
Graphene Oxide-Supported QuEChERS Extraction Coupled with LC-MS/MS for Trace-Level Analysis of Wastewater Pharmaceuticals


Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Standards
2.3. Model Wastewater
2.4. Sample Preparation Extraction and Clean-Up
2.4.1. QuEChERS-Based Extraction
2.4.2. Clean-Up
2.5. LC-MS/MS Conditions
2.6. Method Validation and Matrix Effect
3. Results and Discussion
3.1. LC–MS/MS Optimisation
3.2. Extraction and Clean-Up Optimisation
3.2.1. Extraction
3.2.2. Clean-Up
3.3. Validation Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mravcová, L.; Amrichová, A.; Navrkalová, J.; Hamplová, M.; Sedlář, M.; Gargošová, H.Z.; Fučík, J. Optimization and Validation of Multiresidual Extraction Methods for Pharmaceuticals in Soil, Lettuce, and Earthworms. Environ. Sci. Pollut. Res. 2024, 31, 33120–33140. [Google Scholar] [CrossRef]
- OECD Publishing. Health at a Glance 2021: OECD Indicators; OECD Publishing: Paris, France, 2021; Volume 2. [Google Scholar]
- Pérez-Lucas, G.; Navarro, S. How Pharmaceutical Residues Occur, Behave, and Affect the Soil Environment. J. Xenobiotics 2024, 14, 1343–1377. [Google Scholar] [CrossRef]
- Godfrey, A.R.; Dunscombe, J.; Gravell, A.; Hunter, A.; Barrow, M.P.; van Keulen, G.; Desbrow, C.; Townsend, R. Use of QuEChERS as a Manual and Automated High-Throughput Protocol for Investigating Environmental Matrices. Chemosphere 2022, 308, 136313. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Chu, L.M. Fate of Antibiotics in Soil and Their Uptake by Edible Crops. Sci. Total Environ. 2017, 599–600, 500–512. [Google Scholar] [CrossRef]
- Solliec, M.; Roy-Lachapelle, A.; Gasser, M.O.; Coté, C.; Généreux, M.; Sauvé, S. Fractionation and Analysis of Veterinary Antibiotics and Their Related Degradation Products in Agricultural Soils and Drainage Waters Following Swine Manure Amendment. Sci. Total Environ. 2016, 543, 524–535. [Google Scholar] [CrossRef]
- Wang, L.; Deng, Q.; Hu, H.; Liu, M.; Gong, Z.; Zhang, S.; Xu-Monette, Z.Y.; Lu, Z.; Young, K.H.; Ma, X.; et al. Glyphosate Induces Benign Monoclonal Gammopathy and Promotes Multiple Myeloma Progression in Mice. J. Hematol. Oncol. 2019, 12, 70. [Google Scholar] [CrossRef]
- Choudhury, M.; Adhikari, M.D.; Agarwal, S.; Samanta, P.; Sharma, A.; Kundu, D.; Kumar, S. Pharmaceuticals and Personal Care Products in Soil: Sources, Impacts and Myco-Remediation Strategies. Emerg. Contam. 2025, 11, 100488. [Google Scholar] [CrossRef]
- Zhang, C.; Barron, L.P.; Stürzenbaum, S.R. Pollution of Soil by Pharmaceuticals: Implications for Metazoan and Environmental Health. Annu. Rev. Pharmacol. Toxicol. 2024, 65, 547–565. [Google Scholar] [CrossRef] [PubMed]
- Saeed, H.; Padmesh, S.; Singh, A.; Nandy, A.; Singh, S.P.; Deshwal, R.K. Impact of Veterinary Pharmaceuticals on Environment and Their Mitigation through Microbial Bioremediation. Front. Microbiol. 2024, 15, 1396116. [Google Scholar] [CrossRef] [PubMed]
- Grenni, P.; Visca, A.; Caracciolo, A.B. Antibiotics as Emerging Pollutants of Soil Ecosystems. In Frontier Studies in Soil Science; Springer: Cham, Switzerland, 2024; pp. 21–41. ISBN 978-3-031-50503-4. [Google Scholar]
- Kachhawaha, A.S.; Nagarnaik, P.M.; Jadhav, M.; Pudale, A.; Labhasetwar, P.K.; Banerjee, K. Optimization of a Modified QuEChERS Method for Multiresidue Analysis of Pharmaceuticals and Personal Care Products in Sewage and Surface Water by LC-MS/MS. J. AOAC Int. 2017, 100, 592–597. [Google Scholar] [CrossRef]
- Simarro-Gimeno, C.; Pitarch, E.; Albarrán, F.; Rico, A.; Hernández, F. Ten Years of Monitoring Pharmaceuticals and Pesticides in the Aquatic Environment Nearby a Solid-Waste Treatment Plant: Historical Data, Trends and Risk Assessment. Environ. Pollut. 2025, 366, 125496. [Google Scholar] [CrossRef]
- Li, Z.; Yu, X.; Yu, F.; Huang, X. Occurrence, Sources and Fate of Pharmaceuticals and Personal Care Products and Artificial Sweeteners in Groundwater. Environ. Sci. Pollut. Res. 2021, 28, 20903–20920. [Google Scholar] [CrossRef]
- Peng, Y.; Gautam, L.; Hall, S.W. The Detection of Drugs of Abuse and Pharmaceuticals in Drinking Water Using Solid-Phase Extraction and Liquid Chromatography-Mass Spectrometry. Chemosphere 2019, 223, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Ebele, A.J.; Oluseyi, T.; Drage, D.S.; Harrad, S.; Abou-Elwafa Abdallah, M. Occurrence, Seasonal Variation and Human Exposure to Pharmaceuticals and Personal Care Products in Surface Water, Groundwater and Drinking Water in Lagos State, Nigeria. Emerg. Contam. 2020, 6, 124–132. [Google Scholar] [CrossRef]
