev. Disord. 2014, 44, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Mari, A.; Amati, F.; Mingarelli, R.; Giannotti, A.; Sebastio, G.; Colloridi, V.; Novelli, G.; Dallapiccola, B. Analysis of the elastin gene in 60 patients with clinical diagnosis of Williams syndrome. Hum. Genet. 1995, 96, 444–448. [Google Scholar] [CrossRef]
- Guemann, A.-S.; Andrieux, J.; Petit, F.; Halimi, E.; Bouquillon, S.; Manouvrier-Hanu, S.; Van De Kamp, J.; Boileau, C.; Hanna, N.; Jondeau, G.; et al. ELN gene triplication responsible for familial supravalvular aortic aneurysm. Cardiol. Young 2014, 25, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Karmiloff-Smith, A.; Klima, E.; Bellugi, U.; Grant, J.; Baron-Cohen, S. Is There a Social Module? Language, Face Processing, and Theory of Mind in Individuals with Williams Syndrome. J. Cogn. Neurosci. 1995, 7, 196–208. [Google Scholar] [CrossRef]
- Karmiloff-Smith, A.; Brown, J.H.; Grice, S.; Paterson, S. Dethroning the Myth: Cognitive Dissociations and Innate Modularity in Williams Syndrome. Dev. Neuropsychol. 2003, 23, 227–242. [Google Scholar] [CrossRef]
- Vicari, S.; Caselli, M.C.; Gagliardi, C.; Tonucci, F.; Volterra, V. Language acquisition in special populations: A comparison between Down and Williams syndromes. Neuropsychologia 2002, 40, 2461–2470. [Google Scholar] [CrossRef]
- Volterra, V.; Caselli, M.C.; Capirci, O.; Tonucci, F.; Vicari, S. Early Linguistic Abilities of Italian Children With Williams Syndrome. Dev. Neuropsychol. 2003, 23, 33–58. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.A. The behavioral phenotype of Williams syndrome: A recognizable pattern of neurodevelopment. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2010, 154, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, P.; Menghini, D.; Marotta, L.; De Peppo, L.; Ravà, L.; Salvaguardia, F.; Varuzza, C.; Vicari, S. A comparison between linguistic skills and socio-communicative abilities in Williams syndrome. J. Intellect. Disabil. Res. 2017, 61, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Vivanti, G.; Hamner, T.; Lee, N.R. Neurodevelopmental Disorders Affecting Sociability: Recent Research Advances and Future Directions in Autism Spectrum Disorder and Williams Syndrome. Curr. Neurol. Neurosci. Rep. 2018, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Somerville, M.J.; Mervis, C.B.; Young, E.J.; Seo, E.-J.; Del Campo, M.; Bamforth, S.; Peregrine, E.; Loo, W.; Lilley, M.; Pérez-Jurado, L.A.; et al. Severe Expressive-Language Delay Related to Duplication of the Williams–Beuren Locus. N. Engl. J. Med. 2005, 353, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Stinton, C.; Elison, S.; Howlin, P. Mental Health Problems in Adults With Williams Syndrome. Am. J. Intellect. Dev. Disabil. 2010, 115, 3–18. [Google Scholar] [CrossRef]
- Woodruff-Borden, J.; Kistler, D.J.; Henderson, D.R.; Crawford, N.A.; Mervis, C.B. Longitudinal course of anxiety in children and adolescents with Williams syndrome. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2010, 154, 277–290. [Google Scholar] [CrossRef]
- Alfieri, P.; Scibelli, F.; Digilio, M.C.; Novello, R.L.; Caciolo, C.; Valeri, G.; Vicari, S. Comparison of Adaptive Functioning in Children with Williams Beuren Syndrome and Autism Spectrum Disorder: A Cross-Syndrome Study. 2020; submitted. [Google Scholar]
- Velleman, S.L.; Mervis, C.B. Children With 7q11.23 Duplication Syndrome: Speech, Language, Cognitive, and Behavioral Characteristics and Their Implications for Intervention. Perspect. Lang. Learn. Educ. 2011, 18, 108–116. [Google Scholar] [CrossRef]
- Klein-Tasman, B.P.; Mervis, C.B. Autism Spectrum Symptomatology Among Children with Duplication 7q11.23 Syndrome. J. Autism Dev. Disord. 2018, 48, 1982–1994. [Google Scholar] [CrossRef]
- Mervis, C.B.; Morris, C.A.; Klein-Tasman, B.P.; Velleman, S.; Osborne, L.R. 7q11.23 Duplication Syndrome; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews®; University of Washington: Seattle, WA, USA, 1993–2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK327268/ (accessed on 26 June 2020).
