
Ru(II)-Dppz Derivatives and Their Interactions with DNA: Thirty Years and Counting




Abstract
1. Introduction
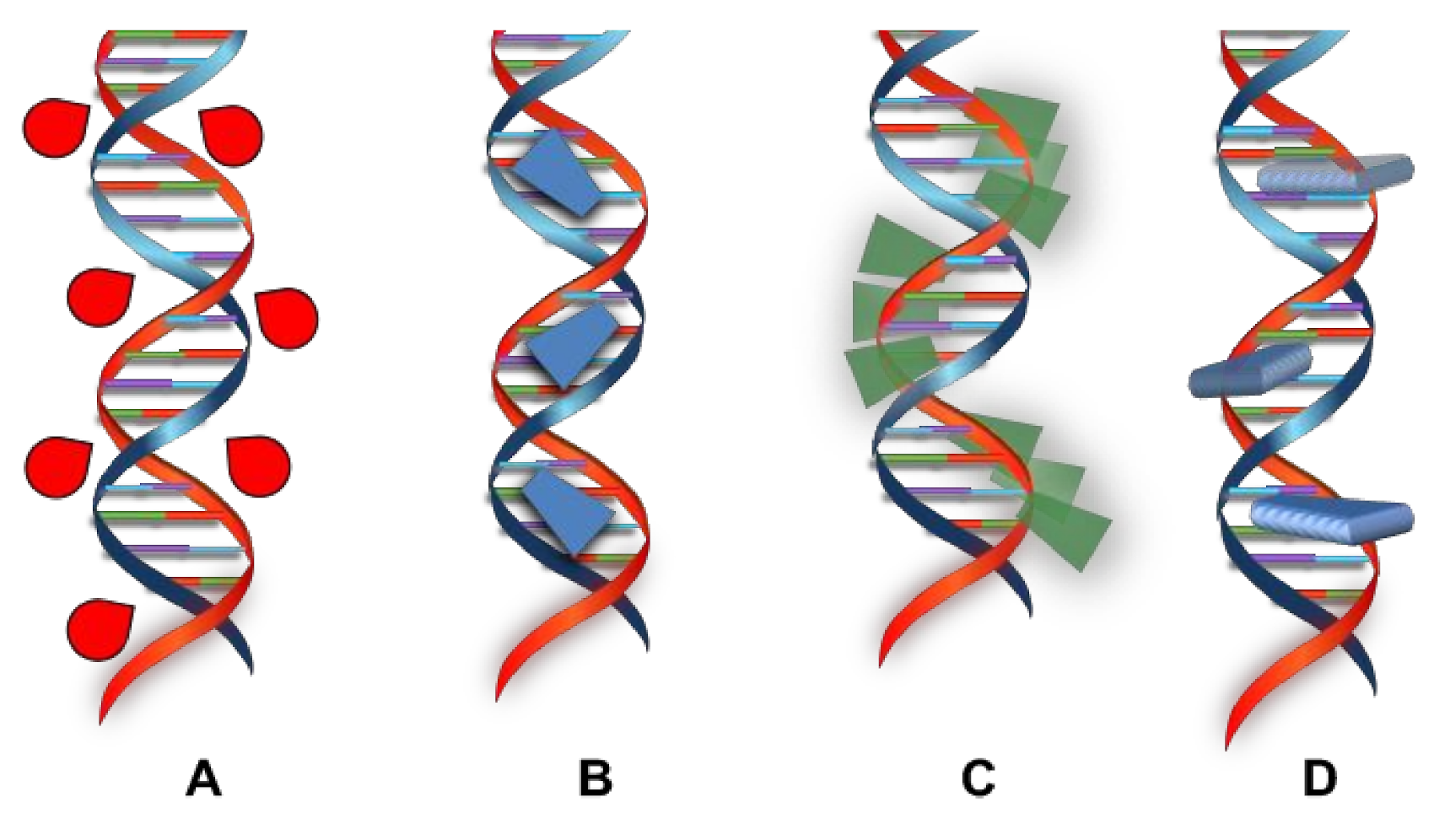
2. DNA Structure
3. DNA Binding
3.1. Interaction of Small Molecules with DNA
- The presence of d orbitals grants more ways (bonds and geometries) such that the species might interact with DNA other than those of organic molecules.
- Steric and electronic properties can be modulated choosing opportune ancillary ligands.
- Bridging ligands of certain dimensions might modify the size of the intercalator.
- It is possible to take advantages from their spectroscopic, magnetic, redox, photophysical, and photochemical properties.
3.2. Ru(II) Complexes as Intercalators
- non-radiative decay, because the intercalated luminophore is surrounded by the DNA hydrophobic environment and is therefore protected from water-induced deactivation;
- intersystem crossing to the 3MC excited state, as the complex is caged into the double helix and its rigidity result is enhanced;
- O2 luminescence quenching.
3.3. Solvent Effects on the Photophysical Properties of the Ru-Dppz Complexes
3.4. Effects of Tuning the Photophyisical and Redox Properties
3.5. Few Examples of Applications: Cellular Imaging, Photodynamic Therapy (PDT), and Sensing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodley, G.A.; Scobie, R.S.; Bates, R.H.; Lewitt, R.M. A possible conformation for double-stranded polynucleotides. Proc. Natl. Acad. Sci. USA 1976, 73, 2959–2963. [Google Scholar] [CrossRef]
- Zeglis, B.M.; Pierre, V.C.; Barton, J.K. Metallo-intercalators and metallo-insertors. Chem. Commun. 2007, 44, 4565–4579. [Google Scholar] [CrossRef]
- Liu, H.K.; Sadler, P.J. Metal Complexes as DNA Intercalators. Acc. Chem. Res. 2011, 44, 349–359. [Google Scholar] [CrossRef]
- Pages, B.J.; Ang, D.L.; Wright, E.P.; Aldrich-Wright, J.R. Metal complex interactions with DNA. Dalton Trans. 2015, 44, 3505–3526. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum Compounds: A New Class of Potent Antitumour Agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct Cellular Responses to Platinum-Induced DNA Damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.K.; Goldberg, J.M.C.; Kumar, V.; Turro, N.J. Binding modes and base specificity of tris(phenanthroline)ruthenium(II) enantiomers with nucleic acids: Tuning the stereoselectivity. J. Am. Chem. Soc. 1986, 108, 2081–2088. [Google Scholar] [CrossRef]
- Kelly, J.M.; Tossi, A.B.; McConnell, D.J.; OhUigin, C. A study of the interactions of some polypyridylruthenium(II) complexes with DNA using fluorescence spectroscopy, topoisomerisation and thermal denaturation. Nucleic Acids Res. 1985, 13, 6017–6034. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, S.; Dabrowiak, J.C.; Chaires, J.B. Tris(phenanthroline)ruthenium(II) enantiomer interactions with DNA: Mode and specificity of binding. Biochemistry 1993, 32, 2573–2584. [Google Scholar] [CrossRef]
- Hiort, C.; Nordén, B.; Rodger, A. Enantiopreferential DNA binding of [ruthenium(II)(1,10-phenanthroline)3]2+ studied with linear and circular dichroism. J. Am. Chem. Soc. 1990, 112, 1971–1982. [Google Scholar] [CrossRef]
- Lincoln, P.; Nordén, B. DNA Binding Geometries of Ruthenium(II) Complexes with 1,10-Phenanthroline and 2,2‘-Bipyridine Ligands Studied with Linear Dichroism Spectroscopy. Borderline Cases of Intercalation. J. Phys. Chem. B 1998, 102, 9583–9594. [Google Scholar] [CrossRef]
- Barton, J.K. Tris(phenanthroline)metal complexes: Probes for DNA helicity. J. Biomol. Struct. Dyn. 1983, 1, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.K.; Dannenberg, J.J.; Raphael, A.L. Enantiomeric selectivity in binding tris(phenanthroline)zinc(II) to DNA. J. Am. Chem. Soc. 1982, 104, 4967–4969. [Google Scholar] [CrossRef]
- Nordén, B.; Tjerneld, F. Binding of inert metal complexes to deoxyribonucleic acid detected by linear dichroism. FEBS Lett. 1976, 67, 368–370. [Google Scholar] [CrossRef]
- Li, G.; Sun, L.; Ji, L.; Chao, H. Ruthenium(II) complexes with dppz: From molecular photoswitch to biological applications. Dalton Trans. 2016, 45, 13261–13276. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Br. J. Nat. 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walters, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Leslie, A.G.; Arnott, S.; Chandrasekaran, R.; Ratliff, R.L. Polymorphism of DNA double helices. J. Mol. Biol. 1980, 143, 49–72. [Google Scholar] [CrossRef]
- Carrier, A.; Le Ber, P.; Auclain, C. Spin-labeled oxazolopyridocarbazole as a probe for studying nonintercalating DNA groove binding ligands. Biochemistry 1990, 29, 6002–6009. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Minagawa, K.; Yoshikawa, K.; Matsumoto, M.; Doi, M. Conformational dynamics of DNA affected by intercalation and minor groove binding: Direct observation of large DNA. Nucleic Acids Symp. Ser. 1991, 25, 131–132. [Google Scholar]
- Hurley, L.H. DNA and associated targets for drug design. J. Med. Chem. 1989, 32, 2027–2033. [Google Scholar] [CrossRef]