- Jiang, X.; Qu, Y.; Zhong, M.; Li, W.; Huang, J.; Yang, H.; Yu, G. Seasonal and Spatial Variations of Pharmaceuticals and Personal Care Products Occurrence and Human Health Risk in Drinking Water—A Case Study of China. Sci. Total Environ. 2019, 694, 133711. [Google Scholar] [CrossRef]
- Muambo, K.E.; Kim, M.G.; Kim, D.H.; Park, S.; Oh, J.E. Pharmaceuticals in Raw and Treated Water from Drinking Water Treatment Plants Nationwide: Insights into Their Sources and Exposure Risk Assessment. Water Res. X 2024, 24, 100256. [Google Scholar] [CrossRef]
- Mazhandu, Z.; Mashifana, T. Active Pharmaceutical Contaminants in Drinking Water: Myth or Fact? DARU J. Pharm. Sci. 2024, 32, 925–945. [Google Scholar] [CrossRef]
- Zanni, S.; Cammalleri, V.; D’Agostino, L.; Protano, C.; Vitali, M. Occurrence of Pharmaceutical Residues in Drinking Water: A Systematic Review. Environ. Sci. Pollut. Res. 2024, 32, 10436–10463. [Google Scholar] [CrossRef]
- Carter, L.J.; Garman, C.D.; Ryan, J.; Dowle, A.; Bergström, E.; Thomas-Oates, J.; Boxall, A.B.A. Fate and Uptake of Pharmaceuticals in Soil-Earthworm Systems. Environ. Sci. Technol. 2014, 48, 5955–5963. [Google Scholar] [CrossRef] [PubMed]
- Kodešová, R.; Klement, A.; Golovko, O.; Fér, M.; Nikodem, A.; Kočárek, M.; Grabic, R. Root Uptake of Atenolol, Sulfamethoxazole and Carbamazepine, and Their Transformation in Three Soils and Four Plants. Environ. Sci. Pollut. Res. 2019, 26, 9876–9891. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Tang, M.; Xu, X. Mechanism of Uptake, Accumulation, Transport, Metabolism and Phytotoxic Effects of Pharmaceuticals and Personal Care Products within Plants: A Review. Sci. Total Environ. 2023, 892, 164413. [Google Scholar] [CrossRef]
- Gao, Q.; Dong, Q.; Wu, L.; Yang, Y.; Hale, L.; Qin, Z.; Xie, C.; Zhang, Q.; Van Nostrand, J.D.; Zhou, J. Environmental Antibiotics Drives the Genetic Functions of Resistome Dynamics. Environ. Int. 2020, 135, 105398. [Google Scholar] [CrossRef]
- Belay, M.H.; Precht, U.; Mortensen, P.; Marengo, E.; Robotti, E. A Fully Automated Online SPE-LC-MS/MS Method for the Determination of 10 Pharmaceuticals in Wastewater Samples. Toxics 2022, 10, 103. [Google Scholar] [CrossRef]
- Mostafa, A.; Shaaban, H.; Alqarni, A.; Al-Ansari, R.; Alrashidi, A.; Al-Sultan, F.; Alsulaiman, M.; Alsaif, F.; Aga, O. Multi-Class Determination of Pharmaceuticals as Emerging Contaminants in Wastewater from Eastern Province, Saudi Arabia Using Eco-Friendly SPE-UHPLC-MS/MS: Occurrence, Removal and Environmental Risk Assessment. Microchem. J. 2023, 187, 108453. [Google Scholar] [CrossRef]
- de la Serna Calleja, M.Á.; Bolado, S.; Jiménez, J.J.; López-Serna, R. Performance Critical Comparison of Offline SPE, Online SPE, and Direct Injection for the Determination of CECs in Complex Liquid Environmental Matrices. Microchem. J. 2023, 187, 108395. [Google Scholar] [CrossRef]
- Guan, J.; Zhang, C.; Wang, Y.; Guo, Y.; Huang, P.; Zhao, L. Simultaneous Determination of 12 Pharmaceuticals in Water Samples by Ultrasound-Assisted Dispersive Liquid–Liquid Microextraction Coupled with Ultra-High Performance Liquid Chromatography with Tandem Mass Spectrometry. Anal. Bioanal. Chem. 2016, 408, 8099–8109. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Hernández-Borges, J.; Borges-Miquel, T.M.; Rodríguez-Delgado, M. ángel Dispersive Liquid-Liquid Microextraction Combined with Ultra-High Performance Liquid Chromatography for the Simultaneous Determination of 25 Sulfonamide and Quinolone Antibiotics in Water Samples. J. Pharm. Biomed. Anal. 2013, 75, 130–137. [Google Scholar] [CrossRef]
- Lemos, V.A.; Barreto, J.A.; Santos, L.B.; dos Santos de Assis, R.; Novaes, C.G.; Cassella, R.J. In-Syringe Dispersive Liquid-Liquid Microextraction. Talanta 2022, 238, 123002. [Google Scholar] [CrossRef] [PubMed]
- Kamal El-Deen, A.; Elmansi, H.; Belal, F.; Magdy, G. Recent Advances in Dispersion Strategies for Dispersive Liquid–Liquid Microextraction from Green Chemistry Perspectives. Microchem. J. 2023, 191, 108807. [Google Scholar] [CrossRef]
