1. Introduction
The development of all-solid-state Li-metal batteries (ASSLBs) has gained significant attention as a promising route toward next-generation energy storage systems with enhanced energy density, safety, and longevity [
1,
2,
3,
4]. Traditional Li-ion batteries (LIBs) are limited by flammable liquid electrolytes and low-capacity graphite anodes, which pose risks of leakage, dendrite formation, and thermal runaway [
5,
6]. In contrast, replacing the liquid electrolyte with a solid-state electrolyte (SSE) and utilizing Li metal as the anode offer substantial improvements in volumetric and gravimetric energy density while greatly reducing safety concerns [
7,
8]. ASSLBs are particularly appealing for high-demand applications such as electric vehicles and grid-level storage, where both energy density and safety are paramount.
Among various solid-state configurations, the LFP/Li
1.3Al
0.3Ti
1.7(PO
4)
3 (LATP)/Li system presents a highly attractive platform [
9,
10,
11]. LFP is a widely studied cathode material due to its exceptional thermal stability, flat voltage profile, long lifecycle, and compatibility with solid-state architectures [
12,
13,
14]. LATP, a NASICON-type solid electrolyte, offers high Li-ion conductivity and relatively good stability against LFP [
15,
16]. Furthermore, Li metal as an anode maximizes energy density. This tri-layer system combines safety, cost-effectiveness, and electrochemical performance, making it a suitable model to explore the fundamental and practical aspects of ASSLBs.
A key challenge in solid-state battery development lies in the solid–solid interface between the electrolyte and electrodes [
17,
18]. Unlike liquid electrolytes that get wet and conform to electrode surfaces, solid electrolytes require intimate physical and chemical contact to enable efficient Li-ion transfer. Interfacial resistance, mechanical mismatch, and chemical reactivity can all hinder ion transport and degrade performance over time. Therefore, understanding and controlling the electrode/electrolyte interface are critical to improving ionic conductivity, cycling stability, and overall device performance in ASSLBs [
19,
20].
Compared to previous reports on Li/LATP/LFP solid-state batteries, the present study offers a distinct approach in both material processing and mechanistic understanding. In contrast to the work by He et al. [
21], which employed a mesoporous LFP–LATP scaffold filled with the MEEP polymer to achieve high areal capacities, our study eliminates the use of polymer fillers entirely, instead relying on a dense LATP pellet with mechanically stable contact. This polymer-free design not only simplifies the cell architecture but also mitigates issues such as polymer degradation or dendrite formation within polymer-rich regions.
While Yao et al. explores Li/LATP/LFP ASSLBs using high-temperature sintered LATP and slurry-cast cathodes with polyvinylidene fluoride (PVDF) binders [
22], our study utilizes an alternative fabrication method—alternating current electrophoretic deposition (AC-EPD)—which enables the uniform and binder-integrated deposition of LFP electrodes directly onto current collectors. This approach simplifies processing while ensuring strong adhesion and consistent morphology. In contrast to the tape-casting or manual mixing methods used in prior studies, AC-EPD offers better control over electrode thickness and composition, contributing to improved interface formation and mechanical integrity—critical factors for solid-state battery performance. Moreover, our study extends beyond the electrochemical testing emphasized in previous studies [
22] by providing a quantitative correlation between Li-ion diffusivity,
b-value analysis (from cyclic voltammetry), and morphological evolution [
22,
23,
24]. For instance, while Mazor et al. focuses on enhancing interfacial contact via chemical strategies [
23], such as coating LFP with ionic conductors or interlayers, our approach addresses interface engineering through controlled electrolyte sintering and the detailed analysis of its impact on ion transport.
In addition, Yan et al. investigates the influence of electrode porosity and processing conditions but lacks a comprehensive discussion of charge storage mechanisms [
24]. Our work bridges this gap by using
b-value analysis to distinguish between diffusion-limited and capacitive charge storage processes across different cell architectures, including comparisons with a liquid electrolyte control system. These insights establish a framework for understanding how interfacial morphology governs charge storage mechanisms in solid-state batteries, further distinguishing this study from the existing literature. While numerous studies focus on maximizing performance in Li/LATP/LFP batteries [
21,
22,
23,
24], the objective of this work is to elucidate the interfacial charge storage mechanism and lithium-ion transport kinetics. The electrochemical performance obtained in this study is on par with or superior to comparable systems reported in the literature with similar cathode mass loading, validating the reliability of our system for interface-sensitive analysis.