- Iida, H.; Jia, G.; Lown, J.W. Rational recognition of nucleic acid sequences. Curr. Opin. Biotechnol. 1999, 10, 29–33. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Bustamante, C.; Collings, P.J.; Giannetto, A.; Gibbs, E.J. Porphyrin assemblies on DNA as studied by a resonance light-scattering technique. J. Am. Chem. Soc. 1993, 115, 5393–5399. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Schaefer, K.F. Resonance Light-Scattering Studies of Porphyrin Diacid Aggregates. Inorg. Chem. 1994, 33, 2062–2065. [Google Scholar] [CrossRef]
- Hudson, B.P.; Sou, J.; Berger, D.J.; McMillin, D.R. Luminescence studies of the intercalation of Cu(TMpyP4) into DNA. J. Am. Chem. Soc. 1992, 114, 8997–9002. [Google Scholar] [CrossRef]
- Santoro, A.; Holub, J.; Fik-Jaskółka, M.A.; Vantomme, G.; Lehn, J.-M. Dynamic Helicates Self-Assembly from Homo- and Heterotopic Dynamic Covalent Ligand Strands. Chem. Eur. J. 2020, 26, 15664–15671. [Google Scholar] [CrossRef] [PubMed]
- Kano, K.; Fukuda, K.; Wakami, H.; Nishiyabu, R.; Pasternack, R.F. Factors Influencing Self-Aggregation Tendencies of Cationic Porphyrins in Aqueous Solution. J. Am. Chem. Soc. 2000, 122, 7494–7502. [Google Scholar] [CrossRef]
- Arena, G.; Monsù Scolaro, L.; Pasternack, R.F.; Romeo, R. Synthesis, Characterization, and Interaction with DNA of the Novel Metallointercalator Cationic Complex (2,2′:6′,2″-terpyridine)methylplatinum(II). Inorg. Chem. 1995, 34, 2994–3002. [Google Scholar] [CrossRef]
- Casamento, M.; Arena, G.E.; Lo Passo, C.; Pernice, I.; Romeo, A.; Monsù Scolaro, L. Interaction of organometallic cationic complex ions containing terpyridine ligands with nucleic acids: An investigation on aggregative phenomena. Inorg. Chim. Acta 1998, 275, 242–249. [Google Scholar] [CrossRef]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A. DNA interaction of platinum(II) complexes with 1,10-phenanthroline and extended phenanthrolines. Inorg. Chem. 2006, 45, 230–235. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Gibbs, E.J.; Villafranca, J.J. Interactions of porphyrins with nucleic acids. Biochemistry 1983, 22, 2406–2414. [Google Scholar] [CrossRef]
- Gibbs, E.J.; Mauer, M.C.; Zhang, H.F.; Reiff, W.M.; Hill, D.T.; Blaszkiewicz, M.M.; Mckinnie, R.E.; Liu, H.-Q.; Pasternack, R.F. Interactions of porphyrins with purified DNA and more highly organized structures. J. Inorg. Biochem. 1988, 32, 39–65. [Google Scholar] [CrossRef]
- Lerman, L.S. Structural considerations in the interaction of DNA and acridines. J. Mol. Biol. 1961, 3, 18–30. [Google Scholar] [CrossRef]
- Haq, I. Thermodynamics of drug-DNA interactions. Arch. Biochem. Biophys. 2002, 403, 1–15. [Google Scholar] [CrossRef]
- Bauer, W.; Vinograd, J. The interaction of closed circular DNA with intercalative dyes. I. The superhelix density of SV40 DNA in the presence and absence of dye. J. Mol. Biol. 1968, 33, 141–171. [Google Scholar] [CrossRef]
- Bauer, W.; Vinograd, J. Interaction of closed circular DNA with intercalative dyes. II. The free energy of superhelix formation in SV40 DNA. J. Mol. Biol. 1970, 47, 419–435. [Google Scholar] [CrossRef]
- Waring, M.J. Variation of the supercoils in closed circular DNA by binding of antibiotics and drugs: Evidence for molecular models involving intercalation. J. Mol. Biol. 1970, 54, 247–279. [Google Scholar] [CrossRef]
- Cairns, J. The application of autoradiography to the study of DNA viruses. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J. DNA Modification and Cancer. Ann. Rev. Biochem. 1981, 50, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Drug-nucleic acid interactions. In Methods in Enzymology; Chairs, J.B., Waring, M.J., Eds.; Academic Press: San Diego, CA, USA, 2001; Volume 340. [Google Scholar]
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry; Freeman, W.H., Ed.; John Wiley & Sons: San Francisco, CA, USA, 1980. [Google Scholar]
- Sauer, K. Methods of Enzymology: Biochemical Spectroscopy; Academic Press: San Diego, CA, USA, 1995; Volume 246. [Google Scholar]
- Utsuno, K.; Tsuboi, M. Degree of DNA unwinding caused by the binding of aclacinomycin A. Chem. Pharm. Bull. 1997, 45, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Crothers, D.M. Calculation of binding isotherms for heterogeneous polymers. Biopolymers 1975, 6, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Kreft, D.; Wang, Y.; Rattay, M.; Toensing, K.; Anselmetti, D. Binding mechanism of anti-cancer chemotherapeutic drug mitoxantrone to DNA characterized by magnetic tweezers. J. Nanobiotechnol. 2018, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, A.; Barattucci, A.; Bonaccorsi, P.; Giannetto, A.; La Ganga, G.; Musarra-Pizzo, M.; Salerno, T.M.G.; Santoro, A.; Sciortino, M.T.; Puntoriero, F.; et al. Carbohydrates and Charges on Oligo(phenylenethynylenes): Towards the Design of Cancer Bullets. Chem. Eur. J. 2018, 24, 16972–16976. [Google Scholar] [CrossRef]
- Barton, D.; Nakanishi, K.; Meth-Cohn, O. DNA Intercalators. In Comprehensive Natural Products Chemistry; Wilson, W.D., Ed.; Pergamon: Oxford, UK, 1999; Volume 427. [Google Scholar]
