Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system is a powerful and widely used tool for genome editing. Recently, it was reported that direct delivery of Cas9-sgRNA ribonucleoproteins (RNPs) reduced off-target effects. Therefore, non-invasive, high-throughput methods are needed for direct delivery of RNPs into cells. Here, we report a novel method for direct delivery of RNPs into cells using a nanostructure with a high-aspect-ratio and uniform nanoneedles. This nanostructure is composed of tens of thousands of nanoneedles laid across a 2D array. Through insertion of the nanoneedle array previously adsorbed with Cas9-sgRNA, it was possible to deliver RNPs directly into mammalian cells for genome editing.
1. Introduction
Genome editing technology has advanced rapidly following the introduction of the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system, which is composed of the Cas9 protein, which cleaves the target DNA and single guide RNA (sgRNA) for recognizing the target DNA sequence [1,2,3,4,5,6,7]. A recent study showed that a minimal protospacer adjacent motif (PAM) sequence (5′-NNG-3′), which is needed for binding Cas9-sgRNA ribonucleoproteins (RNPs) to the target sequence, was sufficient for the activation of Cas9 from Streptococcus canis, indicating the possibility of expanding the target range for genome editing in this system [8]. Although the CRISPR/Cas9 system is a powerful tool for genome engineering, the overexpression of Cas9, derived from a plasmid vector, may cause off-target effects such as the cleavage of non-target sites. In addition, the plasmid-based introduction of Cas9 requires an optimum promoter for gene expression. For Arabidopsis thaliana, a constitutively active promoter from an early embryonic stage can be used to obtain a mutant, because the egg cell-specific expression of Cas9 limits the chance of mutation [9]. This suggests that a promoter that is effective for editing a particular genome should be identified before being used in an organism.
Recently, direct delivery of Cas9-sgRNA RNPs was performed using electroporation, or lipofection using lipid nanoparticles [10,11,12,13]. These studies demonstrated that direct delivery of RNPs may reduce off-target effects compared with plasmid-based DNA delivery. Direct delivery of Cas9 via electroporation leads to Cas9 facilitated cleavage of the genome DNA at the target site almost immediately, and results in the mutation frequency reaching its plateau a day after electroporation [10]. While the direct-delivered Cas9 proteins completely disappear from cells via protein degradation, plasmid DNA continues to constitutively expresses Cas9 for several days, even after mutation frequency at the target site reaches its plateau. Such continuous expression of Cas9 and sgRNA in cells causes off-target mutations. Therefore, introducing only the required quantity of Cas9 protein is important. In addition, direct delivery of Cas9-sgRNA is more versatile, because the use of a specific promoter becomes unnecessary. However, electroporation has disadvantages, such as low cell viability and low throughput. In the case of lipid nanoparticle delivery, many RNPs are needed to obtain a high genome editing efficiency because the ratio of the endosomal escape of Cas9-sgRNA affects its activity in the cells. Therefore, it has become necessary to develop efficient direct Cas9-sgRNA delivery tools that ensure safe genome editing technology.
The delivery of macromolecules into living cells via the insertion of nano-scale acicular materials, such as carbon nanofiber arrays, silicon nanowires, diamond nanoneedle arrays (NNAs), and porous silicon nanoneedle arrays, has been reported as a new delivery platform [14,15,16,17]. Although these studies have demonstrated the usefulness of nanomaterials as a delivery method for nucleic acids, successful delivery of RNPs using acicular materials has not been reported.
We have been developing cell manipulation techniques using 200 nm diameter needle materials, such as the AFM cantilever type nanoneedle (AFM-NN) and the silicon NNA [18,19,20,21]. Our original NNA has several tens of thousands of nanoneedles in a 3 mm square area. Each nanoneedle has a near regular configuration of 200 nm in diameter and 25 μm in length (Figure 1A). Cell doubling time did not change, even after 50 insertions of AFM-NN, and neural stem cells manipulated with an antibody-functionalized NNA were capable of differentiating into neurons [22,23]. These results led us to assume that damage due to the insertion of 200 nm nanoneedles into a cell was negligible. Additionally, nanoneedles were able to access the intracellular space of primary cultured rat hippocampal tissue cells [22], indicating that various types of cells may be targeted via this system. We reported that our NNA device could be used to deliver naked DNA or zinc-finger proteins into cells [19,20]. Therefore, the NNA device may be also applied to efficiently deliver Cas9-sgRNA RNPs into cells. The current study assessed the efficiency of genome editing via RNP delivery using NNA.