- Earhart, B.A.; Williams, M.E.; Zamora, I.; Randolph, L.M.; Votava-Smith, J.K.; Marcy, S.N. Phenotype of 7q11.23 duplication: A family clinical series. Am. J. Med. Genet. Part A 2016, 173, 114–119. [Google Scholar] [CrossRef]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; (WISC-IV); Psychological Corporation: San Antonio, TX, USA, 2003. [Google Scholar]
- Roid, G.H.; Miller, L.J.; Pomplun, M.; Koch, C. Leiter International Performance Scale, 3rd ed.; (Leiter-3); Western Psychological Services: Los Angeles, CA, USA, 2013. [Google Scholar]
- Sparrow, S.S.; Cicchetti, D.; Balla, D.A. Vineland Adaptive Behavior Scales, 2nd ed.; NCS Pearson Inc.: Minneapolis, MN, USA, 2005. [Google Scholar]
- Harrison, P.L.; Oakland, T. ABAS-II Adaptive Behavior Assessment System, 2nd ed.; Harcourt Assessment, Inc.: San Antonio, TX, USA, 2003. [Google Scholar]
- Vicari, S.; Marotta, L.; Luci, A. TFL Test Fono-Lessicale: Valutazione Delle Abilità Lessicali in età Prescolare; Edizioni Erickson: Trento, Italy, 2007. [Google Scholar]
- Kaplan, E.; Goodglass, H.; Weintraub, S. Boston Naming Test; Lea & Febiger: Philadelphia, PA, USA, 1983. [Google Scholar]
- Marini, A.; Marotta, L.; Bulgheroni, S.; Fabbro, F. Batteria per la Valutazione del Linguaggio in Bambini dai 4 ai 12 anni; Giunti OS: Firenze, Italy, 2015. [Google Scholar]
- Bello, A.; Caselli, M.C.; Pettinati, P.; Stefanini, S. PinG: Parole in Gioco; Giunti O.S.: Florence, Italy, 2010. [Google Scholar]
- Dunn, L.M.; Dunn, L.M. Peabody Picture Vocabulary Test, 3rd ed.; American Guidance Services, Inc.: Circle Pines, MN, USA, 1997. [Google Scholar]
- Lancaster, D.R.M. Prove di Valutazione della Comprensione Linguistica PVCL; Giunti Psychometrics: Firenze, Italy, 2007. [Google Scholar]
- Beery, K.E.; Buktenica, N.A. The Beery–Buktenica Developmental Test of Visual–Motor Integration; NCS Pearson: Minneapolis, MN, USA, 1997. [Google Scholar]
- Achenbach, T.M.; Rescorla, L. Manual for the ASEBA School-Age Forms & Profiles; University of Vermont: Burlington, VT, USA, 2001. [Google Scholar]
- Conners, C.K. Conners’ Rating Scales–Revised: User’s Manual; MultiHealth Systems, Incorporated: North Tonawanda, NY, USA, 1997. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar]
- Kaufman, J.; Birmaher, B.; Brent, D.; Rao, U.; Flynn, C.; Moreci, P.; Williamson, D.; Ryan, N. Schedule for Affective Disorders and Schizophrenia for School-Age Children-Present and Lifetime Version (K-SADS-PL): Initial Reliability and Validity Data. J. Am. Acad. Child. Adolesc. Psychiatry 1997, 36, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, D.; Gould, M.S.; Brasic, J.; Ambrosini, P.; Fisher, P.; Bird, H.; Aluwahlia, S. A Children’s Global Assessment Scale (CGAS). Arch. Gen. Psychiatry 1983, 40, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N.; Davis, S.A.; Todd, R.D.; Schindler, M.K.; Gross, M.M.; Brophy, S.L.; Metzger, L.M.; Shoushtari, C.S.; Splinter, R.; Reich, W. Validation of a Brief Quantitative Measure of Autistic Traits: Comparison of the Social Responsiveness Scale with the Autism Diagnostic Interview-Revised. J. Autism Dev. Disord. 2003, 33, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Schopler, E.; Van Bourgondien, M.E.; Wellman, G.J.; Love, S.R. Childhood Autism Rating Scale, 2nd ed.; (CARS2); Western Psychological Services: Los Angeles, CA, USA, 2010. [Google Scholar]