Here, we compared three battery systems: (a) LFP/LATP sintered at 900 °C, (b) LFP/LATP sintered at 1000 °C, and (c) LFP/LiPF6 liquid electrolyte. This comparison enables a systematic investigation of how the interfacial environment affects the microstructure and ionic pathways of LATP and how these changes translate to differences in electrochemical performance. The inclusion of a liquid-electrolyte-based system provides a practical benchmark, highlighting the performance and interfacial characteristics of conventional systems relative to solid-state architectures. In this study, we employed AC-EPD to fabricate uniform LFP composite cathodes for all three battery types. The AC-EPD technique allows for the controlled, reproducible deposition of active materials with consistent film morphology and thickness. Its ability to form uniform cathodes across different substrates (LATP and metal current collectors) enables the direct comparison of electrochemical performance with minimal variation in electrode architecture. This platform is particularly well suited for interface-sensitive studies, as it ensures that differences in performance can be primarily attributed to the electrolyte and interface characteristics rather than inconsistencies in cathode preparation.
3. Results and Discussion
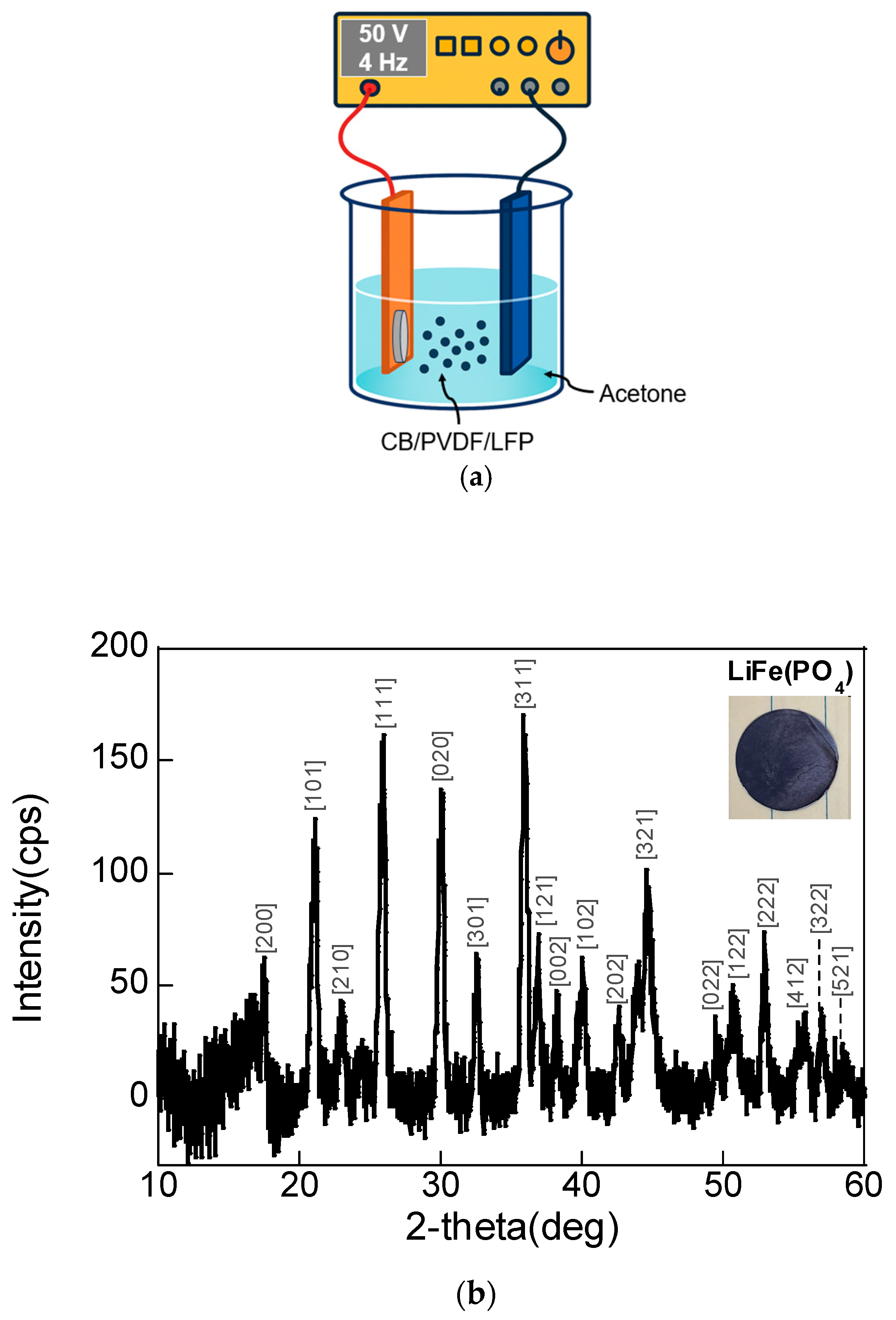
The LFP cathode was deposited on the SS foil using the AC-EPD method using the setup in
Figure 1a. The crystallinity of the LFP film on the SS was confirmed using XRD in
Figure 1b. (Sample 1: Li/LATP (900 °C)/LFP, Sample 2: Li/LATP (1000 °C)/LFP, and Sample 3: Li/LiPF
6 (liquid electrolyte/LFP.) The XRD pattern corresponds to the LFP electrode fabricated by AC-EPD onto an SS foil substrate under the conditions of 50 V and 4 Hz for 10 min. The diffraction peaks match well with the orthorhombic olivine structure of LFP, confirming the successful deposition of phase-pure crystalline LFP on the substrate. The sharp and intense peaks suggest a high degree of crystallinity in the deposited LFP film, with no evidence of secondary phases such as Fe
2O
3 or Li
3PO
4, which could form during improper processing. The absence of significant peak broadening also indicates that the crystallite size is relatively large, and that the EPD process did not degrade the structural integrity of the LFP particles.