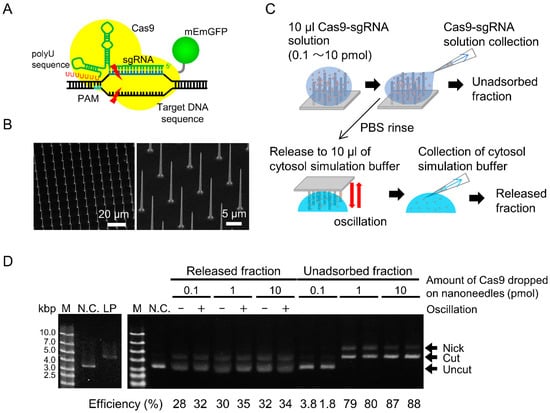
Figure 1.
Estimation of cleavage activity of RNP released from nanoneedle array (NNA). (A) A schematic of Cas9 and poly-U containing single guide RNA (sgRNA) ribonucleoprotein (RNP) cleaving double stranded DNA. (B) Scanning electron microscope images of fabricated NNAs. (C) Schematic of RNP adsorption and release. The RNP solution was poured on the hydrophobized NNA and collected as the unadsorbed fraction. The RNP-adsorbed nanoneedle array was immersed in cytosolic simulation buffer. The buffer was then collected as the released fraction. (D) Electrophoresis of cleaved plasmid DNAs in unadsorbed and released fractions. Super-coiled plasmid DNA coding EGFP (negative control, N.C.) and linearized plasmid (LP) by restriction enzyme were used.
2. Materials and Methods
2.1. Cell Culture
HeLa and HEK293 cells were cultured at 37 °C in a humidified atmosphere with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS) and 2 mM GlutaMAX (Thermo Fisher Scientific, Waltham, MA, USA). FP10SC2 [24] cells were cultured in RPMI 1640 medium (Sigma-Aldrich), supplemented with 2 mM L-glutamine (Wako Pure Chemical Industries, Ltd., Osaka, Japan), 1.5 g/L sodium bicarbonate (Invitrogen, Carlsbad, CA, USA), 2.5 g/L glucose (Wako Pure Chemical Industries), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Sigma-Aldrich), 1 mM sodium pyruvate (Wako Pure Chemical Industries), and 10% FBS (Thermo Fisher Scientific, Inc.). For cell dissociation, cells were treated with 0.25% Trypsin EDTA (Thermo Fisher Scientific) and centrifuged to form a pellet. The resulting pellet was dispersed in phosphate-buffered saline (PBS), and the cells were seeded into culture dishes.
2.2. Preparation of Cas9 Protein
The Cas9 coding gene, derived from S. pyrogenes, was expressed via the pET-28b-Cas9-His plasmid vector (#47327, Addgene, Watertown, MA, USA). The Cas9 protein fused with mEmerald GFP at the C-terminus (Cas9-mEmGFP) was expressed from pET-Cas9-GFP-NLS-His, generated from the pET-28b-Cas9-His. The Cas9 coding vector was transformed into BL21 (DE3) competent cells. The cells were cultured at 37 °C until the OD600 reached 0.6–0.8. To induce expression of Cas9, MgCl2 and isopropyl β-D-1-thiogalactopyranoside solution were added to the culture medium at 1 mM and 0.1 mM, respectively, and the cells were cultured at 20 °C for 6–10 h. Cas9 proteins were collected using Ni-NTA agarose (QIAGEN, Venlo, Netherlands) and stored at −30 °C in HEPES-based buffer (20 mM HEPES, 150 mM NaCl, 1 mM MgCl2, 50% glycerol, pH 7.4) at a concentration of 7.5 µM. The protein concentration was evaluated via densitometry of the CBB-stained band of SDS-PAGE using Image-Pro Plus software (Media Cybernetics, Rockville, MD, USA).