- Castiglia, L.; Husain, R.A.; Marquardt, I.; Fink, C.; Liehr, T.; Serino, D.; Elia, M.; Coci, E.G. 7q11.23 microduplication syndrome: Neurophysiological and neuroradiological insights into a rare chromosomal disorder. J. Intellect. Disabil. Res. 2018, 62, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Marler, J.A.; Sitcovsky, J.L.; Mervis, C.B.; Kistler, D.J.; Wightman, F.L. Auditory function and hearing loss in children and adults with Williams syndrome: Cochlear impairment in individuals with otherwise normal hearing. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154, 249–265. [Google Scholar] [CrossRef]
- Sammour, Z.M.; Gomes, C.M.; De Bessa, J.; Pinheiro, M.S.; Kim, C.A.; Hisano, M.; Bruschini, H.; Srougi, M. Congenital genitourinary abnormalities in children with Williams–Beuren syndrome. J. Pediatr. Urol. 2014, 10, 804–809. [Google Scholar] [CrossRef]
- Jackowski, A.P.; Rando, K.; De Araújo, C.M.; Del Cole, C.G.; Silva, I.; De Lacerda, A.L.T. Brain abnormalities in Williams syndrome: A review of structural and functional magnetic resonance imaging findings. Eur. J. Paediatr. Neurol. 2009, 13, 305–316. [Google Scholar] [CrossRef]
- Association on Intellectual and Developmental Disabilities. Definition of Intellectual Disability. 2010. Available online: https://www.aaidd.org/intellectual-disability/definition#:~:text=Adaptive%20behavior%20is%20the%20collection,people%20in%20their%20everyday%20lives (accessed on 26 October 2020).
- Greer, M.K.; Brown, F.R.; Pai, G.S.; Choudry, S.H.; Klein, A.J. Cognitive, adaptive, and behavioral characteristics of Williams syndrome. Am. J. Med. Genet. 1997, 74, 521–525. [Google Scholar] [CrossRef]
- Mervis, C.B.; Klein-Tasman, B.P.; Mastin, M.E. Adaptive Behavior of 4- Through 8-Year-Old Children With Williams Syndrome. Am. J. Ment. Retard. 2001, 106. [Google Scholar] [CrossRef]
- Mervis, C.B.; Klein-Tasman, B.P. Williams syndrome: Cognition, personality, and adaptive behavior. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 148–158. [Google Scholar] [CrossRef]
- Vicari, S.; Bates, E.; Caselli, M.C.; Pasqualetti, P.; Gagliardi, C.; Tonucci, F.; Evolterra, V. Neuropsychological profile of Italians with Williams syndrome: An example of a dissociation between language and cognition? J. Int. Neuropsychol. Soc. 2004, 10, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Heiz, J.; Barisnikov, K. Visual-motor integration, visual perception and motor coordination in a population with Williams syndrome and in typically developing children. J. Intellect. Disabil. Res. 2016, 60, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B.; Procyshyn, T.L. Williams syndrome deletions and duplications: Genetic windows to understanding anxiety, sociality, autism, and schizophrenia. Neurosci. Biobehav. Rev. 2017, 79, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Carter, C.S.; Ying, J.; Bellugi, U.; Pournajafi-Nazarloo, H.; Korenberg, J.R. Oxytocin and Vasopressin Are Dysregulated in Williams Syndrome, a Genetic Disorder Affecting Social Behavior. PLoS ONE 2012, 7, e38513. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Ferris, C.; Van De Kar, L.; Coccaro, E.F. Cerebrospinal fluid oxytocin, life history of aggression, and personality disorder. Psychoneuroendocrinology 2009, 34, 1567–1573. [Google Scholar] [CrossRef]
- Beitchman, J.H.; Zai, C.C.; Muir, K.; Berall, L.; Nowrouzi, B.; Choi, E.; Kennedy, J.L. Childhood aggression, callous-unemotional traits and oxytocin genes. Eur. Child. Adolesc. Psychiatry 2012, 21, 125–132. [Google Scholar] [CrossRef]