The inset image in
Figure 1a shows the surface of the EPD-deposited LFP film, which appears dense and uniform, further supporting the effectiveness of the AC-EPD method in forming a high-quality electrode layer. Overall, the XRD analysis in
Figure 1b confirms that the AC-EPD process preserved the crystal structure of LFP and enabled its direct deposition onto conductive substrates for solid-state or conventional LIB applications.
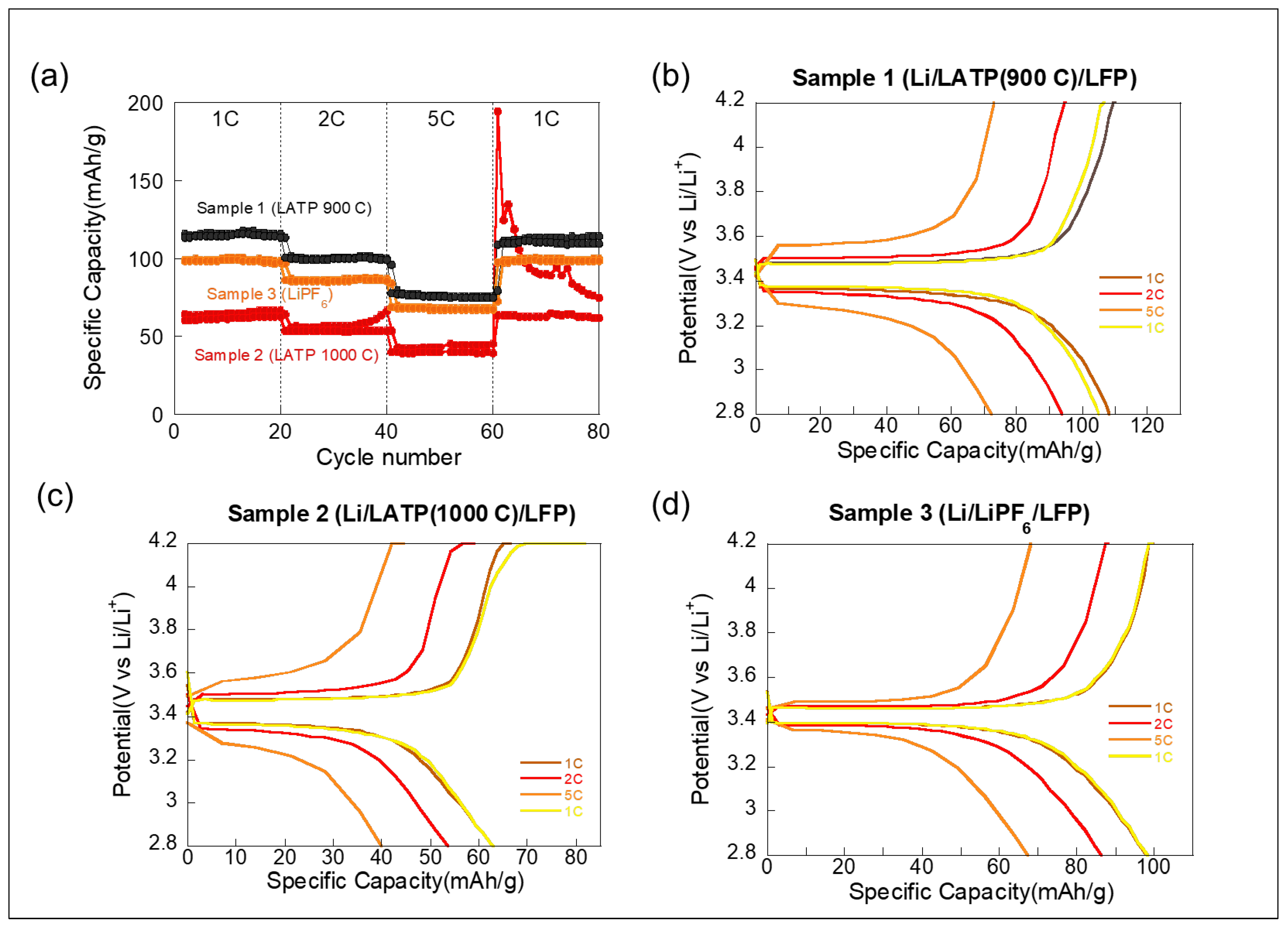
The rate performance of the three samples was compared across a range of current densities from 1 C to 5 C in
Figure 2, demonstrating its excellent high-rate capability in Sample 1. The rate performance of the three LFP-based cells—each interfaced with a different electrolyte environment—is presented in
Figure 2a, with the corresponding charge–discharge voltage profiles shown in
Figure 2b–d. These results clearly highlight the effect of the electrolyte interface and LATP sintering temperature on the electrochemical behavior of the LFP electrodes under various current rates. Sample 1, using LATP sintered at 900 °C, demonstrates the best rate performance and stability among the three configurations. As shown in
Figure 2a, it maintains a specific capacity of approximately 80–120 mAh∙g
−1 across a wide range of current rates (1 C to 5 C), and nearly full capacity is recovered upon returning to 1 C. The corresponding voltage profiles in
Figure 2b display well-defined charge–discharge plateaus, even at high rates, suggesting low interfacial resistance and effective Li
+ transport across the LFP/LATP interface. This performance indicates favorable interfacial contact and mechanical compliance between the LFP and LATP processed at this temperature.
In contrast, Sample 2, which used LATP sintered at 1000 °C, shows significantly lower capacity and poor rate capability. The capacity drops below 50 mAh∙g