- Balzani, V.; Credi, A.; Venturi, M. Photochemistry and photophysics of coordination compounds: An extended view. Coord. Chem. Rev. 1998, 171, 3–16. [Google Scholar] [CrossRef]
- Turro, N.J. From molecular chemistry to supramolecular chemistry to superdupermolecular chemistry. Controlling covalent bond formation through non-covalent and magnetic interactions. Chem. Commun. 2002, 2279–2292. [Google Scholar] [CrossRef]
- Sun, S.S.; Lees, A.J. Transition metal based supramolecular systems: Synthesis, photophysics, photochemistry and their potential applications as luminescent anion chemosensors. Coord. Chem. Rev. 2002, 230, 171–192. [Google Scholar] [CrossRef]
- Balzani, V.; Bergamini, G.; Campagna, S.; Puntoriero, F. Photochemistry and Photophysics of Coordination Compounds I; Balzani, V., Campagna, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 1–36. [Google Scholar]
- Puntoriero, F.; Arrigo, A.; Santoro, A.; La Ganga, G.; Tuyèras, F.; Campagna, S.; Dupeyre, G.; Lainé, P.P. Photoinduced Intercomponent Processes in Selectively Addressable Bichromophoric Dyads Made of Linearly Arranged Ru(II) Terpyridine and Expanded Pyridinium Components. Inorg. Chem. 2019, 58, 5807–5817. [Google Scholar] [CrossRef]
- Santoni, M.-P.; Santoro, A.; Salerno, T.M.G.; Puntoriero, F.; Nastasi, F.; Di Pietro, M.L.; Galletta, M.; Campagna, S. Photoinduced Charge Separation in a Donor–Spacer–Acceptor Dyad with N-Annulated Perylene Donor and Methylviologen Acceptor. ChemPhysChem 2015, 15, 3147–3150. [Google Scholar] [CrossRef] [PubMed]
- Jenette, K.W.; Lippard, S.J.; Vassiliades, G.A.; Bauer, W.R. Metallointercalation Reagents. 2-Hydroxyethanethiolato(2,2′,2″-terpyridine)-platinum(II) Monocation Binds Strongly to DNA By Intercalation. Proc. Natl. Acad. Sci. USA 1974, 71, 3839–3843. [Google Scholar] [CrossRef]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A.; Nicolò, F.; Rotondo, E. Noncovalent Interactions of Platinum(II) Square Planar Complexes Containg Ligands Out-of-Plane with DNA. Inorg. Chem. 1998, 37, 563–568. [Google Scholar] [CrossRef]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A. Stacking Surface Effect in the DNA Intercalation of Some Polypiridine Platinum(II) Complexes. Inorg. Chem. 1999, 38, 1754–1758. [Google Scholar] [CrossRef]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A.; Vainiglia, P.A. The intercalation to DNA of bipyridyl complexes of platinum(II) with thioureas. J. Inorg. Biochem. 2005, 99, 560–565. [Google Scholar] [CrossRef]
- Marverti, G.; Cusumano, M.; Ligabue, A.; Di Pietro, M.L.; Vainiglia, P.A.; Ferrari, A.; Bergomi, M.; Moruzzi, M.S.; Frassineti, C. Studies on the anti-proliferative effects of novel DNA-intercalating bipyridyl–thiourea–Pt(II) complexes against cisplatin-sensitive and -resistant human ovarian cancer cells. J. Inorg. Biochem. 2008, 102, 699–712. [Google Scholar] [CrossRef]
- Marverti, G.; Ligabue, A.; Montanari, M.; Guerrieri, D.; Cusumano, M.; Di Pietro, M.L.; Troiano, L.; Di Vono, E.; Iotti, S.; Farruggia, G.; et al. Characterization of the cell growth inhibitory effects of a novel DNA-intercalating bipyridyl-thiourea-Pt(II) complex in cisplatin-sensitive and—Resistant human ovarian cancer cells. Investig. New Drugs 2011, 29, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Campagna, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Photochemistry and Photophysics of Coordination Compounds: Ruthenium. Top. Curr. Chem. 2007, 280, 117–214. [Google Scholar]
- Santoni, M.-P.; Hanan, G.S.; Hasenknopf, B.; Proust, A.; Nastasi, F.; Serroni, S.; Campagna, S. Dinuclear Ru(II) complexes of bis-(dipyrid-2-yl)triazine (bis-dpt) ligands as efficient electron reservoirs. Chem. Commun. 2011, 47, 3586–3588. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.V.; Barton, J.K.; Turro, N.J. Photophysics of ruthenium complexes bound to double helical DNA. J. Am. Chem. Soc. 1985, 107, 5518–5523. [Google Scholar] [CrossRef]
- Barton, J.K.; Danishefsky, A.T.; Goldberg, J.M. Tris(phenanthroline)ruthenium(II): Stereoselectivity in binding to DNA. J. Am. Chem. Soc. 1984, 106, 2172–2176. [Google Scholar] [CrossRef]
- Rehmann, J.P.; Barton, J.K. 1H NMR studies of tris(phenanthroline) metal complexes bound to oligonucleotides: Characterization of binding modes. Biochemistry 1990, 29, 1701–1709. [Google Scholar] [CrossRef]
- Lippard, S.J.; Bond, P.J.; Wu, K.C.; Bauer, W.R. Stereochemical requirements for intercalation of platinum complexes into double-stranded DNA’s. Science 1976, 194, 726–728. [Google Scholar] [CrossRef]
- Wang, A.H.J.; Nathans, J.; van der Marel, G.; van Boom, J.H.; Rich, A. Molecular structure of a double helical DNA fragment intercalator complex between deoxy CpG and a terpyridine platinum compound. Nature 1978, 276, 471–474. [Google Scholar] [CrossRef]
- Mei, H.-Y.; Barton, J.K. Chiral probe for A-form helixes of DNA and RNA: Tris(tetramethylphenanthroline)ruthenium(II). J. Am. Chem. Soc. 1986, 108, 7414–7416. [Google Scholar] [CrossRef]
- Jenkins, Y.; Friedman, A.E.; Turro, N.J.; Barton, J.K. Characterization of dipyridophenazine complexes of ruthenium(II): The light switch effect as a function of nucleic acid sequence and conformation. Biochemistry 1992, 31, 10809–10816. [Google Scholar] [CrossRef]