- Malik, A.I.; Zai, C.C.; Abu, Z.; Nowrouzi-Kia, B.; Beitchman, J.H. The role of oxytocin and oxytocin receptor gene variants in childhood-onset aggression. Genes Brain Behav. 2012, 11, 545–551. [Google Scholar] [CrossRef]
- Thompson, R.J.; Parker, K.J.; Hallmayer, J.F.; Waugh, C.E.; Gotlib, I.H. Oxytocin receptor gene polymorphism (rs2254298) interacts with familial risk for psychopathology to predict symptoms of depression and anxiety in adolescent girls. Psychoneuroendocrinology 2011, 36, 144–147. [Google Scholar] [CrossRef]
- Klein-Tasman, B.P.; Van Der Fluit, F.; Mervis, C.B. Autism Spectrum Symptomatology in Children with Williams Syndrome Who Have Phrase Speech or Fluent Language. J. Autism Dev. Disord. 2018, 48, 3037–3050. [Google Scholar] [CrossRef]
- Klein-Tasman, B.P.; Phillips, K.D.; Lord, C.E.; Mervis, C.B.; Gallo, F.J. Overlap With the Autism Spectrum in Young Children With Williams Syndrome. J. Dev. Behav. Pediatr. 2009, 30, 289–299. [Google Scholar] [CrossRef]
- Lord, C.; Rutter, M.; DiLavore, P.; Risi, S.; Gotham, K.; Bishop, S. Autism Diagnostic Observation Schedule, 2nd ed.; (ADOS-2); Western Psychological Corporation: Los Angeles, CA, USA, 2012. [Google Scholar]
- Randall, M.; Egberts, K.J.; Samtani, A.; Scholten, R.J.; Hooft, L.; Livingstone, N.; Sterling-Levis, K.; Woolfenden, S.; Williams, K. Diagnostic tests for autism spectrum disorder (ASD) in preschool children. Cochrane Database Syst. Rev. 2018, 7, CD009044. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient N | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gender | M | M | F | M | F | M | F | M | M | M | 7M; 3F |
| Current Age (Years) | 7.7 | 9.10 | 5.7 | 5.6 | 4.9 | 12.3 | 9.2 | 7.5 | 7.8 | 11.3 | M: 8; SD: 2.6 |
| Age at diagnosis (Years) | 3 | 6 | 3 | 3 | 1.6 | 8.6 | 7.6 | 3.6 | 4 | 11 | M: 6.4; SD: 3.1 |
| CGH array (start and end point) | 7q11.23(72,726,578–74,119,570)x3 | 7q11.23(72,726,578–74,139,390)x3 | 7q11.23(72,726,578–74,139,390)x3 | 7q11.23(72,726,578–74,339,044)x3 | 7q11.23(72,726,578–74,139,390)x3 | 7q11.22q11.23(72,044,007–74,139,390)x3 | 7q11.23(72,283,565–74,134,911)x3 | 7q11.23(72,726,578–74,339,044)x3 | 7q11.23(72,726,578–74,119,570)x3 | 7q11.23(72,726,578–74,139,390)x3 | |
| Length of duplicated region | 1.4 Mb | 1.4 Mb | 1.4 Mb | 1.6 Mb | 1.4 Mb | 2.1 Mb | 1.9 Mb | 1.6 Mb | 1.4 Mb | 1.4 Mb | 1.4–2.1 Mb |
| Inheritance | de novo | maternal | NA | paternal | de novo | de novo | de novo | NA | paternal | de novo | 5/8 de novo; 3/8 inherited |
| Growth at last evaluation (centile) | weight <3rd; height 3rd–10th; OFC 75th | weight >97th; height 90th–97th; OFC 50–75th | weight 10th; height 10th; OFC 25th | weight: 10th; height 97th; OFC >97th | Weight 50th–75th; height 25th–50th; OFC 75th–97th | Weight 50th– 75th; height 10th–25th | weight 50th-75th; height 25th–50th, OFC 50th | Weight:75th–97th; Height 25th–50th OFC 75th | weight: 75th; height 97th; OFC 75th | weight: 97th; height 50–75th | |
| Facial | |||||||||||
| Macrocephaly | relative | no | no | yes | relative | NA | no | no | no | NA | 3/8 |
| Brachycephaly | no | yes | no | no | no | NA | no | yes | no | NA | 2/8 |
| Prominent forehead | yes | yes | no | yes | no | NA | yes | yes | yes | NA | 6/8 |
| Elongated palpebral fissures | yes | no deep set eyes | no | yes palpebral ptosis | no downslanting palpebral fissures | Yes | yes | yes deep set eyes | yes | yes palpebral ptosis | 7/10 (2/10 ptosis; 2/10 deep set eyes) |