2.3. Preparation of sgRNA
Because the addition of 7 uracil bases to sgRNA slightly improved the cleavage activity of Cas9, sgRNA containing a poly-U sequence was used (Figure 1A). A DNA fragment with a T7 promoter binding site and a sgRNA sequence was obtained from 2 oligo DNAs, which contained a 23 base complementary sequence via T7 RNA polymerase reaction. The oligo named sgRNA-(7U), which has a polyadenine sequence, was mixed with T7sgRNA-GFP-g1 (R332) or T7sgRNA-mNes (F461) oligo which has a T7 promoter sequence (Table 1). The two oligo DNAs were annealed and polymerized in a 5′ to 3′ direction by Klenow Fragment. Next, sgRNA was transcribed using the synthesized DNA fragment and RiboMax™ Large Scale RNA Production Systems (Promega, Madison, WI, USA). Transcribed sgRNA was isolated using ISOGEN II (NIPPON GENE, Tokyo, Japan). A sgRNA has a 7 bases poly-U sequence after the 3ʹ-end of sgRNA.

Table 1.
Sequence of oligo DNA and sgRNA.
2.4. Evaluation of the Cleavage Activity of RNP
The RNP complexes were constructed by mixing 1 µl of Cas9 (0.1, 1, 10 pmol), 1 µL of sgRNA (sgRNA_GFPg1-7U, 10 pmol), 1 µL of 10 × Cas9 buffer (New England Biolabs Inc., Ipswich, MA, USA), and 7 µL of UltraPure™ DNase/RNase-Free Distilled Water (Thermo Fisher Scientific), and incubating at room temperature (RT) for 10 min. We adsorbed Cas9-sgRNA RNPs on the surface of NNA containing tens of thousands of nanoneedles as follows. First, the NNA, in which needles were arrayed squarely at 10 µm intervals in a 3 mm square area, was cleaned with an O2 plasma asher, JPA300 (J-SCIENCE Lab Co. Ltd., Kyoto, Japan), at 300 W for 10 min. Then, the NNA was immersed in 1% hydrogen fluoride (HF) solution for 1 min. The NNA was rinsed with ultrapure water, and the RNP solution was poured on it, followed by incubation at 4 °C for 30 min. Finally, the unadsorbed fraction of the RNP solution left on the NNA was collected, and the NNA was rinsed with PBS and fitted to the array manipulator (detailed information is provided in [19]). Next, the NNA was soaked in 10 μL of cytosolic simulation buffer (25 mM HEPES, 115 mM CH3COOK, 5 mM MgCl2, 20 v/v% polyethylene glycol 8000) and oscillated to release the RNPs from the NNA surface using fluidic shear force (condition described in the Section 2.6). Following oscillation, we collected the solution as a released fraction. Substrate plasmid DNA containing EGFP (pEGFP-C1, GenBank accession number U55763) was used to evaluate the cleavage activity of RNP. Then 50 ng of plasmid DNA was added to the unadsorbed fraction and the released fraction, followed by incubation at 37 °C for 60 min. The cleaved plasmid DNAs were electrophoresed using a 1% agarose gel in Tris-acetate EDTA buffer and stained with GelGreen Nucleic Acid Gel Stain (Biotium, Fremont, CA, USA). The brightness of the obtained band was measured using Image-Pro Plus (Media Cybernetics). The efficiency was calculated using the following formula:
Cutting efficiency (%) = 100 × {1 − (brightness of uncut band/total brightness of cut, nick and uncut band)}
2.5. Fluorescent Observation of Cas9 Delivery to Cell
HEK293 cells were inoculated on a collagen-coated glass bottom dish and incubated overnight in a CO2 incubator. After washing the cells with PBS, 1.5 pmol Cas9-mEmGFP fusion protein, or RNP, prepared using 15 pmol sgRNA in FBS and phenol red-free DMEM (total 100 μL) was added to the dish, and the cells were immediately observed using FV-300 and IX-71 confocal laser scanning microscopes (CLSM, Olympus, Tokyo, Japan). In order to evaluate the nucleus insertion efficiency of the nanoneedle, HEK293 cells expressing the DsRed2-nuclear export signal (NES) [18] were used to visualize the cytosol. An NNA adsorbed with Cas9-mEmGFP was inserted into the DsRed2-NES expressing cells, and images were obtained via CLSM. When one or more fluorescent spots derived from Cas9-mEmGFP adsorbed needles were observed in the nucleus, that cell was defined as a nucleus inserted cell. Nucleus insertion efficiency was calculated as a percentage of nucleus inserted cells (the number of nucleus-inserted cells in total cells (n = 52)).