- Díaz-Álvarez, M.; Martín-Esteban, A. Preparation and Further Evaluation of L-Menthol-Based Natural Deep Eutectic Solvents as Supported Liquid Membrane for the Hollow Fiber Liquid-Phase Microextraction of Sulfonamides from Environmental Waters. Adv. Sample Prep. 2022, 4, 100047. [Google Scholar] [CrossRef]
- Es’haghi, Z. Determination of Widely Used Non-Steroidal Anti-Inflammatory Drugs in Water Samples by in Situ Derivatization, Continuous Hollow Fiber Liquid-Phase Microextraction and Gas Chromatography-Flame Ionization Detector. Anal. Chim. Acta 2009, 641, 83–88. [Google Scholar] [CrossRef]
- Mlunguza, N.Y.; Ncube, S.; Mahlambi, P.N.; Chimuka, L.; Madikizela, L.M. Determination of Selected Antiretroviral Drugs in Wastewater, Surface Water and Aquatic Plants Using Hollow Fibre Liquid Phase Microextraction and Liquid Chromatography—Tandem Mass Spectrometry. J. Hazard. Mater. 2020, 382, 121067. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Saldaña, V.; Castro-García, C.; Rodríguez-Maese, R.; Leal-Quezada, L.O. Advances in the Extraction Methods for the Environmental Analysis of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs). TrAC Trends Anal. Chem. 2023, 169, 117409. [Google Scholar] [CrossRef]
- Avino, P.; Notardonato, I.; Passarella, S.; Vincenzo Russo, M. Determination of Non-Steroidal Anti-Inflammatory Drugs in Animal Urine Samples by Ultrasound Vortex-Assisted Dispersive Liquid-Liquid Microextraction and Gas Chromatography Coupled to Ion Trap-Mass Spectrometry. Appl. Sci. 2020, 10, 5441. [Google Scholar] [CrossRef]
- Arghavani-Beydokhti, S.; Rajabi, M.; Asghari, A. Coupling of Two Centrifugeless Ultrasound-Assisted Dispersive Solid/Liquid Phase Microextractions as a Highly Selective, Clean, and Efficient Method for Determination of Ultra-Trace Amounts of Non-Steroidal Anti-Inflammatory Drugs in Complicated Matrices. Anal. Chim. Acta 2018, 997, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Nciri, N. A Swift, Straightforward, and Innovative Approach for Detecting Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Aquatic Matrices Using Direct Immersion (DI) Three-Phase Single-Drop Microextraction (SDME) Coupled In-Line with Capillary Electrophoresis CE. In Trends in Environmental Sustainability and Green Energy—CGEEE 2023: Springer Proceedings in Earth and Environmental Sciences; Kim, J., Chen, Z., Eds.; Springer: Cham, Switzerland, 2024; pp. 27–40. ISBN 978-3-031-52330-4. [Google Scholar]
- Kiszkiel-Taudul, I.; Starczewska, B. Single Drop Microextraction Coupled with Liquid Chromatography-Tandem Mass Spectrometry (SDME-LC-MS/MS) for Determination of Ranitidine in Water Samples. Microchem. J. 2019, 145, 936–941. [Google Scholar] [CrossRef]
- Yıldırım, S.; Cocovi-Solberg, D.J.; Uslu, B.; Solich, P.; Horstkotte, B. Lab-In-Syringe Automation of Deep Eutectic Solvent-Based Direct Immersion Single Drop Microextraction Coupled Online to High-Performance Liquid Chromatography for the Determination of Fluoroquinolones. Talanta 2022, 246, 123476. [Google Scholar] [CrossRef]
- Szarka, A.; Vnuková, L.; Keršňáková, Z.; Viktoryová, N.; Hrouzková, S. Contamination with Pharmaceuticals in Aquatic Environment: Focus on Analytical Methodologies. Appl. Sci. 2024, 14, 8645. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and Easy Multiresidue Method Employing Acetonitrile Extraction/Partitioning and “Dispersive Solid-Phase Extraction” for the Determination of Pesticide Residues in Produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, S.H.; Eun, H.R.; Kim, S.M.; Jeong, M.J.; Baek, J.W.; Lee, Y.H.; Noh, H.H.; Shin, Y. Enhancement of Tricyclazole Analysis Efficiency in Rice Samples Using an Improved QuEChERS and Its Application in Residue: A Study from Unmanned Arial Spraying. Appl. Sci. 2024, 14, 5607. [Google Scholar] [CrossRef]
- Nardelli, V.; D’amico, V.; Ingegno, M.; Della Rovere, I.; Iammarino, M.; Casamassima, F.; Calitri, A.; Nardiello, D.; Li, D.; Quinto, M. Pesticides Contamination of Cereals and Legumes: Monitoring of Samples Marketed in Italy as a Contribution to Risk Assessment. Appl. Sci. 2021, 11, 7283. [Google Scholar] [CrossRef]
- Wenio, I.; Derewiaka, D.; Majewska, E.; Bartosiewicz, I.; Ryszkowska, E. Development of a Rapid Method for Simultaneous Determination of Pesticides in Plant Oil Using GC-MS/MS. Appl. Sci. 2024, 14, 4923. [Google Scholar] [CrossRef]
- Jo, H.W.; Park, M.G.; Jeon, H.J.; Moon, J.K.; Lee, S.E. Analysis of Multiresidue Pesticides in Agricultural Paddy Soils near Industrial Areas in Korea by GC–MS/MS and LC–MS/MS Using QuEChERS Extraction with DSPE Clean-up. Appl. Sci. 2021, 11, 8415. [Google Scholar] [CrossRef]