| Nose | high nasal bridge | high nasal bridge | high nasal bridge with bulbous nose | high broad nasal bridge | normal | NA | bulbous nose | prominent, high nasal bridge | bulbous nose | NA | 5/8 high nasal bridge 3/8 bulbous nose |
| Dysmorphic ears | additional crus of the antihelix, abnormally folded helix | horizontal crus of helix | asymmetric low set and posteriorly rotated | low-set posteriorly rotated ears with thickened earlobes | thickened helix | NA | no | no | thickened helix large ears | NA | 6/8 |
| Short philtrum | no | yes | no | yes | no | prominent | yes | yes | yes | NA | 6/9 |
| Microstomia | yes | no | no | yes | yes | NA | no | NA | no | NA | 3/7 |
| Thin lips | yes | yes | yes | yes | yes, everted upper lip | No | no | no | yes | NA | 6/9 |
| Other | NA | telecanthus, medial flaring of the eyebrow, exophoria | horizontal eyebrow, synophrys, short neck, pectus excavatum | prognathism, long chin with horizontal crease | micrognathia | NA | horizontal eyebrow; short neck, widely spaced teeth | overhanging columella | synophrys, hypertelorism, divergent strabismus thickened nostrils | large incisors, short neck, low anterior and posterior hairline | |
| Hearing loss | no | no | no | no | no | mild conductive (tubal stenosis) | no recurrent otitis media | no | no | no | 1/10 |
| Cardiac malformation | no | mild ascending aortic dilatation | no | no | PDA, pulmonary valve dysplasia | No | PDA, mild tricuspid insufficiency | no | PDA(spontaneously resolved) | left ventricular hypertrabeculation, aortic insufficiency | 3/10 PDA; 1/10 ascending aortic dilatation |
| Cryptorchidism | no | yes bilateral | NA | yes bilateral | NA | no | NA | no | no | no | 2/7 |
| Hypotonia | yes | no | yes | no | no | no | yes | no | no | yes | 4/10 |
| Joint laxity | yes | no | yes | no | no | no | yes | no | no | yes | 4/10 |
| Pes planus-valgus | yes | yes | yes | no | no | yes | no | no | no | no | 4/10 |
| Neurological | |||||||||||
| Brain MRI findings | NA | Cerebral ultrasound: enlargement of periencephalic cerebral spaces | atrophy of EC with enlargement of the 3rd and lateral ventricles, thin corpus callosum | NA | NA | NA | normal | NA | normal | thin corpus callosum, arachnoid cyst | 2/5 thin corpus callosum 3/5 aspecific abnormalities |
| Epilepsy EEG | normal | normal | abnormal | NA | NA | NA | normal | normal | normal | normal | 1/7 abnormal |
| Sleep pattern | regular | regular | regular | NA | NA | irregular until 4 years | regular | regular | NA | NA | 1/6 irregular |
| Other | periodic migraine episodes. hepatic steatosis | asthmatic bronchitis | Adenoidectomy; bronchospasm; pharmacological therapy (risperdone 0.25 mg; sertraline 0.5 mL). | hypermetropia astigmatism | pharmacological therapy risperdone 0.25 mg (2 times per day) | periodic migraine episodes, pelvis dilatation | 2/10 migraine 1/10 hepatic steatosis |
| Test | P1 | P2 | P3 | P4 | P6 | P7 | P8 | P9 | P10 | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| Age in years | 7.7 | 9.10 | 5.7 | 5.6 | 12.3 | 9.2 | 7.5 | 7.8 | 11.3 | M: 8.5; SD: 2.4 |
| Cognitive level | 85 a | 47 b | 82 a | 81 a | 75 b | 81 a | 65 b | 62 a | 68 a | M: 71.77; SD: 12.39 |
| Adaptive Composite Score | 71 c | 51 | 57 c | 54 c | 62 c | 85 c | 45 c | 38 c | 40 d | M: 55.88; SD: 15.13 |
| Communication Domain | 55 c | 56 | 45 c | 51 c | 64 c | 71 c | 45 c | 40 c | 49 d | M: 52.88; SD: 9.84 |