−1 at high rates and exhibits abnormal decay in the charging capacity when cycled back to 1 C. This is further supported by the voltage curves in
Figure 2c, which reveal distorted and less-defined plateaus, indicating sluggish kinetics and possible interfacial degradation. The higher sintering temperature likely led to a denser LATP microstructure with reduced surface compliance, impairing interfacial contact and increasing resistance during cycling, which will be discussed later.
Sample 3, which employed a liquid electrolyte (1 M LiPF
6), exhibits moderate performance. The initial capacity is around 100 mAh·g
−1 at 1 C and gradually declines with the increasing rate, but the capacity largely recovers when the current is reduced back to 1 C.
Figure 2d shows smoother voltage profiles than Sample 2. While the liquid electrolyte allows better wetting and ion transport than poorly sintered LATP, it still falls short of the well-optimized solid interface seen in Sample 1. Overall, these results demonstrate that the sintering condition of LATP has a substantial impact on the performance of all-solid-state batteries, with optimized conditions (900 °C) enabling superior rate capability and cycling stability through improved electrode–electrolyte interfacial contact.
From the comparison of the cycling performance in the
Supporting Information (Figure S1), the LATP 900 °C and 1000 °C samples both exhibit stable cycling behavior over repeated cycles (500 cycles), although the 900 °C sample shows a higher initial capacity. This difference is likely due to better interfacial contact and lower resistance in the 900 °C-sintered LATP, which enhances lithium-ion transport. In contrast, the 1000 °C sample, while more densely sintered, may have poorer interfacial compliance, resulting in lower capacity. However, both samples maintain similar long-term stability, suggesting that their bulk structures remain intact and chemically stable throughout cycling.