- Mahdavi, V.; Heris, M.E.S.; Dastranj, M.; Farimani, M.M.; Eslami, Z.; Aboul-Enein, H.Y. Assessment of Pesticide Residues in Soils Using a QuEChERS Extraction Procedure and LC-MS/MS. Water. Air. Soil Pollut. 2021, 232, 1–10. [Google Scholar] [CrossRef]
- Acosta-Dacal, A.; Rial-Berriel, C.; Díaz-Díaz, D.; Bernal-Suárez, M.; Luzardo, O.P. Optimization and Validation of a QuEChERS-Based Method for the Simultaneous Environmental Monitoring of 218 Pesticide Residues in Clay Loam Soil. Sci. Total Environ. 2021, 753, 142015. [Google Scholar] [CrossRef]
- Boti, V.; Kobothekra, V.; Albanis, T.; Konstantinou, I. Quechers-Based Methodology for the Screening of Alkylphenols and Bisphenol a in Dairy Products Using LC-LTQ/Orbitrap Ms. Appl. Sci. 2021, 11, 9358. [Google Scholar] [CrossRef]
- Areo, O.M.; Abafe, O.A.; Gbashi, S.; Njobeh, P.B. Detection of Multi-Mycotoxins in Rooibos and Other Consumed Teas in South Africa by a Modified QuEChERS Method and Ultra-High Performance Liquid Chromatography Tandem Mass Spectrometry. Food Control 2023, 143, 109255. [Google Scholar] [CrossRef]
- González-Jartín, J.M.; Rodríguez-Cañás, I.; Alfonso, A.; Sainz, M.J.; Vieytes, M.R.; Gomes, A.; Ramos, I.; Botana, L.M. Multianalyte Method for the Determination of Regulated, Emerging and Modified Mycotoxins in Milk: QuEChERS Extraction Followed by UHPLC–MS/MS Analysis. Food Chem. 2021, 356, 129647. [Google Scholar] [CrossRef] [PubMed]
- Casado, N.; Morante-Zarcero, S.; Sierra, I. Application of the QuEChERS Strategy as a Useful Sample Preparation Tool for the Multiresidue Determination of Pyrrolizidine Alkaloids in Food and Feed Samples: A Critical Overview. Appl. Sci. 2022, 12, 4325. [Google Scholar] [CrossRef]
- Temerdashev, Z.A.; Ovsepyan, S.K.; Musorina, T.N.; Vasileva, L.V.; Vasilev, A.M.; Korpakova, I.G. QuEChERS Extraction of PAHs from Various Soils and Sediments Followed by Chromatographic Determination. J. Anal. Chem. 2023, 78, 1159–1173. [Google Scholar] [CrossRef]
- Alexandre, B.; Barbara, G.; Laure, W.; Bruno, D.; Adriana, G.O.; Emmanuelle, V. Development of a Multiple-Class Analytical Method Based on the Use of Synthetic Matrices for the Simultaneous Determination of Commonly Used Commercial Surfactants in Wastewater by Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. A 2016, 1450, 64–75. [Google Scholar] [CrossRef]
- Surma, M.; Sznajder-Katarzyńska, K.; Wiczkowski, W.; Zieliński, H. Assessment of Bioactive Surfactant Levels in Selected Cereal Products. Appl. Sci. 2022, 12, 5242. [Google Scholar] [CrossRef]
- Ofman, P.; Skoczko, I. PAH Removal Effectiveness Comparison from Hydraulic Fracturing Model Wastewater in SBR Reactors with Granular and Flocked Activated Sludge. Desalin. Water Treat. 2018, 134, 41–51. [Google Scholar] [CrossRef]
- Kaczynski, P.; Hrynko, I.; Lozowicka, B. Evolution of Novel Sorbents for Effective Clean-up of Honeybee Matrix in Highly Toxic Insecticide LC/MS/MS Analysis. Ecotoxicol. Environ. Saf. 2017, 139, 124–131. [Google Scholar] [CrossRef]
- Anagnostopoulos, C.J.; Aplada Sarli, P.; Liapis, K.; Haroutounian, S.A.; Miliadis, G.E. Validation of Two Variations of the QuEChERS Method for the Determination of Multiclass Pesticide Residues in Cereal-Based Infant Foods by LC-MS/MS. Food Anal. Methods 2012, 5, 664–683. [Google Scholar] [CrossRef]
- Walorczyk, S. Validation and Use of a QuEChERS-Based Gas Chromatographic-Tandem Mass Spectrometric Method for Multiresidue Pesticide Analysis in Blackcurrants Including Studies of Matrix Effects and Estimation of Measurement Uncertainty. Talanta 2014, 120, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Yum, H.; Jeong, S.; Jang, M.; Moon, S.; Kang, M.; Kim, B.; Kim, D.; Choe, S.; Yang, W.; Kim, J.; et al. Fast and Reliable Analysis of Veterinary Metomidate and Etomidate in Human Blood Samples by Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS) in a Postmortem Case. J. Forensic Sci. 2021, 66, 2532–2538. [Google Scholar] [CrossRef] [PubMed]