| Daily Living Skills Domain | 82 c | 49 | 73 c | 58 c | 75 c | 107 c | 44 c | 42 c | 40 d | M: 63.33; SD: 22.61 |
| Socialization Domain | 87 c | 65 | 81 c | 71 c | 59 c | 82 c | 67 c | 54 c | 53 d | M: 68.78; SD: 12.46 |
| Motor Skills Domain | - | - | 54 c | 65 c | - | - | - | - | - | M: 59.5; SD: 7.78 |
| Lexical Production | severely impaired | severely impaired | severely impaired | severely impaired | spared | slightly impaired | slightly impaired | severely impaired | severely impaired | 6/9 severely impaired 2/9 slightly impaired 1/9 spared |
| Lexical Comprehension | severely impaired | slightly impaired | severely impaired | severely impaired | spared | severely impaired | severely impaired | severely impaired | severely impaired | 7/9 severely impaired 1/9 slightly impaired 1/9 spared |
| Grammar Comprehension | severely impaired | slightly impaired | severely impaired | severely impaired | severely impaired | severely impaired | severely impaired | severely impaired | severely impaired | 8/9 severely impaired 1/9 slightly impaired 0/9 spared |
| Visuo-motor skills | 73 | 74 | 57 | 111 | 58 | 92 | 89 | 63 | 60 | M: 75.22; SD: 18.59 |
| Test/Participants | P1 | P2 | P3 | P4 | P6 | P7 | P8 | P9 | P10 |
|---|---|---|---|---|---|---|---|---|---|
| CARS 2 | 22 | NA | 22.5 | 36.5 * | 25.5 | 17 | 20.5 | 26 | NA |
| SRS | Not clinical | NA | Non clinical | Social Awareness Social Cognition Social Communication Social Motivation Mannerisms SRS Total Score | Social Awareness Social Cognition Social Communication Social Motivation Mannerisms SRS Total Score | Social Awareness | Social Awareness Social Cognition Social Communication Social Motivation Mannerisms SRS Total Score | Social Awareness Social Cognition Social Communication Social Motivation Mannerisms SRS Total Score | NA |
| CBCL | Not clinical | NA | Not clinical | Attention Problem | Withdrawn/Depressed; Social Problems; Internalizing, Externalizing and Total Problems | Not clinical | Anxious/Depressed; Withdrawn/Depressed; Somatic Complaints; Social Problems; Thought Problems; Rule-Breaking Behavior; Aggressive Behavior; Internalizing, Externalizing and Total Problems | Anxious/Depressed; Withdrawn/Depressed; Social Problems; Thought Problems; Aggressive Behavior; Internalizing, Externalizing and Total Problems | Anxious/Depressed; withdrawn/Depressed; Somatic Complaints; Thought Problems; Internalizing, and Total Problems |
| CPRS-R-L | Not clinical | Cognitive problems/inattention; Anxious–shy; Social problems; Psychosomatic; DSM–IV inattentive; DSM–IV total | Not clinical | Not clinical | Cognitive problems/inattention; Anxious–shy; Perfectionism; Social problems; ADHD index; CGI restless–impulsive; CGI emotional lability; CGI total; DSM–IV inattentive; DSM–IV total | Perfectionism | Oppositional; Cognitive problems/inattention; Hyperactivity; Anxious–shy; Perfectionism; Social problems; Psychosomatic; ADHD index; CGI restless–impulsive; CGI emotional lability; CGI total; DSM–IV inattentive; DSM–IV hyperactive–impulsive; DSM–IV total | Oppositional; Cognitive Problems/inattention; Hyperactivity; Anxious–shy; Perfectionism; Social problems; Psychosomatic; ADHD index; CGI restless–impulsive; CGI emotional lability; CGI total; DSM–IV inattentive; DSM–IV hyperactive–impulsive; DSM–IV total | NA |