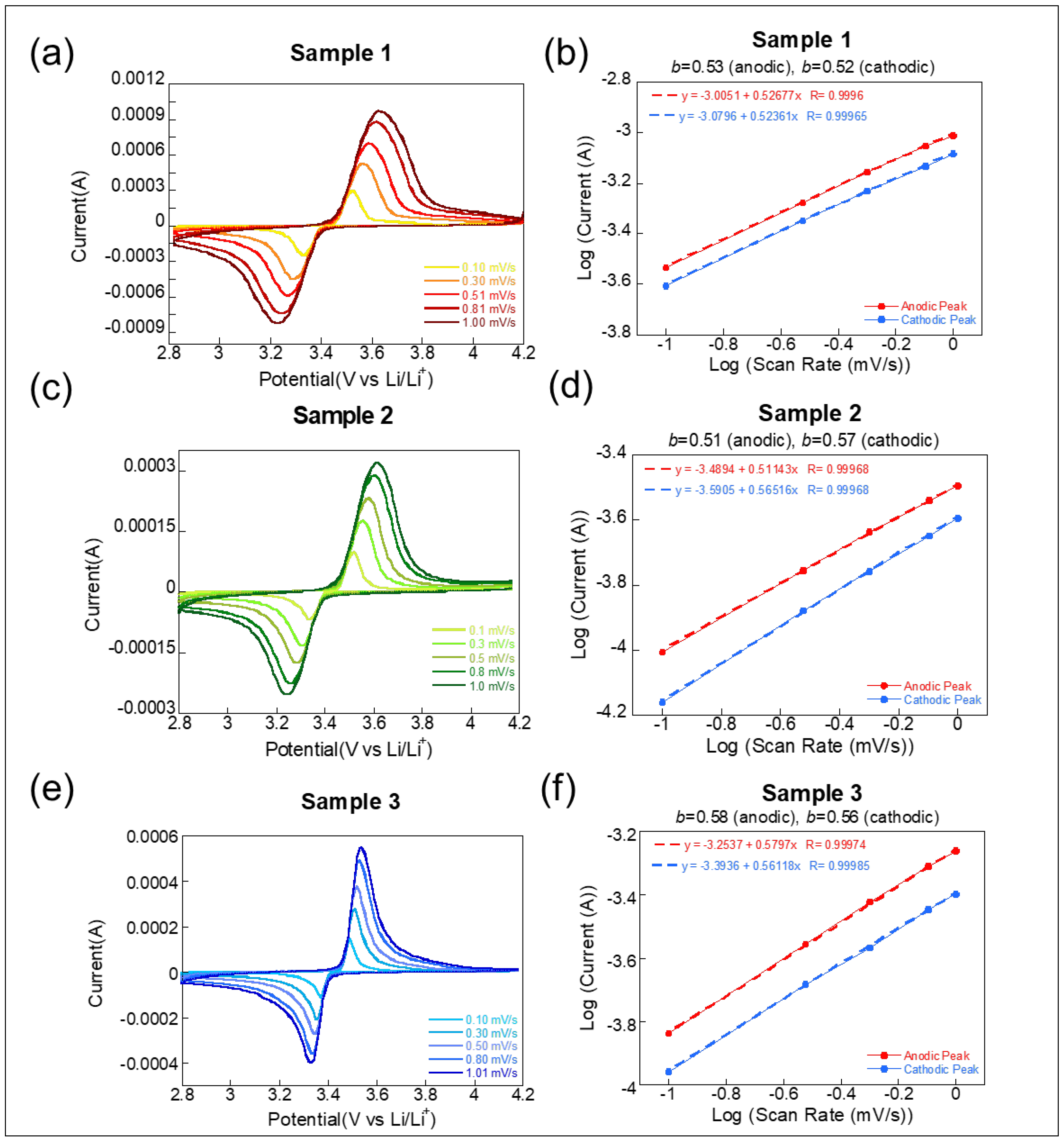
The comparison of
b-values for the three LFP-based samples, derived from CV at various scan rates, provides insight into the dominant charge storage mechanisms operating in each system [
27,
28]. The
b-value, obtained from the slope of log(current) versus log(scan rate), indicates whether the process is diffusion-controlled (
b ≈ 0.5) or surface-capacitive (
b ≈ 1.0). Sample 1, which uses LATP sintered at 900 °C, shows
b-values of 0.53 (anodic) and 0.52 (cathodic), as seen in
Figure 3a,b. These values are very close to 0.5, indicating that the redox process is predominantly diffusion-controlled. The near-symmetric CV curves and stable peak separation further suggest efficient Li
+ intercalation/deintercalation and good electrode–electrolyte interfacial contact. This result aligns well with the excellent rate performance observed earlier, confirming the favorable ion transport dynamics in this solid-state configuration.