- Friedman, A.E.; Chambron, J.C.; Sauvage, J.-P.; Turro, N.J.; Barton, J.K. A molecular light switch for DNA: Ru(bpy)2(dppz)2+. J. Am. Chem. Soc. 1990, 112, 4960–4962. [Google Scholar] [CrossRef]
- Hiort, C.H.; Lincoln, P.; Nordén, B. DNA binding of DELTA- and LAMBDA-[Ru(phen)2DPPZ]2+. J. Am. Chem. Soc. 1993, 115, 3448–3454. [Google Scholar] [CrossRef]
- Moucheron, C.; Kirsch-De Mesmaeker, A. New DNA-binding ruthenium(II) complexes as photo-reagents for mononucleotides and DNA. J. Phys. Org. Chem. 1998, 11, 577–583. [Google Scholar] [CrossRef]
- Hartshorn, R.M.; Barton, J.K. Novel dipyridophenazine complexes of ruthenium(II): Exploring luminescent reporters of DNA. J. Am. Chem. Soc. 1992, 114, 5919–5925. [Google Scholar] [CrossRef]
- Önfelt, B.; Lincoln, P.; Nordén, B. A Molecular Staple for DNA: Threading Bis-intercalating [Ru(phen)2dppz]2+ Dimer. J. Am. Chem. Soc. 1999, 121, 10846–10847. [Google Scholar] [CrossRef]
- Önfelt, B.; Lincoln, P.; Nordén, B. Enantioselective DNA Threading Dynamics by Phenazine-Linked [Ru(phen)2dppz]2+ Dimers. J. Am. Chem. Soc. 2001, 123, 3630–3637. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, C.; Adams, H.; Haq, I.; Thomas, J.A. A ruthenium dipyridophenazine complex that binds preferentially to GC sequences. Chem. Commun. 2003, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Ihara, T.; Okada, K.; Tsujimura, Y.; Shirasaka, Y.; Tazaki, M.; Jyo, A. Asymmetric cooperativity in tandem hybridization of enantiomeric metal complex-tethered short fluorescent DNA probes. Chem. Commun. 2005, 4523–4525. [Google Scholar] [CrossRef]
- Monczak, K.; Peuntinger, K.; Sorsche, D.; Heinemann, F.W.; Guldi, D.M.; Rau, S. Synthesis and Characterization of a Trisheteroleptic RuII-Based Molecular Switch. Chem. Eur. J. 2014, 20, 15426–15433. [Google Scholar] [CrossRef]
- Metcalfe, C.; Webb, M.; Thomas, J.A. A facile synthetic route to bimetallic ReI complexes containing two dppz DNA intercalating ligands. Chem. Commun. 2002, 2026–2027. [Google Scholar] [CrossRef] [PubMed]
- Olmon, E.D.; Sontz, P.A.; Blanco-Rodríguez, A.M.; Towrie, M.; Clark, I.P.; Vlček, A., Jr.; Barton, J.K. Charge Photoinjection in Intercalated and Covalently Bound [Re(CO)3(dppz)(py)]+–DNA Constructs Monitored by Time-Resolved Visible and Infrared Spectroscopy. J. Am. Chem. Soc. 2011, 133, 13718–13730. [Google Scholar] [CrossRef]
- Thorp-Greenwood, F.L.; Coogan, M.P.; Mishra, L.; Kumari, N.; Rai, G.; Saripella, S. The importance of cellular localisation of probes: Synthesis, photophysical properties, DNA interactions and cellular imaging properties of rhenium dppz complexes with known cellular localisation vectors. New J. Chem. 2012, 36, 64–72. [Google Scholar] [CrossRef]
- Fumanal, M.; Vela, S.; Gattuso, H.; Monari, A.; Daniel, C. Absorption Spectroscopy and Photophysics of a ReI-dppz Probe for DNA-Mediated Charge Transport. Chem. Eur. J. 2018, 24, 14425–14435. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.L.; Reddy, Y.H.K.; Kumar, K.A.; Vidhisha, S.; Satyanarayana, S. Synthesis, characterization, DNA-binding, and DNA-photocleavage properties of [Co(bpy)2(7-NO2-dppz)]3+, [Co(dmb)2(7-NO2-dppz)]3+, and [Co(phen)2(7-NO2-dppz)]3+ complexes: (7-nitro-dppz = 7-nitrodipyrido[3,2-a:2′-3′-c]phenazine; bpy = 2,2′-bipyridine; dmb = 4,4′-dimethyl-2,2′-bipyridine; phen = 1,10-phenanthroline) and their toxicity on different microorganisms. Nucleosides Nucleotides Nucleic Acids 2009, 28, 204–219. [Google Scholar] [PubMed]
- Shahabadi, N.; Kashanian, S.; Mahdavi, M. DNA interaction studies of cobalt (II) mixed-ligand complexes containing dimethyl-1,10-phenanthroline and dipyrido[3,2-a:2′,3′-c]phenazine: The role of methyl substitutions on the mode of binding. DNA Cell Biol. 2011, 30, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Dhar, S.; Nethaji, M.; Chakravarty, A.R. Bis(dipyridophenazine)copper(II) complex as major groove directing synthetic hydrolase. Dalton Trans 2004, 12, 1896–1900. [Google Scholar] [CrossRef]
- Madureira, J.; Ramos, C.I.V.; Marques, M.; Maia, C.; de Sousa, B.; Campino, L.; Santana-Marques, M.G.; Farrell, N. Nonclassic metallointercalators with dipyridophenazine: DNA interaction studies and leishmanicidal activity. Inorg. Chem. 2013, 52, 8881–8894. [Google Scholar] [CrossRef] [PubMed]
- Angeles-Boza, A.M.; Bradley, P.M.; Fu, P.K.-L.; Wicke, S.E.; Bacsa, J.; Dunbar, K.R.; Turro, C. DNA binding and photocleavage in vitro by new dirhodium(II) dppz complexes: Correlation to cytotoxicity and photocytotoxicity. Inorg. Chem. 2004, 43, 8510–8519. [Google Scholar] [CrossRef]
- Menon, E.L.; Perera, R.; Navarro, M.; Kuhn, R.J.; Morrison, H. Phototoxicity against tumor cells and sindbis virus by an octahedral rhodium bisbipyridyl complex and evidence for the genome as a target in viral photoinactivation. Inorg. Chem. 2004, 43, 5373–5381. [Google Scholar] [CrossRef]
- Petitjean, A.; Puntoriero, F.; Campagna, S.; Juris, A.; Lehn, J.-M. Multicomponent supramolecular devices: Synthesis, optical, and electronic properties of bridged bis-dirhodium and -diruthenium complexes. Eur. J. Inorg. Chem. 2006, 19, 3878–3892. [Google Scholar] [CrossRef]