- Christophoridis, C.; Veloutsou, S.; Mitsika, E.; Zacharis, C.K.; Christia, C.; Raikos, N.; Fytianos, K. Determination of Illicit Drugs and Psychoactive Pharmaceuticals in Wastewater from the Area of Thessaloniki (Greece) Using LC–MS/MS: Estimation of Drug Consumption. Environ. Monit. Assess. 2021, 193, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Canela, C.; Edo, S.; Rodríguez, N.; Gotor, G.; Lacorte, S. Comprehensive Characterization of 76 Pharmaceuticals and Metabolites in Wastewater by Lc-Ms/Ms. Chemosensors 2021, 9, 273. [Google Scholar] [CrossRef]
- Morsy, M.I.; Nouman, E.G.; Abdallah, Y.M.; Zainelabdeen, M.A.; Darwish, M.M.; Hassan, A.Y.; Gouda, A.S.; Rezk, M.R.; Abdel-Megied, A.M.; Marzouk, H.M. A Novel LC-MS/MS Method for Determination of the Potential Antiviral Candidate Favipiravir for the Emergency Treatment of SARS-CoV-2 Virus in Human Plasma: Application to a Bioequivalence Study in Egyptian Human Volunteers. J. Pharm. Biomed. Anal. 2021, 199, 114057. [Google Scholar] [CrossRef]
- Turković, L.; Bočkor, L.; Ekpenyong, O.; Silovski, T.; Lovrić, M.; Crnković, S.; Nigović, B.; Sertić, M. Development and Validation of a Novel LC-MS/MS Method for the Simultaneous Determination of Abemaciclib, Palbociclib, Ribociclib, Anastrozole, Letrozole, and Fulvestrant in Plasma Samples: A Prerequisite for Personalized Breast Cancer Treatment. Pharmaceuticals 2022, 15, 614. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yu, S.; Chai, G.; Liu, J.; Zhou, Q. (Tony) An LC-MS/MS Method for Simultaneous Analysis of the Cystic Fibrosis Therapeutic Drugs Colistin, Ivacaftor and Ciprofloxacin. J. Pharm. Anal. 2021, 11, 732–738. [Google Scholar] [CrossRef]
- Habler, K.; Brügel, M.; Teupser, D.; Liebchen, U.; Scharf, C.; Schönermarck, U.; Vogeser, M.; Paal, M. Simultaneous Quantification of Seven Repurposed COVID-19 Drugs Remdesivir (plus Metabolite GS-441524), Chloroquine, Hydroxychloroquine, Lopinavir, Ritonavir, Favipiravir and Azithromycin by a Two-Dimensional Isotope Dilution LC–MS/MS Method in Human Serum. J. Pharm. Biomed. Anal. 2021, 196, 113935. [Google Scholar] [CrossRef]
- Sadutto, D.; Picó, Y. Sample Preparation to Determine Pharmaceutical and Personal Care Products in an All-Water Matrix: Solid Phase Extraction. Molecules 2020, 25, 5204. [Google Scholar] [CrossRef]
- Salvia, M.V.; Vulliet, E.; Wiest, L.; Baudot, R.; Cren-Olivé, C. Development of a Multi-Residue Method Using Acetonitrile-Based Extraction Followed by Liquid Chromatography-Tandem Mass Spectrometry for the Analysis of Steroids and Veterinary and Human Drugs at Trace Levels in Soil. J. Chromatogr. A 2012, 1245, 122–133. [Google Scholar] [CrossRef]
- Rutkowska, E.; Kaczyński, P.; Iwaniuk, P.; Łozowicka, B.; Hrynko, I.; Jankowska, M.; Konecki, R.; Rogowska, W.; Rusiłowska, J.; Pietkun, M.; et al. An Extensive Pesticide Residue Study in Minor Polish Vegetables Based on Critical Consumer Diets. Food Control 2025, 176, 111383. [Google Scholar] [CrossRef]
- Zaidon, S.Z.; Ho, Y.B.; Hamsan, H.; Hashim, Z.; Saari, N.; Praveena, S.M. Improved QuEChERS and Solid Phase Extraction for Multi-Residue Analysis of Pesticides in Paddy Soil and Water Using Ultra-High Performance Liquid Chromatography Tandem Mass Spectrometry. Microchem. J. 2019, 145, 614–621. [Google Scholar] [CrossRef]
- Lafay, F.; Daniele, G.; Fieu, M.; Pelosi, C.; Fritsch, C.; Vulliet, E. Ultrasound-Assisted QuEChERS-Based Extraction Using EDTA for Determination of Currently-Used Pesticides at Trace Levels in Soil. Environ. Sci. Pollut. Res. 2022, 1–15. [Google Scholar] [CrossRef]
- Rodríguez-Llorente, D.; Hernández, E.; Gutiérrez-Sánchez, P.; Navarro, P.; Ismael Águeda, V.; Álvarez-Torrellas, S.; García, J.; Larriba, M. Extraction of Pharmaceuticals from Hospital Wastewater with Eutectic Solvents and Terpenoids: Computational, Experimental, and Simulation Studies. Chem. Eng. J. 2023, 451, 138544. [Google Scholar] [CrossRef]
- Lehotay, S.J.; Son, K.A.; Kwon, H.; Koesukwiwat, U.; Fu, W.; Mastovska, K.; Hoh, E.; Leepipatpiboon, N. Comparison of QuEChERS Sample Preparation Methods for the Analysis of Pesticide Residues in Fruits and Vegetables. J. Chromatogr. A 2010, 1217, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Lehotay, S.J.; O’Neil, M.; Tully, J.; García, A.V.; Contreras, M.; Mol, H.; Heinke, V.; Anspach, T.; Lach, G.; Fussell, R.; et al. Determination of Pesticide Residues in Foods by Acetonitrile Extraction and Partitioning with Magnesium Sulfate: Collaborative Study. J. AOAC Int. 2007, 90, 485–520. [Google Scholar] [CrossRef]
- CEN-CENELEC. Available online: https://www.cencenelec.eu/ (accessed on 12 April 2025).