Sample 2, paired with LATP sintered at 1000 °C, exhibits similar
b-values of 0.51 (anodic) and 0.57 (cathodic) in
Figure 3c,d. Although still diffusion-dominated, the cathodic value is marginally higher, indicating a slightly more capacitive contribution during discharge. In contrast, Sample 3, using a liquid electrolyte (1 M LiPF
6), shows the highest
b-values—0.58 (anodic) and 0.56 (cathodic)—as shown in
Figure 3e,f. These values reflect a more significant contribution from surface capacitive processes, likely due to enhanced ion mobility and electrolyte wetting at the interface. The sharper, more symmetric CV peaks support this interpretation, pointing to fast surface redox activity and a more pronounced pseudocapacitive effect. In other words, all three systems show charge storage predominantly governed by diffusion, but to varying extents. Sample 1 demonstrates the most ideal solid-state interfacial kinetics, Sample 2 reveals limitations due to high-temperature sintering, and Sample 3 benefits from surface-dominated processes in liquid electrolytes. This comparison underscores how electrolyte type and interface quality critically influence the charge storage behavior in LFP electrodes.
It is also important to note that the broader redox peaks observed in CV curves for the solid-state cells (Samples 1 and 2) relative to the liquid electrolyte cell (Sample 3) arise from a combination of factors including higher interfacial resistance, lower ionic conductivity of LATP, and limited solid–solid contact at the LFP/LATP interface. These factors result in slower ion transport and more distributed electrochemical reactions, leading to peak broadening and increased polarization
The diffusivity analysis presented in
Figure 4 compares the Li-ion diffusion behavior of three LFP-based electrodes interfaced with different electrolytes and processing conditions. The current response at anodic and cathodic peaks plotted against the square root of the scan rate (
Figure 4a–c) was used to extract diffusion coefficients based on the Randles–Sevcik equation. The resulting diffusivity values are summarized in the bar graph shown in
Figure 4d, providing insights into how interface characteristics affect Li-ion transport kinetics.
Sample 1, which uses LATP sintered at 900 °C, exhibits the highest diffusivity values for both anodic and cathodic processes, in the order of ~10−9 cm2∙s−1. This suggests that the interface between LFP and LATP is well formed, likely due to optimal sintering that balances densification and ionic conductivity. The high diffusion coefficient is consistent with the previously observed strong rate capability and minimal polarization in Sample 1, indicating efficient Li+ transport across the electrode–electrolyte interface. In contrast, Sample 2, based on LATP sintered at 1000 °C, shows the lowest diffusivity among the three. Both anodic and cathodic values drop significantly, approaching ~10−10 cm2∙s−1. This reduction reflects poor Li+ transport, which may be attributed to excessive grain growth or reduced surface compliance in the over-sintered LATP, leading to inferior contact with the LFP cathode. The compromised interface in Sample 2 results in sluggish ion kinetics, which aligns with its poor capacity retention and distorted voltage profiles seen in rate performance measurements. Sample 3, using a liquid electrolyte (1 M LiPF6), shows intermediate diffusivity values between Samples 1 and 2. Although not as high as Sample 1, the liquid electrolyte provides sufficient ion mobility and wetting, facilitating moderate diffusion (~10−9 cm2∙s−1). This is reflected in the CV symmetry and decent capacity recovery in rate cycling tests. However, limitations such as SEI formation and side reactions may hinder performance at higher current rates. The diffusivity results reinforce that interfacial quality is a critical factor in determining Li-ion transport in LFP electrodes. Properly sintered LATP at 900 °C (Sample 1) yields the most favorable conditions for fast and reversible diffusion, whereas excessive sintering (Sample 2) degrades ion transport. The liquid system (Sample 3) performs reasonably well, but lacks the structural robustness and long-term stability of an optimized solid-state interface.
The relationship between diffusivity and b-value across the three LFP-based samples reveals how ion transport kinetics and charge storage mechanisms are closely interconnected. In Sample 1, which uses LATP sintered at 900 °C, the b-values are close to 0.5 for both anodic and cathodic peaks, indicating a diffusion-controlled charge storage process. This is consistent with the high Li-ion diffusivity measured for this sample (~10−9 cm2∙s−1), suggesting efficient Li+ transport through the electrode and across the solid-state interface. The well-sintered LATP structure likely enables good contact and facilitates rapid bulk diffusion, which dominates the electrochemical behavior. In Sample 2, where LATP was sintered at a higher temperature of 1000 °C, the diffusivity is the lowest among the three samples (~10−10 cm2∙s−1), reflecting sluggish Li-ion transport. Although the b-values still hover near 0.5–0.57, indicating primarily diffusion-controlled behavior, the slightly higher cathodic b-value suggests a minor increase in surface-related contributions, potentially due to the poor interfacial contact that limits bulk diffusion. The degraded diffusivity is likely the result of reduced interface compliance and increased resistance, caused by excessive LATP grain growth or densification at elevated sintering temperatures. Sample 3, which uses a liquid electrolyte (1 M LiPF6), shows the highest b-values (~0.58) for both anodic and cathodic peaks, pointing to a stronger contribution from surface capacitive mechanisms. The liquid electrolyte facilitates faster ion movement at the electrode–electrolyte interface, enhancing pseudocapacitive and non-faradaic processes. Although its diffusivity is not as high as Sample 1, it remains sufficient (~10−9 cm2∙s−1) to support fast surface redox reactions. The relatively higher b-values in this case reflect the dominance of interfacial kinetics over bulk diffusion, which is typical in liquid systems.