2.6. Knockout of EGFP Gene in Reporter Cells
EGFP reporter cells were constructed by transforming cells with an EGFP-coding plasmid and selecting cells using hygromycin for 1 month [25]. The reporter cells (1 × 104 cells) were seeded on 3 mm square area in a 40 mm culture dish (TPP Techno Plastic Products AG, Trasadingen, Switzerland) and cultured overnight. Next, 15 pmol Cas9 and 10 pmol sgRNA were used for RNP complex formation. The preparation of RNP-adsorbed NNA was as described above. The RNP-adsorbed NNA was moved towards the cells while being observed via an inverted microscope (GX71, Olympus). Following insertion into the cells, the NNA was oscillated at an amplitude of 1.0 µm and a frequency of approximately 5 kHz for 1 min. The cells were then rinsed with PBS twice and cultured in fresh DMEM for 48 h. In order to introduce RNPs via lipofection, 1 × 104 reporter cells were seeded on a 48 well plate in DMEM containing 10% FBS and penicillin/amphotericin. Then, 15 pmol Cas9 protein and 10 pmol sgRNA were mixed in 10 µL Cas9 buffer and incubated at RT for 10 min. Next, 1 µL of Lipofectamine 2000 (Thermo Fisher Scientific) was added to the RNP solution and the final volume was adjusted to 25 µL with Opti-MEM (Thermo Fisher Scientific). Following 15 min of incubation at RT, the lipofection solution was added to each well. The medium was replaced with fresh medium following 16–20 h of incubation. The cells were incubated for 48 h before being subjected to fluorescence imaging.
To evaluate EGFP knockout efficiency, we determined an area of cells from bright field images and that of EGFP expressing cells from fluorescent images. The fluorescence intensity in an area due to the absence of EGFP expressing cells was used as a threshold value to determine areas of EGFP expressing cells. The EGFP knockout efficiency was calculated based on the area of EGFP disrupted cells relative to the total area of cells as seen in the bright field image, using Image-Pro Plus (Media Cybernetics). At least 2 × 103 cells were present in the area approached by the Si-NNA.
2.7. Knockout of Nestin Gene in Mouse Breast Cancer Cells
The sgRNA sequence for knockout of the intermediate filament protein, nestin, was designed by Benchling (Biology Software, (2017); retrieved from https://benchling.com). RNPs, prepared using 5 pmol Cas9-mEmGFP and 15 pmol sgRNA, were added onto the NNA, which had been previously treated with asher and 1% HF. Then, the array was completely dried via evaporation for 30 min.
FP10SC2 mouse breast cancer cells (1 × 104 cells) were seeded on a laminin-coated glass-bottom dish. The method used for delivering RNPs into the cells using the NNA was described in the previous section. After culturing the cells for 120 h, nestin knockout in the cells was evaluated by immunostaining. The cells were washed with PBS 3 times for 15 min each and fixed by incubation with 4% paraformaldehyde for 15 min. Following permeabilization with 0.1% Triton X-100 for 2 min, the cells were washed with a blocking solution composed of 0.4% Block Ace (DS Pharma Biomedical, Osaka, Japan) in PBS. The cells were then subjected to immunostaining with 500-fold diluted anti-nestin antibodies (Rat-401; BioLegend, San Diego, CA, USA) for 1 h, following which the cells were washed 3 times with 0.4% Block Ace buffer, and incubated with 1000-fold diluted Alexa Fluor 488-conjugated goat anti-mouse IgG antibodies (A-11001; Invitrogen) for 1 h. After washing three times with 0.4% Block Ace buffer, the cells were incubated with 1000-fold diluted DAPI (4′,6-diamidino-2-phenylindole, R415; Thermo Fisher Scientific) for 1 h to stain nuclei. After removing excess DAPI by washing, fluorescence images of the cells were acquired using a CCD camera attached to a fluorescence microscope (DP30/IX70, Olympus).
The fluorescence intensity of at least 30 cells in the cell population was measured manually using the image analyzing software ImageJ (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, MD, USA, https://imagej.nih.gov/ij/, 1997–2018.). The threshold value for fluorescence intensities of knockout cells was set at the average plus five SD of the nestin knockout cell line which had been obtained previously [26]. Knockout efficiency was calculated as a percentage of the number of cells exhibiting a lower fluorescence intensity than the threshold value.