- Pailler, J.Y.; Krein, A.; Pfister, L.; Hoffmann, L.; Guignard, C. Solid Phase Extraction Coupled to Liquid Chromatography-Tandem Mass Spectrometry Analysis of Sulfonamides, Tetracyclines, Analgesics and Hormones in Surface Water and Wastewater in Luxembourg. Sci. Total Environ. 2009, 407, 4736–4743. [Google Scholar] [CrossRef]
- Lindsey, M.E.; Meyer, M.; Thurman, E.M. Analysis of Trace Levels of Sulfonamide and Tetracycline Antimicrobials in Groundwater and Surface Water Using Solid-Phase Extraction and Liquid Chromatography/Mass Spectrometry. Anal. Chem. 2001, 73, 4640–4646. [Google Scholar] [CrossRef]
- Paíga, P.; Santos, L.H.M.L.M.; Delerue-Matos, C. Development of a Multi-Residue Method for the Determination of Human and Veterinary Pharmaceuticals and Some of Their Metabolites in Aqueous Environmental Matrices by SPE-UHPLC–MS/MS. J. Pharm. Biomed. Anal. 2017, 135, 75–86. [Google Scholar] [CrossRef]
- Kaczyński, P.; Łozowicka, B. A Novel Approach for Fast and Simple Determination Pyrrolizidine Alkaloids in Herbs by Ultrasound-Assisted Dispersive Solid Phase Extraction Method Coupled to Liquid Chromatography–Tandem Mass Spectrometry. J. Pharm. Biomed. Anal. 2020, 187, 113351. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, C.; Hong, Y.; Lee, W.; Lee, S.; Chung, H.; Kim, H.; Jeong, D.H. Determination of Pharmaceuticals in Solid Samples in Municipal Wastewater Treatment Plants by Online SPE LC–MS/MS Using QuEChERS Extraction. Environ. Monit. Assess. 2021, 193, 1–14. [Google Scholar] [CrossRef]
- Kaczyński, P. Large-Scale Multi-Class Herbicides Analysis in Oilseeds by Rapid One-Step QuEChERS-Based Extraction and Cleanup Method Using Liquid Chromatography–Tandem Mass Spectrometry. Food Chem. 2017, 230, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Adulkar, T.V.; Rathod, V.K. Pre-Treatment of High Fat Content Dairy Wastewater Using Different Commercial Lipases. Desalin. Water Treat. 2015, 53, 2450–2455. [Google Scholar] [CrossRef]
- Adulkar, T.V.; Rathod, V.K. Ultrasound Assisted Enzymatic Pre-Treatment of High Fat Content Dairy Wastewater. Ultrason. Sonochem. 2014, 21, 1083–1089. [Google Scholar] [CrossRef]
- Ekka, B.; Mieriņa, I.; Juhna, T.; Turks, M.; Kokina, K. Quantification of Different Fatty Acids in Raw Dairy Wastewater. Clean. Eng. Technol. 2022, 7, 100430. [Google Scholar] [CrossRef]
- Mao, F.; Kong, Q.; Ni, W.; Xu, X.; Ling, D.; Lu, Z.; Li, J. Melting Point Distribution Analysis of Globally Approved and Discontinued Drugs: A Research for Improving the Chance of Success of Drug Design and Discovery. ChemistryOpen 2016, 5, 357–368. [Google Scholar] [CrossRef]
- Tetko, I.V.; Sushko, Y.; Novotarskyi, S.; Patiny, L.; Kondratov, I.; Petrenko, A.E.; Charochkina, L.; Asiri, A.M. How Accurately Can We Predict the Melting Points of Drug-like Compounds? J. Chem. Inf. Model. 2014, 54, 3320–3329. [Google Scholar] [CrossRef]
- Ji, B.; Yang, L.; Ren, C.; Xu, X.; Zhao, W.; Yang, Y.; Xu, G.; Zhao, D.; Bai, Y. A Modified QuEChERS Method Based on a Reduced Graphene Oxide-Coated Melamine Sponge for Multiresidue Analysis of Veterinary Drugs in Mutton by UPLC–MS/MS. Food Chem. 2024, 433, 137376. [Google Scholar] [CrossRef]
- Hrynko, I.; Łozowicka, B.; Kaczyński, P. Development of Precise Micro Analytical Tool to Identify Potential Insecticide Hazards to Bees in Guttation Fluid Using LC–ESI–MS/MS. Chemosphere 2021, 263, 128143. [Google Scholar] [CrossRef]
- Yadav, S.; Goel, N.; Kumar, V.; Singhal, S. Graphene Oxide as Proficient Adsorbent for the Removal of Harmful Pesticides: Comprehensive Experimental Cum DFT Investigations. Anal. Chem. Lett. 2019, 9, 291–310. [Google Scholar] [CrossRef]