The electrochemical performance of the three LFP-based systems can be comprehensively understood by analyzing the correlation between rate capability, b-values, and Li-ion diffusivity. Sample 1, which incorporates LATP sintered at 900 °C, exhibits the highest specific capacity across all tested C-rates, maintaining stable performance even at 5 C. This excellent rate capability is supported by b-values close to 0.5, indicating a diffusion-controlled charge storage mechanism. The high Li-ion diffusivity (~10−9 cm2∙s−1) measured in this sample further confirms efficient bulk ion transport and low interfacial resistance, enabling fast and reversible electrochemical reactions during high-rate cycling.
In contrast, Sample 2, based on LATP sintered at 1000 °C, shows significantly poorer rate performance, with lower capacities at all current densities and incomplete capacity recovery when the current returns to 1 C. Although the b-values (~0.51 anodic, ~0.57 cathodic) suggest the process is still largely diffusion-controlled, the much lower diffusivity (~10−10 cm2∙s−1) indicates sluggish ion transport. The compromised interface due to over-sintering likely results in poor contact between LFP and LATP, which hinders Li-ion movement and reduces the cell’s ability to perform under high current conditions.
Sample 3, which uses a liquid electrolyte (1 M LiPF
6), demonstrates intermediate rate performance—better than Sample 2 but slightly inferior to Sample 1 at higher C-rates. This behavior is consistent with its relatively higher
b-values (~0.58), suggesting a greater contribution from surface capacitive processes. The measured diffusivity (~10
−9 cm
2∙s
−1) is sufficient to support fast interfacial reactions, though it does not fully compensate for limitations in bulk ion transport. The liquid electrolyte provides good ionic conductivity and wetting, which enhances surface-driven processes but may still be constrained by interfacial stability or side reactions during extended cycling. Sample 1 achieves superior rate performance due to its high diffusivity and well-formed solid-state interface that supports diffusion-controlled kinetics. Sample 2 is limited by low diffusivity and poor interfacial contact, while Sample 3 benefits from surface capacitive effects and moderate diffusivity but lacks the structural stability of an optimized solid-state system. These findings collectively demonstrate that high-rate capability in LFP electrodes depends on a delicate balance between bulk diffusion, interfacial contact, and surface kinetics, as summarized in
Table 1.
The SEM images in
Figure 5a–d illustrate the morphological evolution of AC-EPD LFP electrodes under various cycling and interfacial conditions. In
Figure 5a, the as-deposited LFP electrode before cycling shows a loosely packed morphology with distinguishable primary particles. The structure appears porous, with noticeable interparticle spacing, indicating a typical surface formed by AC-EPD without compaction from cycling-induced stress or interface interactions.
After cycling in contact with a liquid electrolyte (1 M LiPF
6), as shown in
Figure 5b, the morphology significantly changes. The surface becomes smoother and more compact, suggesting surface reconstruction and particle merging likely caused by electrolyte-induced side reactions or binder/electrolyte degradation. The reduction in contrast and particle definition may also point to the formation of a surface film or decomposition products, such as SEI-like layers, which can passivate the electrode surface.
In contrast, LFP electrodes cycled in contact with solid-state LATP electrolytes sintered at different temperatures show distinct morphological behavior. In
Figure 5c, the interface with LATP sintered at 900 °C preserves a relatively granular texture, with LFP particles still visible and partially interconnected. This suggests moderate interfacial contact and mechanical pressure during cycling, likely promoting Li transport while avoiding extensive surface degradation.