3. Results and Discussion
3.1. Cleavage Activity and Amount of Released RNP
We examined whether the Cas9-sgRNA RNPs could be released from the NNA (Figure 1B). RNPs were adsorbed on the hydrophobized NNA and the unadsorbed fraction was collected. The RNPs adsorbed on the NNA were then released by oscillating the NNA in cytosolic simulation buffer. The solution was collected as the released fraction (Figure 1C). The substrate plasmids were added to both unadsorbed and released fractions, and were allowed to react for 60 min. The cleavage efficiency of the unadsorbed fraction was considered as saturated when the amount of applied Cas9 exceeded 1 pmol (Figure 1D; right). Moreover, most of the DNAs were not cut when a Cas9 threshold of 0.1 pmol was used, indicating that almost all of the RNPs were adsorbed. The number of Cas9 packed on the NNA, as estimated by dividing the NNA area by the area occupied by a single Cas9 molecule, was approximately 1 pmol. Therefore, the actual surface capacity of NNA to adsorb RNP would be between 0.1 and 1 pmol.
On the other hand, the cleavage activities of the released fractions were clearly evident (Figure 1D; left). The results indicated that cleavage activity of a Cas9-sgRNA RNP was retained even after adsorption onto the silicon surface. The cleavage efficiencies were almost similar, ranging between 30–35%, under all conditions. When 0.1 and 1.0 pmol of intact RNP were directly mixed into the plasmid solution, the cleavage efficiencies were 34% and 100%, respectively (data not shown). These results indicated that the maximum level of RNP adsorption onto NNA was approximately 0.1 pmol and that almost all RNPs could be released from the NNA, where oscillation had a slight effect on RNP release. Consequently, the amount of RNPs released from a single nanoneedle is estimated to be 0.1 amol. In an RNP delivery test, we used 10 pmol of RNP or more, and performed oscillation expecting assured adsorption and release of RNPs.
3.2. Delivery of Cas9-EmGFP to HEK293 Cell
Cas9 protein contains an arginine-rich α-helix domain that interacts with sgRNA [27]. Since the cationic domain was expected to electrostatically interact with the negatively charged cell surface, the behavior of the Cas9-mEmGFP fusion protein was verified with or without sgRNA. The addition of the Cas9-mEmGFP solution to HEK293 cells in a culture dish resulted in the immediate adsorption of Cas9-mEmGFP onto the cell surface (Figure 2A,B). By contrast, Cas9-mEmGFP forming an RNP complex with sgRNA at a ratio of 1:10 was not adsorbed onto the cell surface. While the adsorption of RNPs onto the cell surface was observed after 15 min, the adsorption of Cas9 was inhibited by interaction with sgRNA, which was possibly due to the neutralizing of positive charges derived from the arginine-rich domain. Because the total time taken by the NNA for the insertion of RNPs was less than 1 min, the adsorption of RNPs on cells during nanoneedle insertion ceased to be a factor. Cas9-mEmGFP proteins with or without sgRNA were incorporated into the cytosol after 22 h (Figure 2C), indicating that protein adsorption onto the cell surface progressed via endocytosis. Due to difficulties associated with escape from endosomes, most delivered molecules are degraded, indicating that direct delivery of Cas9 into the nucleus maybe preferable for genome editing purposes. Nanoneedles on the NNA are shown, indicating that at least one nanoneedle was inserted into a red fluorescent HEK293 cell (Figure 2D; green dots). Dark areas in the cells without red fluorescence are nuclei. The probability of the nucleus contacting nanoneedles was estimated to be about 70%, and the percentage of cells that contacted multiple needles was about 10%. Moreover, fluorescence intensities of the nanoneedles decreased after retraction from the cells, indicating that Cas9-mEmGFP had been successfully released from the surface of the nanoneedles (Figure 2E). Therefore, we concluded that our NNA system may be used for direct delivery of Cas9-sgRNA RNPs into living cells.
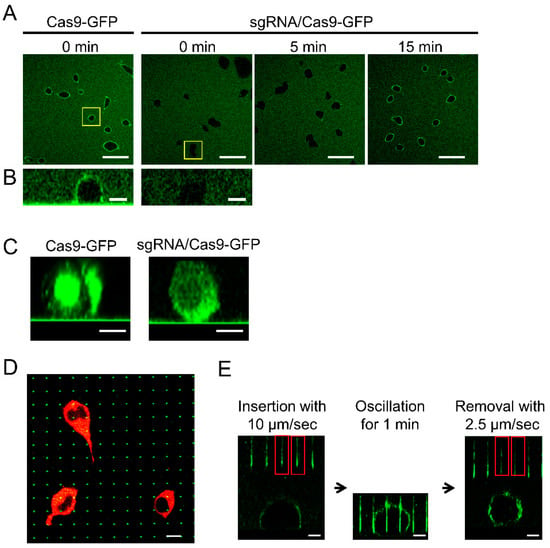
Figure 2.