- Tanveer, Z.I.; Ahmad, K.; Dong, Z.; Chen, Y.; Liu, X.; Wu, Y.; Xu, T. Evaluation of Reduced Graphene Oxide-Based Nanomaterial as Dispersive Solid Phase Extraction Sorbent for Isolation and Purification of Aflatoxins from Poultry Feed, Combined with UHPLC–MS/MS Analysis. Food Addit. Contam. Part A 2023, 40, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Gupta, V. Methods for the Determination of Limit of Detection and Limit of Quantitation of the Analytical Methods. In Proceedings of the Chronicles of Young Scientists; Medknow Publications and Media Pvt. Ltd.: Mumbai, India, 2011; Volume 2, p. 21. [Google Scholar]
- Medina-Pastor, P.; Valverde, A.; Pihlström, T.; Masselter, S.; Gamon, M.; Mezcua, M.; Rodríguez-Torreblanca, C.; Fernández-Alba, A.R. Comparative Study of the Main Top-down Approaches for the Estimation of Measurement Uncertainty in Multiresidue Analysis of Pesticides in Fruits and Vegetables. J. Agric. Food Chem. 2011, 59, 7609–7619. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound and CAS Number | Molecular Formula | Structural Formula | Molar Mass [g⋅mol−1] | Bioavailability | Lowest PNECs Freshwater [µg⋅L−1] | pKa | Water Solubility [mg⋅mL−1] | logP | Melting Point [°C] |
|---|---|---|---|---|---|---|---|---|---|
| Angiotensin II receptor antagonists | |||||||||
| candesartan 139481-59-7 | C24H20N6O3 |  | 440.5 | 15% | 100 | 3.44/1.5 | >0.01 | 6.1 | 183–185 |
| valsartan 137862-53-4 | C24H29N5O3 |  | 435.5 | 25% | 560 | 4.7 | 0.02 | 1.5 | 116–117 |
| Angiotensin-converting enzyme inhibitors | |||||||||
| ramipril 87333-19-5 | C23H32N2O5 |  | 416.5 | 28% | 1000 | 3.75/5.2 | 0.04 | 2.9 | 109 |
| Antiarrhythmics | |||||||||
| propafenone 54063-53-5 | C21H27NO3 |  | 341.5 | nd | 0.85 | 14.09/9.63 | >0.01 | 3.2 | 173 |
| Antibiotics | |||||||||
| ciprofloxacin 85721-33-1 | C17H18FN3O3 | 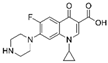 | 331.3 | 70% | 0.064 | 6.1 | 1.35 | 0.3 | 255–257 |
| doxycycline 564-25-0 | C22H24N2O8 |  | 444.4 | ~100% | 2 | 3.09 | 50 | 0.6 | 201 |
| Antidepressants | |||||||||
| duloxetine 116539-59-4 | C18H19NOS |  | 297.4 | 32–80% | 0.43 | 9.7 | >0.01 | 4 | 164 |
| escitalopram 128196-01-0 | C20H21FN2O |  | 324.4 | ~80% | 2.68 | 9.5 | >0.01 | 1.3 | 147–152 |
| fluoxetine 54910-89-3 | C17H18F3NO |  | 309.3 | 60–80% | 0.1 | 9.8 | >0.01 | 4.1 | 193–197 |
| sertraline 79617-96-2 | C17H17Cl2N |  | 306.2 | 44% | 0.0094 | 9.16 | 3.8 | 5.1 | 245–246 |
| venlafaxine 93413-69-5 | C17H27NO2 |  | 277.4 | ~42% | 0.88 | 14.42/8.91 | 572 | 2.7 | 240 |
| Antidiabetics | |||||||||
| metformin 657-24-9 | C4H11N5 |  | 129.2 | 50–60% | 160 | 12.4 | 1.38 | −2.6 | 223–226 |
| Beta-blockers | |||||||||
| atenolol 29122-68-7 | C14H22N2O3 |  | 266.3 | 50–60% | 150 | 9.6 | 0.7 | 0.2 | 158–160 |
| bisoprolol 66722-44-9 | C18H31NO4 |  | 325.5 | >90% | 92 | 9.5 | 0.07 | 2.2 | 100–103 |
| metoprolol 51384-51-1 | C15H25NO3 |  | 267.4 | 50–70% | 8.6 | 9.7 | 0.40 | 2.2 | 120 |
| Diuretics | |||||||||
| indapamide 26807-65-8 | C16H16ClN3O3S |  | 365.8 | nd | 1.29 | 8.8 | 0.03 | 2.5 | 160–162 |
| torasemide 56211-40-6 | C16H20N4O3S |  | 348.4 | 80–90% | nd | 7.1 | 0.06 | 3.4 | 163–164 |
| Hormones | |||||||||
| levonorgestrel 797-63-7 | C21H28O2 |  | 312.5 | 85–100% | 0.00001 | 17.91/−1.5 | >0.01 | 3.8 | 232–239 |
| Trial | Extraction Solvent | Buffer | Additives |
|---|---|---|---|
| 1 | acetonitrile | - | - |
| 2 | acetate | - | |
| 3 | citrate | - | |
| 4 | - | formic acid 1 | |
| 5 | - | 0.2 M Na2EDTA | |
| 6 | citrate | 0.2 M Na2EDTA | |
| 7 | ethyl acetate | - | - |
| 8 | citrate | - | |
| 9 | - | 0.2 M Na2EDTA | |
| 10 | citrate | 0.2 M Na2EDTA |
| Pharmaceutical or Metabolite | Retention Time (min) | Quantification | Confirmation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| MRM Transition m/z | EP (V) | CE (V) | CXP (V) | MRM Transition m/z | EP (V) | CE (V) | CXP (V) | ||
| Atenolol | 2.35 | 267.3 > 267 | 5 | 35 | 2 | 267.3 > 133 | 2 | 39 | 4 |
| Bisoprolol | 3.7 | 326 > 116 | 10 | 27 | 8 | 326 > 74 | 10 | 43 | 12 |
| Candesartan | 5.4 | 441.2 > 63.2 | 10 | 74 | 17 | 441.2 > 207.2 | 12 | 54 | 10 |