- Liang, H.; Hao, T.; Yin, C.; Yang, X.; Fu, H.; Zheng, X.; Li, R.; Xiao, D.; Chen, H. Cyclometalated Rhodium(III) Complexes Based on Substituted 2-Phenylpyridine Ligands: Synthesis, Structures, Photophysics, Electrochemistry, and DNA-Binding Properties. Eur. J. Inorg. Chem. 2017, 36, 4149–4157. [Google Scholar] [CrossRef]
- Trovato, E.; Di Pietro, M.L.; Puntoriero, F. Shining a new light on an old game—An OsII-based near-IR light switch. Eur. J. Inorg. Chem. 2012, 3984–3988. [Google Scholar] [CrossRef]
- Wragg, A.; Gill, M.R.; Hill, C.J.; Su, X.; Meijer, A.J.H.M.; Smythe, C.; Thomas, J.A. Dinuclear osmium (II) probes for high-resolution visualisation of cellular DNA structure using electron microscopy. Chem. Commun. 2014, 50, 14494–14497. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Che, C.M.; Yan, S.C. Platinum (II) complexes with dipyridophenazine ligands as human telomerase inhibitors and luminescent probes for G-quadruplex DNA. J. Am. Chem. Soc. 2009, 131, 1835–1846. [Google Scholar] [CrossRef]
- Puntoriero, F.; Campagna, S.; Di Pietro, M.L.; Giannetto, A.; Cusumano, M. Luminescence of a Pt(II) complex in the presence of DNA. Dependence of luminescence changes on the interaction binding mode. Photochem. Photobiol. Sci. 2007, 6, 357–360. [Google Scholar] [CrossRef]
- Cusumano, M.; Di Pietro, M.L.; Giannetto, A.; Nicolò, F.; Nordén, B.; Lincoln, P. Ambivalent Intercalators for DNA: L-Shaped Platinum(II) Complexes. Inorg. Chem. 2004, 43, 2416–2421. [Google Scholar] [CrossRef]
- Jacques, A.; Kirsch-De Mesmaeker, A.; Elias, B. Selective DNA Purine Base Photooxidation by Bis-terdentate Iridium(III) Polypyridyl and Cyclometalated Complexes. Inorg. Chem. 2014, 53, 1507–1512. [Google Scholar] [CrossRef]
- Sreedharan, S.; Sinopoli, A.; Jarman, P.J.; Robinson, D.; Clemmet, C.; Scattergood, P.A.; Rice, C.R.; Smythe, C.G.W.; Thomas, J.A.; Elliott, P.I.P. Mitochondria-localising DNA-binding biscyclometalated phenyltriazole iridium(III) dipyridophenazene complexes: Syntheses and cellular imaging properties. Dalton Trans. 2018, 47, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Dreyse, P.; Santander-Nelli, M.; Zambrano, D.; Rosales, L.; Sanhueza, L. Electron-donor substituents on the dppz-based ligands to control luminescence from dark to bright emissive state in Ir(III) complexes. Int. J. Quantum. Chem. 2020, 120, e26167. [Google Scholar] [CrossRef]
- Olson, E.J.C.; Hu, D.; Hörmann, A.; Jonkman, A.M.; Arkin, M.R.; Stemp, E.D.A.; Barton, J.K.; Barbara, P.F. First Observation of the Key Intermediate in the “Light-Switch” Mechanism of [Ru(phen)2dppz]2+. J. Am. Chem. Soc. 1997, 119, 11458–11467. [Google Scholar] [CrossRef]
- Coates, C.G.; Olofsson, J.; Coletti, M.; McGarvey, J.J.; Önfelt, B.; Lincoln, P.; Nordén, B.; Tuite, E.; Matousek, P.; Parker, A.W. Picosecond Time-Resolved Resonance Raman Probing of the Light-Switch States of [Ru(Phen)2dppz]2+. J. Phys. Chem. B 2001, 105, 12653–12664. [Google Scholar] [CrossRef]
- Olofsson, J.; Onfelt, B.; Lincoln, P. Three-State Light Switch of [Ru(phen)2dppz]2+: Distinct Excited-State Species with Two, One, or No Hydrogen Bonds from Solvent. J. Phys. Chem. A 2004, 108, 4391–4398. [Google Scholar] [CrossRef]
- Onfelt, B.; Olofsson, J.; Lincoln, P.; Norden, B. Picosecond and Steady-State Emission of [Ru(phen)2dppz]2+ in Glycerol: Anomalous Temperature Dependence. J. Phys. Chem. A 2003, 107, 1000–1009. [Google Scholar] [CrossRef]
- Olofsson, J.; Wilhelmsson, L.M.; Lincoln, P. Effects of Methyl Substitution on Radiative and Solvent Quenching Rate Constants of [Ru(phen)2dppz]2+ in Polyol Solvents and Bound to DNA. J. Am. Chem. Soc. 2004, 126, 15458–15465. [Google Scholar] [CrossRef]
- Pourtois, G.; Beljonne, D.; Moucheron, C.; Schumm, S.; Kirsch-De Mesmaeker, A.; Lazzaroni, R.; Bredas, J.L. Photophysical properties of ruthenium(II) polyazaaromatic compounds: A theoretical insight. J. Am. Chem. Soc. 2004, 126, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Brennaman, M.K.; Meyer, T.J.; Papanikolas, J.M. [Ru(bpy)2dppz]2+ Light-Switch Mechanism in Protic Solvents as Studied through Temperature-Dependent Lifetime Measurements. J. Phys. Chem. A 2004, 108, 9938–9944. [Google Scholar] [CrossRef]
- Nair, R.B.; Cullum, B.M.; Murphy, C.J. Optical Properties of [Ru(phen)2dppz]2+ as a Function of Nonaqueous Environment. Inorg. Chem. 1997, 36, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; VCH Publishers: Weinheim, Germany, 1988. [Google Scholar]
- Stimpson, S.; Jenkinson, D.R.; Sadler, A.; Latham, M.; Wragg, A.; Meijer, A.J.H.M.; Thomas, J.A. Tuning the Excited State of Water-Soluble IrIII-Based DNA Intercalators that are Isostructural with [RuII(NN)2(dppz)] Light-Switch Complexes. Angew. Chem. Int. Ed. 2015, 54, 3000–3003. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.B.; Yeung, L.K.; Murphy, C.J. Synthesis and Solvent-Dependent Properties of Ru(acac)2dppz. Inorg. Chem. 1999, 38, 2536–2538. [Google Scholar] [CrossRef]