Adsorption of Cas9-mEmGFP on the cell surface and insertion of NNA into the cells. (A) Confocal laser scanning microscopes (CLSM) images of HEK293 cells that were incubated with Cas9-mEmGFP with or without sgRNA. Scale bar: 50 μm. (B) Side view of HEK293 cells immediately following the addition of the Cas9-mEmGFP or RNP complex with sgRNA. Scale bar: 10 μm. (C) Side view of HEK293 cells 22 h following the addition of Cas9-mEmGFP or RNP. Scale bar: 20 μm. (D) Confocal XY slice image of HEK293 cells expressing DsRed2-NES for the visualization of the cytosol. The NNA was adsorbed with Cas9-mEmGFP interacting with sgRNA and inserted into HEK293 cells. Scale bar: 10 μm. (E) Confocal XZ cross section images of the nanoneedle insertion process into a cell. The red rectangles indicate nanoneedles inserted into the cell. Scale bar: 10 μm.
3.3. EGFP Knockout by Direct RNP Delivery
Next, we delivered Cas9-sgRNA into cells using the NNA. HeLa cells that stably expressed EGFP were used for the assay (Figure 3A). The NNA, on which RNPs were adsorbed, was moved towards the cells using an array manipulator while being observed with an inverted microscope. Following insertion, the NNA was oscillated for 1 min. Some cells did not express EGFP after 48 h of culturing (Figure 3B). Although the mock treatment of cells inserted by bare NNA showed that 4 ± 1% (average ± SD) of the total cells showed deficient EGFP expression, non-treated reporter cells were also found to include 4% of EGFP negative cell. This indicated that the insertion of the nanoneedle had no effect on gene expression. When RNPs were delivered using the NNA, the ratio of EGFP-depressed cells was 32 ± 11%. On the other hand, EGFP depression efficiency of lipofection was 57 ± 3%, which was rather high compared to that obtained for NNA. Although the same amount of RNP solution, containing 15 pmol Cas9 and 10 pmol sgRNA, was used in both delivery processes, the amount of RNP adsorbed onto the NNA surface was approximately 0.1 pmol, as stated above. We surmised that such a large difference in knockout efficiency was due to the difference in the effective amount of RNP delivered to cells.
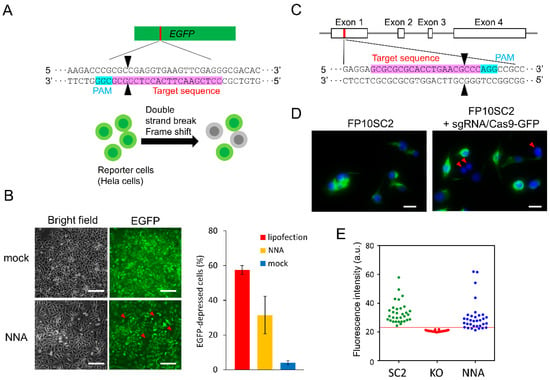
Figure 3.
Gene disruption in mammalian cells by direct delivery of the RNP complex. (A) Schematic of the EGFP disruption. (B) EGFP disruption in reporter cells (n = 3). Left panel shows bright field and fluorescence images of the portion of cells that were cultured for 48 h after the insertion of bare NNA (mock) or RNP delivery using NNA. Scale bar: 200 μm. Right panel shows the ratio of EGFP-depressed cells. RNP was introduced by lipofection (red) or NNA (yellow), respectively and the mock-treated cells was showed in blue. (C) Schematic representation of the mouse nestin gene and the sgRNA targeting sequence. (D) Immuno-stained FP10SC2 cells after delivery of RNP with the NNA (right) and negative control (left). Green indicates nestin, and blue indicates the nucleus stained with DAPI. Scale bar: 20 μm. (E) Fluorescence intensity threshold for estimating nestin deficient cells. Parental cells (SC2; green; positive control), nestin knockout cell line 5NK-H10 (H10; red; negative control), and RNP introduced cells by NNA (blue). Red line indicates the average fluorescence intensity of H10 plus five SD. The demonstrated plot and threshold were based on an experiment replicated 7 times. The target sequence of sgRNA and protospacer adjacent motif (PAM) are depicted in pink and light blue. Black arrowheads indicate cleavage sites. Red arrowheads indicate target-protein-deficient cells.