| Ciprofloxacin | 3.05 | 332 > 288 | 7 | 25 | 8 | 332 > 314 | 7 | 29 | 8 |
| Doxycycline | 5.25 | 445.1 > 428 | 10 | 29 | 13 | 445.1 > 321 | 10 | 42 | 13 |
| Duloxetine | 4.05 | 298.1 > 44.1 | 3 | 33 | 4 | 298.1 > 154.2 | 3 | 11 | 4 |
| Escitalopram | 2.2 | 325.1 > 143 | 15 | 53 | 10 | 325.1 > 109.1 | 15 | 30 | 10 |
| Fluoxetine | 4.25 | 310.1 > 148.1 | 11 | 15 | 4 | 310.3 > 91.2 | 5 | 25 | 2 |
| Indapamide | 3.55 | 366.1 > 132.2 | 10 | 20 | 7 | 366.1 > 147.1 | 12 | 18 | 5 |
| Levovorgestrel | 4.75 | 313.3 > 245.3 | 10 | 25 | 12 | 313.3 > 96.3 | 8 | 28 | 10 |
| Metformin | 1.55 | 130.1 > 71 | 4 | 32 | 2 | 130.1 > 60 | 4 | 19 | 2 |
| Metoprolol | 2.75 | 268 > 191 | 10 | 27 | 12 | 268 > 145 | 10 | 35 | 4 |
| Propafenone | 3.95 | 342.4 > 116.1 | 8 | 32 | 7 | 342.4 > 98.1 | 12 | 29 | 6 |
| Ramipril | 4.2 | 417.2 > 234.2 | 5 | 30 | 3 | 417.2 > 117.1 | 5 | 57 | 2 |
| Sertraline | 4.75 | 306.1 > 159.1 | 7 | 35 | 4 | 306.1 > 275.1 | 5 | 17 | 4 |
| Torasemide | 3.6 | 349.1 > 264.1 | 5 | 25 | 3 | 349.1 > 168.1 | 5 | 66 | 2 |
| Valsartan | 4.65 | 436 > 235.2 | 7 | 37 | 11 | 436 > 291.2 | 7 | 23 | 6 |
| Venlafaxine | 3.55 | 278.2 > 58.1 | 8 | 37 | 4 | 278.2 > 260.2 | 8 | 17 | 4 |
| Venlafaxine N-oxide | 3.3 | 294 > 121 | 4 | 30 | 12 | 295 > 178 | 6 | 24 | 16 |
| Venlafaxine O-desmethyl | 3.1 | 264.1 > 58.1 | 11 | 31 | 4 | 264.1 > 246.4 | 11 | 17 | 4 |
| Pharmaceutical or Metabolite | LOD (μg mL−1) | LOQ (μg mL−1) | Recovery (RSD) (%) | U (%) | ME (%) | ||
|---|---|---|---|---|---|---|---|
| 0.001 (μg⋅mL−1) | 0.01 (μg⋅mL−1) | 0.1 (μg⋅mL−1) | |||||
| Atenolol | 0.05 | 0.15 | 96 (8) | 97 (4) | 98 (4) | 11 | 4 |
| Bisoprolol | 0.06 | 0.20 | 71 (8) | 70 (4) | 75 (6) | 14 | −2 |
| Candesartan | 0.20 | 0.65 | 85 (5) | 86 (5) | 84 (7) | 11 | 4 |
| Ciprofloxacin | 0.09 | 0.30 | 81 (4) | 80 (9) | 79 (11) | 13 | 8 |
| Doxycycline | 0.15 | 0.50 | 97 (16) | 98 (5) | 95 (5) | 11 | −10 |
| Duloxetine | 0.13 | 0.45 | 96 (12) | 94 (13) | 96 (12) | 20 | −1 |
| Escitalopram | 0.15 | 0.50 | 90 (6) | 88 (9) | 90 (9) | 21 | 7 |
| Fluoxetine | 0.10 | 0.35 | 95 (6) | 96 (8) | 99 (4) | 14 | −2 |
| Indapamide | 0.15 | 0.50 | 75 (6) | 73 (3) | 75 (6) | 11 | 12 |
| Levonorgestrel | 0.15 | 0.50 | 87 (6) | 84 (6) | 87 (9) | 17 | 15 |
| Metformin | 0.10 | 0.35 | 85 (10) | 84 (13) | 82 (6) | 26 | 8 |
| Metoprolol | 0.08 | 0.25 | 84 (7) | 73 (9) | 74 (6) | 20 | −9 |
| Propafenone | 0.08 | 0.25 | 82 (5) | 80 (5) | 82 (5) | 8 | −7 |
| Ramipril | 0.09 | 0.30 | 72 (7) | 70 (3) | 73 (5) | 9 | −11 |
| Sertraline | 0.13 | 0.45 | 72 (10) | 74 (7) | 76 (6) | 15 | 7 |
| Torasemide | 0.19 | 0.65 | 90 (11) | 92 (4) | 94 (6) | 14 | 2 |
| Valsartan | 0.15 | 0.50 | 97 (6) | 95 (11) | 93 (8) | 22 | −6 |
| Venlafaxine | 0.22 | 0.70 | 74 (9) | 73 (5) | 74 (6) | 9 | 2 |
| Venlafaxine N-oxide | 0.20 | 0.65 | 73 (8) | 71 (6) | 73 (4) | 20 | 12 |
| Venlafaxine-O-desmethyl | 0.07 | 0.25 | 83 (11) | 82 (3) | 84 (6) | 7 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogowska, W.; Kaczyński, P. Graphene Oxide-Supported QuEChERS Extraction Coupled with LC-MS/MS for Trace-Level Analysis of Wastewater Pharmaceuticals. Appl. Sci. 2025, 15, 8441. https://doi.org/10.3390/app15158441
Rogowska W, Kaczyński P. Graphene Oxide-Supported QuEChERS Extraction Coupled with LC-MS/MS for Trace-Level Analysis of Wastewater Pharmaceuticals. Applied Sciences. 2025; 15(15):8441. https://doi.org/10.3390/app15158441
Chicago/Turabian StyleRogowska, Weronika, and Piotr Kaczyński. 2025. "Graphene Oxide-Supported QuEChERS Extraction Coupled with LC-MS/MS for Trace-Level Analysis of Wastewater Pharmaceuticals" Applied Sciences 15, no. 15: 8441. https://doi.org/10.3390/app15158441
APA StyleRogowska, W., & Kaczyński, P. (2025). Graphene Oxide-Supported QuEChERS Extraction Coupled with LC-MS/MS for Trace-Level Analysis of Wastewater Pharmaceuticals. Applied Sciences, 15(15), 8441. https://doi.org/10.3390/app15158441