- Keller, C.E.; Pollard, C.; Yeung, L.K.; Plessinger, W.D.; Murphy, C.J. Optical sensing properties of [Ru (CN)4dppz]2− (dppz = dipyrido [3,2-a:2,3-c] phenazine). Inorg. Chim. Acta 2000, 298, 209–215. [Google Scholar] [CrossRef]
- Liu, J.G.; Zhang, Q.L.; Shi, X.F.; Ji, L.N. Interaction of [Ru(dmp)2(dppz)]2+ and [Ru(dmb)2(dppz)]2+ with DNA: Effects of the Ancillary Ligands on the DNA-Binding Behaviors. Inorg. Chem. 2001, 40, 5045–5050. [Google Scholar] [CrossRef] [PubMed]
- Greguric, A.; Greguric, I.D.; Hambley, T.W.; Aldrich-Wright, J.R.; Collins, J.G. Minor groove intercalation of Δ-[Ru(Me2phen)2dppz]2+ to the hexanucleotide d(GTCGAC)2. J. Chem. Soc. Dalton Trans. 2002, 849–855. [Google Scholar] [CrossRef]
- Moucheron, C; Kirsch-De Mesmaeker, A; Choua, S; Photophysics of Ru(phen)2(PHEHAT)2+: A Novel “Light Switch” for DNA and Photo-oxidant for Mononucleotides. Inorg. Chem. 1997, 36, 584–592. [CrossRef]
- Boisdenghien, A.; Moucheron, C.; Kirsch-De Mesmaeker, A. Ru(phen)2(PHEHAT)]2+ and [Ru(phen)2(HATPHE)]2+: Two Ruthenium(II) Complexes with the Same Ligands but Different Photophysics and Spectroelectrochemistry. Inorg. Chem. 2005, 44, 7678–7685. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, L.; Kirsch-De Mesmaeker, A. Spectroelectrochemical Characteristics and Photophysics of a Series of RuII Complexes with 1,4,5,8,9,12-hexaatatriphenylene: Effects of Polycomplexation. J. Chem. Soc. Faraday Trans. 1992, 88, 2471–2480. [Google Scholar] [CrossRef]
- Masschelein, A.; Jacquet, L.; Kirsch-DeMesmaeker, A.; Nasielski, J. Ruthenium Complexes with 1,4,5,8-Tetraazaphenanthrene. Unusual Photophysical Behavior of the Tris-Homoleptic Compound. Inorg. Chem. 1990, 29, 855–860. [Google Scholar] [CrossRef]
- Ortmans, I.; Elias, B.; Kelly, J.M.; Moucheron, C.; Kirsch-DeMesmaeker, A. [Ru(TAP)2(dppz)]2+: A DNA intercalating complex, which luminesces strongly in water and undergoes photo-induced proton-coupled electron transfer with guanosine-5′-monophosphate. Dalton Trans. 2004, 668–676. [Google Scholar] [CrossRef]
- Nair, R.B.; Teng, E.S.; Kirkland, S.L.; Murphy, C.J. Synthesis and DNA-Binding Properties of [Ru(NH3)4dppz]2+. Inorg. Chem. 1998, 37, 139–141. [Google Scholar] [CrossRef]
- Liu, J.-G.; Ye, B.-H.; Li, H.; Zhen, Q.-X.; Ji, L.-N.; Fu., Y.-H. Polypyridyl ruthenium(II) complexes containing intramolecular hydrogen-bond ligand: Syntheses, characterization, and DNA-binding properties. J. Inorg. Biochem. 1999, 76, 265–271. [Google Scholar] [CrossRef]
- Schwalbe, M.; Karnahl, M.; Tschierlei, S.; Uhlemann, U.; Schmitt, M.; Dietzek, B.; Popp, J.; Groake, R.; Vos, J.G.; Rau, S. The switch that wouldn’t switch–unexpected luminescence from a ruthenium(II)-dppz-complex in water. Dalton Trans. 2010, 39, 2768–2771. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, T.J.; White, T.A.; Turro, C. New Ligand Design Provides Delocalization and Promotes Strong Absorption throughout the Visible Region in a Ru(II) Complex. J. Am. Chem. Soc. 2018, 140, 229–234. [Google Scholar] [CrossRef]
- Toupin, N.P.; Nadella, S.; Steinke, S.J.; Turro, C.; Kodanko, J. Dual-Action Ru(II) Complexes with Bulky π-Expansive Ligands: Phototoxicity without DNA Intercalation. J. Inorg. Chem. 2020, 59, 3919–3933. [Google Scholar] [CrossRef]
- Foxon, S.P.; Green, C.; Walker, M.G.; Wragg, A.; Adams, H.; Weinstein, J.A.; Parker, S.C.; Meijer, A.J.H.M.; Thomas, J.A. Synthesis, Characterization, and DNA Binding Properties of Ruthenium(II) Complexes Containing the Redox Active Ligand Benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine-11,16-quinone. Inorg. Chem. 2012, 51, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Puckett, C.A.; Barton, J.K. Fluorescein Redirects a Ruthenium-Octaarginine Conjugate to the Nucleus. J. Am. Chem. Soc. 2009, 131, 8738–8739. [Google Scholar] [CrossRef] [PubMed]
- Puckett, C.A.; Barton, J.K. Targeting a ruthenium complex to the nucleus with short peptides. Bioorg. Med. Chem. 2010, 18, 3564–3569. [Google Scholar] [CrossRef]
- Liu, Y.; Chouai, A.; Degtyareva, N.N.; Lutterman, D.A.; Dunbar, K.R.; Turro, C. Chemical Control of the DNA Light Switch: Cycling the Switch ON and OFF. J. Am. Chem. Soc. 2005, 127, 10796–10797. [Google Scholar] [CrossRef]
- Bolger, J.; Gourdon, A.; Ishow, E.; Launay, J.-P. Electronic structure of the “molecular light switch” bis(bipyridine)dipyrido[3,2-a:2′,3′-c]phenazineruthenium(2+). Cyclic voltammetric, UV/visible and EPR/ENDOR study of multiply reduced complexes and ligands. Inorg. Chem. 1996, 35, 2937–2944. [Google Scholar] [CrossRef]
- Shade, C.M.; Kennedy, R.D.; Rouge, J.L.; Rosen, M.S.; Wang, M.X.; Seo, S.E.; Clingerman, D.J.; Mirkin, C.A. Duplex-Selective Ruthenium-Based DNA Intercalators. Chem. Eur. J. 2015, 21, 10983–10987. [Google Scholar] [CrossRef]
- Boynton, A.N.; Marcélis, L.; Barton, J.K. [Ru(Me4phen)2dppz]2+, a Light Switch for DNA Mismatches. J. Am. Chem. Soc. 2016, 138, 5020–5023. [Google Scholar] [CrossRef]
- Campagna, S.; Cavazzini, M.; Cusumano, M.; Di Pietro, M.L.; Giannetto, A.; Puntoriero, F.; Quici, S. Luminescent Ir(III) Complex Exclusively Made of Polypyridine Ligands Capable of Intercalating into Calf-Thymus DNA. Inorg. Chem. 2011, 50, 10667–10672. [Google Scholar] [CrossRef]