3.4. Nestin Knockout by Direct RNP Delivery
To further demonstrate the usefulness of our method of delivering Cas9-sgRNA via an NNA, we performed a gene knockout targeting the type VI intermediate filament nestin in a chromosome of FP10SC2 mouse breast cancer cells [24]. An RNP complex solution, consisting of Cas9-mEmGFP and sgRNA, targeting the rod domain of nestin, was poured on the NNA and allowed to dry completely to avoid loss of RNP (Figure 3C). Nestin deficient cells were counted according to their fluorescence intensity after immunostaining (Figure 3D). A previously established nestin knockout cell line 5NK-H10 (H10) was used as the negative control [26] and an average fluorescence intensity of H10 cells plus five SD was set as a threshold to determine nestin negative cells. Neither H10 cells with a fluorescence higher than the threshold nor wild type cells with a fluorescence lower than the threshold were detected (Figure 3E). NNA-treated cells with a fluorescence intensity under the threshold value were considered as nestin deficient cells. The results indicated that the nestin disruption efficiency of this method was 15.4 ± 8.2% (n = 7), while transfection of a plasmid vector expressing Cas9 and similar amounts of sgRNA resulted in a disruption efficiency of 20.3 ± 4.7% (n = 3) [26]. Moreover, we also detected nestin gene disruption in genomic DNA via a conventional T7E1 assay (data not shown). We selected 5 loci similar to the nestin target sequence with higher than 75% homologies and investigated off-target gene disruption, and determined that neither insertion nor deletion occurred at all in the candidate sites of even knockout cells generated by the plasmid-based method. This indicated that the sgRNA sequence used in this study recognized target sequence by high specificity. In further studies, off-target effect must be studied.
When all RNPs were retained on the NNA surface via drying, the gene disruption efficiency of this method reached levels comparable to that of plasmid-based genome editing. Although the efficiency of RNP introduction needs to be improved, we were able to successfully demonstrate genome editing by the direct delivery of Cas9-sgRNA using NNA. The lower efficiency of this method compared to introduction via lipofection is presumed to be due to the loss of RNPs on the NNA surface and the insertion efficiency of nanoneedles into cells. In a previous study, we succeeded in ensuring one-to-one contact between the nanoneedles and cells by preparing a cell array which aligned cells in a configuration that was identical to that of the 10,000 nanoneedles in the array [23]. Under optimized conditions, the efficiency of genome editing using NNA may be considered superior to that of the classical method.
4. Conclusions
We delivered the Cas9-sgRNA RNP complex directly into cells using the NNA system. It was confirmed that we could adsorb and release Cas9-sgRNA from the NNA surface. The efficiencies of gene disruption in HeLa cells and mouse breast cancer cells that were achieved using this method were approximately 32% and 16%, respectively. Although its efficiency needs to be improved and the contribution to the reduction of off-target effects must be clarified in further studies, this method of delivery may be applied to gene knock-in via homology-directed repair by a 2-step protocol, where the first step involves donor plasmid introduction and the second step involves the introduction of RNP, because we have also succeeded in introducing plasmid DNA using NNA [19].