| C-GAS | Variable functioning (60) | NA | NA | NA | Moderate degree of interference in functioning (50) | Moderate degree of interference in functioning (50) | Moderate degree of interference in functioning in most social areas (47) | NA | Major impairment of functioning in several areas (40) |
| Best Estimate Consensus Diagnosis | Anxiety Disorder NOS (criteria not reached for Separation Anxiety Disorder and Social Phobia) | Anxiety Disorder NOS (criteria not reached for Generalized Anxiety Disorder) | Anxiety Disorder NOS (criteria not reached for Social Phobia) | Disruptive Behavior Disorder NOS | Disruptive Behavior Disorder NOS; Dysthymic Disorder | Generalized Anxiety Disorder (criteria not reached for Social Phobia) | Generalized Anxiety Disorder | Generalized Anxiety Disorder (criteria not reached for Social Phobia) | Generalized Anxiety Disorder (criteria not reached for Separation Anxiety Disorder and Social Phobia) |
| Other Clinical Features | Specific phobia (birds); stereotypes (hand flapping); Sporadic enuresis episodes; bruxism | Stuttering; Repetitive Movements | Selective Mutism Inhibition | Emotional dysregulation; hyperfagia; sleep talking; autistic features; alternate inhibition and social disinhibition (hugging non familiar people) | History of inhibition (actually social disinhibition); problem in motor regulation; repetitive behaviors; anxiety | Insistence on sameness; enuresis; emotional dysregulation; specific phobias; aggressive behaviors; social disinhibition (inappropriate sexual behavior; showing genitals); enuresis | Social disinhibition (hugging non-familiar people); emotional dysregulation; aggressive behaviors. | Behavioral oddities; Insistence on sameness; compulsive behaviors; imaginary friend |
| P1 | P2 | P3 | P4 | P6 | P7 | P8 | P9 | P10 | T | |
|---|---|---|---|---|---|---|---|---|---|---|
| Anxiety/Specific Phobias | + | - | - | - | + | + | + | + | + | 6/9 |
| Separation Anxiety/Social Inhibition | + | + | + | + | + | + | + | + | + | 9/9 |
| Aggressive Behaviors | - | - | - | + | + | - | + | + | - | 4/9 |
| Repetitive Behaviors/Movements | + | + | - | - | - | - | - | + | + | 4/9 |
| Social Disinhibition | - | - | - | + | + | - | + | + | - | 4/9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dentici, M.L.; Bergonzini, P.; Scibelli, F.; Caciolo, C.; De Rose, P.; Cumbo, F.; Alesi, V.; Capolino, R.; Zanni, G.; Sinibaldi, L.; et al. 7q11.23 Microduplication Syndrome: Clinical and Neurobehavioral Profiling. Brain Sci. 2020, 10, 839. https://doi.org/10.3390/brainsci10110839
Dentici ML, Bergonzini P, Scibelli F, Caciolo C, De Rose P, Cumbo F, Alesi V, Capolino R, Zanni G, Sinibaldi L, et al. 7q11.23 Microduplication Syndrome: Clinical and Neurobehavioral Profiling. Brain Sciences. 2020; 10(11):839. https://doi.org/10.3390/brainsci10110839
Chicago/Turabian StyleDentici, Maria Lisa, Paola Bergonzini, Francesco Scibelli, Cristina Caciolo, Paola De Rose, Francesca Cumbo, Viola Alesi, Rossella Capolino, Ginevra Zanni, Lorenzo Sinibaldi, and et al. 2020. "7q11.23 Microduplication Syndrome: Clinical and Neurobehavioral Profiling" Brain Sciences 10, no. 11: 839. https://doi.org/10.3390/brainsci10110839
APA StyleDentici, M. L., Bergonzini, P., Scibelli, F., Caciolo, C., De Rose, P., Cumbo, F., Alesi, V., Capolino, R., Zanni, G., Sinibaldi, L., Novelli, A., Tartaglia, M., Digilio, M. C., Dallapiccola, B., Vicari, S., & Alfieri, P. (2020). 7q11.23 Microduplication Syndrome: Clinical and Neurobehavioral Profiling. Brain Sciences, 10(11), 839. https://doi.org/10.3390/brainsci10110839