- Di Pietro, M.L.; Puntoriero, F.; Tuyéras, F.; Ochsenbein, P.; Lainé, P.P.; Campagna, S. Photochemically driven intercalation of small molecules into DNA by in situ irradiation. Chem. Commun. 2010, 46, 5169–5171. [Google Scholar] [CrossRef] [PubMed]
- Trovato, E.; Di Pietro, M.L.; Giannetto, A.; Dupeyre, G.; Lainé, P.P.; Nastasi, F.; Puntoriero, F.; Campagna, S. Designing expanded bipyridinium as redox and optical probes for DNA. Photochem. Photobiol. Sci. 2020, 19, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.G.; Kelly, J.M. Ruthenium polypyridyl chemistry; from basic research to applications and back again. Dalton Trans. 2006, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.M.; McConnell, D.J.; OhUigin, C.; Tossi, B.; Kirsch-De Maesmaeker, A.; Masschelein, A.; Nasielski, J. Ruthenium polypyridyl complexes; their interaction with DNA and their role as sensitisers for its photocleavage. J. Chem. Soc. Chem. Commun. 1987, 1821–1823. [Google Scholar] [CrossRef]
- Blasius, R.; Moucheron, C.; Kirsch-De Mesmaeker, A. Photoadducts of Metallic Compounds with Nucleic Acids−Role Played by the Photoelectron Transfer Process and by the TAP and HAT Ligands in the RuII Complexes. Eur. J. Inorg. Chem. 2004, 3971–3979. [Google Scholar] [CrossRef]
- Le Gac, S.; Foucart, M.; Gerbaux, P.; Defrancq, E.; Moucheron, C.; Kirsch-De Mesmaeker, A. Photo-reactive RuII-oligonucleotide conjugates: Influence of an intercalating ligand on the inter- and intra-strand photo-ligation processes. Dalton Trans. 2010, 9672–9683. [Google Scholar] [CrossRef]
- Keane, P.M.; Poynton, F.E.; Hall, J.P.; Sazanovich, I.V.; Towrie, M.; Gunnlaugsson, T.; Quinn, S.J.; Cardin, C.J.; Kelly, J.M. Reversal of a Single Base-Pair Step Controls Guanine Photo-Oxidation by an Intercalating Ruthenium(II) Dipyridophenazine Complex. Angew. Chem. Int. Ed. 2015, 127, 8484–8488. [Google Scholar] [CrossRef]
- Vanderlinden, W.; Blunt, M.; David, C.C.; Moucheron, C.; Kirsch-De Mesmaeker, A.; De Feyter, S. Mesoscale DNA Structural Changes on Binding and Photoreaction with Ru[(TAP)2PHEHAT]2+. J. Am. Chem. Soc. 2012, 134, 10214–10221. [Google Scholar] [CrossRef]
- Blasius, R.; Nierengarten, H.; Luhmer, M.; Constant, J.F.; Defrancq, E.; Dumy, P.; van Dorsselaer, A.; Moucheron, C.; Kirsch-De Mesmaeker, A. Photoreaction of [Ru(HAT)2phen]2+ with Guanosine-5′-Monophosphate and DNA: Formation of New Types of Photoadducts. Chem. Eur. J. 2005, 11, 1507–1517. [Google Scholar] [CrossRef]
- Shi, S.; Geng, X.; Zhao, J.; Yao, T.; Wang, C.; Yang, D.; Zheng, L.; Ji, L. Interaction of [Ru(bpy)2(dppz)]2+ with human telomeric DNA: Preferential binding to G-quadruplexes over i-motif. Biochimie 2010, 92, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhao, J.; Geng, X.; Yao, T.; Huang, H.; Liu, T.; Zheng, L.; Li, Z.; Yang, D.; Ji, L. Molecular “light switch” for G-quadruplexes and i-motif of human telomeric DNA: [Ru(phen)2(dppz)]2+. Dalton Trans. 2010, 39, 2490–2493. [Google Scholar] [CrossRef] [PubMed]
- Wachter, E.; Moyá, D.; Parkin, S.; Glazer, E.C. Ruthenium Complex “Light Switches” that are Selective for Different G-Quadruplex Structures. Chem. Eur. J. 2016, 22, 550–559. [Google Scholar] [CrossRef]
- Zeraati, M.; Langley, D.B.; Schofield, P.; Moye, A.L.; Rouet, R.; Hughes, W.E.; Bryan, T.M.; Dinger, M.E.; Christ, D. I-motif DNA structures are formed in the nuclei of human cells. Nat. Chem. 2018, 10, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Gueron, M.; Leroy, J.L. The i-motif in nucleic acids. Curr. Opin. Struct. Biol. 2000, 10, 326–331. [Google Scholar] [CrossRef]
- Baptista, R.; Devereux, S.J.; Gurung, S.P.; Hall, J.P.; Sazanovich, I.V.; Towrie, M.; Cardin, C.J.; Brazier, J.A.; Kelly, J.M.; Quinn, S.J. The influence of loops on the binding of the [Ru(phen)2dppz]2+ light-switch compound to i-motif DNA structures revealed by time-resolved spectroscopy. Chem. Commun. 2020, 56, 9703–9706. [Google Scholar] [CrossRef]
- Devereux, S.J.; Poynton, F.E.; Baptista, F.R.; Gunnlaugsson, T.; Cardin, C.J.; Sazanovich, I.V.; Towrie, M.; Kelly, J.M.; Quinn, S.J. Caught in the Loop: Binding of the [Ru(phen)2(dppz)]2+ Light-Switch Compound to Quadruplex DNA in Solution Informed by Time-Resolved Infrared Spectroscopy. Chem. Eur. J. 2020, 26, 17103–17109. [Google Scholar] [CrossRef]
- Fang, Y.-Q.; Taylor, N.J.; Hanan, G.S.; Loiseau, F.; Passalacqua, R.; Campagna, S.; Nierengarten, H.; Van Dorsselaer, A. A Strategy for Improving the Room-Temperature Luminescence Properties of Ru(II) Complexes with Tridentate Ligands. J. Am. Chem. Soc. 2002, 124, 7912–7913. [Google Scholar] [CrossRef]
- Fang, Y.-Q.; Taylor N., J.; Laverdière, F.; Hanan, G.S.; Loiseau, F.; Nastasi, F.; Campagna, S.; Nierengarten, H.; Leize, E.; Van Dorsselaer, A. Ruthenium(II) Complexes with Improved Photophysical Properties Based on Planar 4′-(2-Pyrimidinyl)-2,2′:6′,2″-terpyridine Ligands. Inorg. Chem. 2007, 46, 2854–2863. [Google Scholar] [CrossRef]