Author Contributions
C.N. conceived of the methods and organized the research. A.Y. and D.M. wrote the manuscript. A.Y., D.M., Y.H., and M.M. performed the experiments and data analysis. Y.K. designed and constructed sgRNA and reporter cells for the Cas9 test. F.I developed the array manipulator. T.K. designed the protocol to fabricate the NNA. All authors read and approved the final manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 26249127, 17H03471 and 17K13026.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.L.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.B.; Jiang, W.Y.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Jakimo, N.; Jacobson, J.M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Higashiyama, T. pKAMA-ITACHI vectors for highly efficient CRISPR/Cas9-mediated gene knockout in Arabidopsis thaliana. Plant Cell Physiol. 2017, 58, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zuris, J.A.; Meng, F.T.; Rees, H.; Sun, S.; Deng, P.; Han, Y.; Gao, X.; Pouli, D.; Wu, Q.; et al. Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Nat. Acad. Sci. USA 2016, 113, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liang, X.Q.; Xie, H.M.; Kumar, S.; Ravinder, N.; Potter, J.; du Jeu, X.D.; Chesnut, J.D. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol. Lett. 2016, 38, 919–929. [Google Scholar] [CrossRef] [PubMed]
- McKnight, T.E.; Melechko, A.V.; Griffin, G.D.; Guillorn, M.A.; Merkulov, V.I.; Serna, F.; Hensley, D.K.; Doktycz, M.J.; Lowndes, D.H.; Simpson, M.L. Intracellular integration of synthetic nanostructures with viable cells for controlled biochemical manipulation. Nanotechnology 2003, 14, 551–556. [Google Scholar] [CrossRef]
- Krivitsky, V.; Hsiung, L.C.; Lichtenstein, A.; Brudnik, B.; Kantaev, R.; Elnathan, R.; Pevzner, A.; Khatchtourints, A.; Patolsky, F. Si nanowires forest-based on-chip biomolecular filtering, separation and preconcentration devices: Nanowires do it all. Nano Lett. 2012, 12, 4748–4756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Y.; Yan, L.; Kwok, S.Y.; Li, W.; Wang, Z.; Zhu, X.; Zhu, G.; Zhang, W.; Chen, X.; et al. Poking cells for efficient vector-free intracellular delivery. Nat. Commun. 2014, 5, 4466. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, C.; De Rosa, E.; Martinez, J.O.; Liu, X.; Steele, J.; Stevens, M.M.; Tasciotti, E. Biodegradable silicon nanoneedles delivering nucleic acids intracellularly induce localized in vivo neovascularization. Nat. Mater. 2015, 14, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Obataya, I.; Nakamura, C.; Han, S.; Nakamura, N.; Miyake, J. Nanoscale operation of a living cell using an atomic force microscope with a nanoneedle. Nano Lett. 2005, 5, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, D.; Rao Sathuluri, R.; Kato, Y.; Silberberg, Y.R.; Kawamura, R.; Iwata, F.; Kobayashi, T.; Nakamura, C. Oscillating high-aspect-ratio monolithic silicon nanoneedle array enables efficient delivery of functional bio-macromolecules into living cells. Sci. Rep. 2015, 5, 15325. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, D.; Nishio, M.; Kato, Y.; Yoshida, W.; Abe, K.; Fukazawa, K.; Ishihara, K.; Iwata, F.; Ikebukuro, K.; Nakamura, C. ATP-mediated release of a DNA-binding protein from a silicon nanoneedle array. Electrochemistry 2016, 84, 305–307. [Google Scholar] [CrossRef]
- Matsumoto, D.; Yamagishi, A.; Saito, M.; Sathuluri, R.R.; Silberberg, Y.R.; Iwata, F.; Kobayashi, T.; Nakamura, C. Mechanoporation of living cells for delivery of macromolecules using nanoneedle array. J. Biosci. Bioeng. 2016, 122, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Mieda, S.; Amemiya, Y.; Kihara, T.; Okada, T.; Sato, T.; Fukazawa, K.; Ishihara, K.; Nakamura, N.; Miyake, J.; Nakamura, C. Mechanical force-based probing of intracellular proteins from living cells using antibody-immobilized nanoneedles. Biosens. Bioelectron. 2012, 31, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, R.; Miyazaki, M.; Shimizu, K.; Matsumoto, Y.; Silberberg, Y.R.; Sathuluri, R.R.; Iijima, M.; Kuroda, S.; Iwata, F.; Kobayashi, T.; et al. A new cell separation method based on antibody-immobilized nanoneedle arrays for the detection of intracellular markers. Nano Lett. 2017, 17, 7117–7124. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Kurabayashi, A.; Akimitsu, N.; Furihata, M. Expression of cadherin-17 promotes metastasis in a highly bone marrow metastatic murine breast cancer model. BioMed Res. Int. 2017, 2017, 8494286. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Kato, Y.; Miyagishi, M.; Takagi, Y.; Taira, K. Small-interfering-RNA expression in cells based on an efficiently constructed dumbbell-shaped DNA. Angew. Chem. Int. Ed. 2004, 43, 3160–3163. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, A.; Kato, Y.; Meguro, K.; Yamagishi, A.; Nakamura, C.; Uyeda, T.Q.P. A genome editing vector that enables easy selection and identification of knockout cells. Plasmid 2018, 98, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).