However, in
Figure 5d, where LATP was sintered at 1000 °C, the LFP morphology becomes more densely packed and featureless. The clear particle boundaries visible in
Figure 5a,c are significantly diminished, implying enhanced densification or possible interfacial reactions at elevated sintering-induced interface conditions. The higher sintering temperature likely increased LATP grain boundary conductivity and mechanical stiffness, leading to stronger physical contact and potentially more intense interfacial stress or reactions during cycling. The dense and featureless morphology observed in
Figure 5d for LFP cycled with LATP sintered at 1000 °C may be attributed to mechanical stress accumulation at the rigid LATP interface, leading to particle compaction. Additionally, potential interfacial reactions—such as elemental diffusion from LATP (e.g., Ti or P)—could chemically modify the LFP surface, suppressing the original granular texture. These mechanisms, though not directly confirmed here, are consistent with the reported degradation pathways in high-temperature-processed solid-state interfaces and warrant further investigation using post-mortem structural and compositional analyses.
Although cross-sectional SEM/EDS analysis would provide the direct evidence of the LFP/LATP interface structure, attempts to prepare intact cross-sections were hindered by interfacial delamination and cracking during mechanical polishing, due to the brittle nature of sintered LATP. The surface SEM images indicate significant grain coarsening in LATP sintered at 1000 °C, consistent with excessive densification. This structural evolution is likely to reduce interfacial compliance and limit Li-ion transport, as also supported by the reduced diffusivity values. These findings align with those of the previous reports which state that excessive sintering can suppress beneficial grain boundary conduction and lead to brittle interfaces [
29,
30]. Further investigation using cross-sectional SEM or focused ion beam analysis is planned to validate this hypothesis.
In summary, the SEM images reveal that a moderately porous, well-adhered interface (Sample 1) supports fast Li-ion diffusion and diffusion-limited kinetics, leading to high-rate performance. Excessively dense morphology (Sample 2) impairs interfacial transport, lowering diffusivity and electrochemical efficiency. Meanwhile, surface smoothing and interphase formation (Sample 3) encourage surface-driven kinetics, reflected in higher b-values and acceptable diffusivity, albeit with less structural integrity than solid-state counterparts. Thus, morphology is a direct indicator of electrochemical performance, especially in systems where solid–solid interfaces govern charge transport.
Overall, these images highlight the critical role of the electrode/electrolyte interface—whether liquid or solid—and processing temperature on the structural integrity and surface stability of LFP electrodes. Solid-state interfaces, especially with optimized LATP processing, may offer improved morphological stability compared to liquid electrolytes, though high-temperature sintering can also induce unintended surface modifications. In this study, LATP pellets were sintered at 900 °C and 1000 °C to intentionally produce two distinct interfacial microstructures for comparative analysis. The lower temperature (900 °C) typically leads to less-densified LATP with higher interfacial resistance, while the higher temperature (1000 °C) may result in coarsened grains and potential interface degradation. These boundary conditions were selected to represent limiting cases rather than an exhaustive sintering profile, aiming to elucidate the impact of interface quality on electrochemical performance. Further work including intermediate sintering temperatures (e.g., 950 °C) is ongoing to extend the observed trends.
The electrochemical performance of our AC-EPD-fabricated LFP electrode is comparable to previously reported Li/LATP/LFP systems with similar mass loading [
21,
22,
23,
24]. This validates that our system offers a suitable performance baseline for probing interfacial kinetics and charge storage mechanisms, which is the central focus of this study. Further, to evaluate the scalability of the AC-EPD method, we also tested LFP electrodes with a mass loading of ~2 mg∙cm
−2 using a liquid electrolyte, as shown in
Figure S2 (Supporting Information). These higher-mass-loading electrodes demonstrated stable cycling and good rate performance, confirming that the interface-sensitive design remains effective under more practical loading conditions. Unlike prior works that primarily emphasized performance optimization, our platform enables mechanistic analysis by combining a reproducible AC-EPD fabrication process with interface-sensitive evaluation methods.
It is also important to emphasize that, in the liquid electrolyte configuration, the EC/DEC solvent system can decompose on the LFP surface to form a cathode–electrolyte interphase (CEI), typically composed of LiF, Li2CO3, and organic moieties. While this CEI can protect the electrode, it may also increase interfacial resistance over time. In contrast, the solid-state LATP interface does not undergo similar solvent-driven decomposition; however, it is subject to mechanical and chemical interfacial instability, especially under high-temperature sintering or prolonged cycling. For example, interfacial reactions such as Ti reduction or phosphate interdiffusion can occur, which degrade ionic conductivity and cycle stability. Therefore, although solid-state systems can suppress solvent-driven SEI formation, maintaining interfacial integrity requires the careful control of interface chemistry and processing conditions.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}