- Liu, Y.; Hammitt, R.; Lutterman, D.A.; Thummel, R.P.; Turro, C. Marked Differences in Light-Switch Behavior of Ru(II) Complexes Possessing a Tridentate DNA Intercalating Ligand. Inorg. Chem. 2007, 46, 6011–6021. [Google Scholar] [CrossRef] [PubMed]
- Kober, E.M.; Caspar, J.V.; Sullivan, B.P.; Meyer, T. Synthetic routes to new polypyridyl complexes of osmium(II). Inorg. Chem. 1988, 27, 4587–4598. [Google Scholar] [CrossRef]
- Holmlin, R.E.; Stemp, E.D.A.; Barton, J.K. Os(phen)2dppz2+ in Photoinduced DNA-Mediated Electron Transfer Reactions. J. Am. Chem. Soc. 1996, 118, 5236–5244. [Google Scholar] [CrossRef]
- Holmlin, R.E.; Dandliker, P.J.; Barton, J.K. Charge Transfer through the DNA Base Stack. Angew. Chem. 1997, 109, 2830–2848, Angew. Chem. Int. Ed. Engl.1997, 36, 2714–2730. [Google Scholar] [CrossRef]
- Petralia, S.; Castagna, M.E.; Cappello, E.; Puntoriero, F.; Trovato, E.; Gagliano, A.; Conoci, S. A miniaturized silicon based device for nucleic acids electrochemical detection. Sens. Bio-Sens. Res. 2015, 6, 90–94. [Google Scholar] [CrossRef][Green Version]
- Petralia, S.; Sciuto, E.L.; Di Pietro, M.L.; Zimbone, M.; Grimaldi, M.G.; Conoci, S. An innovative chemical strategy for PCR-free genetic detection of pathogens by an integrated electrochemical biosensor. Analyst 2017, 142, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, B.C.; Estalayo-Adrián, S.; Blasco, S.; Bright, S.A.; Kelly, J.M.; Williams, D.C.; Gunnlaugsson, T. Luminescent ruthenium polypyridyl complexes with extended ‘dppz’ like ligands as DNA targeting binders and cellular agents. Dalton Trans. 2016, 45, 18208–18220. [Google Scholar] [CrossRef]
- Neugebauer, U.; Pellegrin, Y.; Devocelle, M.; Forster, R.J.; Signac, W.; Moran, N.; Keyes, T.E. Ruthenium polypyridyl peptide conjugates: Membrane permeable probes for cellular imaging. Chem. Commun. 2008, 2, 5307–5309. [Google Scholar] [CrossRef]
- Brennaman, M.K.; Alstrum-Acevedo, J.H.; Fleming, C.N.; Jang, P.; Meyer, T.J.; Papanikolas, J.M. Turning the [Ru(bpy)2(dppz)]2+ Light-Switch On and Off with Temperature. J. Am. Chem. Soc. 2002, 124, 15094–15098. [Google Scholar] [CrossRef]
- Gill, M.R.; Thomas, J.A. Ruthenium(II) polypyridyl complexes and DNA–from structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012, 41, 3179–3192. [Google Scholar] [CrossRef]
- Chambron, J.-C.; Sauvage, J.-P. Ru(bipy)2dppz2+: A highly sensitive luminescent probe for micellar sodium dodecyl sulfate solutions. Chem. Phys. Lett. 1991, 182, 603–607. [Google Scholar] [CrossRef]
- De la Cadena, A.; Pascher, T.; Davydova, D.; Akimov, D.; Herrmann, F.; Presselt, M.; Wächtler, M.; Dietzek, B. Intermolecular exciton-exciton annihilation in phospholipid vesicles doped with [Ru(bpy)2dppz]2+. Chem. Phys. Lett. 2016, 644, 56–61. [Google Scholar] [CrossRef]
- Schatzschneider, U.; Niesel, J.; Ott, I.; Gust, R.; Alborzinia, H.; Wölfl, S. Cellular uptake, cytotoxicity, and metabolic profiling of human cancer cells treated with ruthenium(II) polypyridyl complexes [Ru(bpy)2(N-N)]Cl2 with N-N = bpy, phen, dpq, dppz, and dppn. ChemMedChem 2008, 3, 1104–1109. [Google Scholar] [CrossRef]
- Byrne, A.; Burke, C.S.; Keye, T.E. Precision targeted ruthenium(II) luminophores; highly effective probes for cell imaging by stimulated emission depletion (STED) microscopy. Chem. Sci. 2016, 7, 6551–6562. [Google Scholar] [CrossRef]
- Puckett, C.A.; Barton, J.K. Methods to Explore Cellular Uptake of Ruthenium Complexes. J. Am. Chem. Soc. 2007, 129, 46–47. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Elmes, R.B.P.; Erby, M.L.; Bright, S.A.; Poynton, F.E.; Nolan, D.E.; Quinn, S.J.; Gunnlaugsson, T.; Williams, D.C. Detailed Biological Profiling of a Photoactivated and Apoptosis Inducing pdppz Ruthenium(II) Polypyridyl Complex in Cancer Cells. J. Med. Chem. 2015, 58, 4494–4505. [Google Scholar] [CrossRef]
- Jia, F.; Wang, S.; Man, Y.; Kumar, P.; Liu, B. Recent Developments in the Interactions of Classic Intercalated Ruthenium Compounds: [Ru(bpy)2dppz]2+ and [Ru(phen)2dppz]2+ with a DNA Molecule. Molecules 2019, 24, 769. [Google Scholar] [CrossRef]
- Matson, M.; Svensson, F.R.; Nordén, B.; Lincoln, P. Correlation Between Cellular Localization and Binding Preference to RNA, DNA, and Phospholipid Membrane for Luminescent Ruthenium(II) Complexes. J. Phys. Chem. B 2011, 115, 1706–1711. [Google Scholar] [CrossRef]
- Svensson, F.R.; Abrahamsson, M.; Strömberg, N.; Ewing, A.G.; Lincoln, P.J. Ruthenium(II) Complex Enantiomers as Cellular Probes for Diastereomeric Interactions in Confocal and Fluorescence Lifetime Imaging Microscopy. Phys. Chem. Lett. 2011, 2, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Pierroz, V.; Joshi, T.; Leonidova, A.; Mari, C.; Schur, J.; Ott, I.; Spiccia, L.; Ferrari, S.; Gasser, G. Molecular and Cellular Characterization of the Biological Effects of Ruthenium(II) Complexes Incorporating 2-Pyridyl-2-pyrimidine-4-carboxylic Acid. J. Am. Chem. Soc. 2012, 134, 20376–20387. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Joyce, L.E.; Dickson, N.M.; Turro, C. Efficient DNA photocleavage by [Ru(bpy)2(dppn)]2+ with visible light. Chem. Commun. 2010, 46, 2426–2428. [Google Scholar] [CrossRef] [PubMed]
- Pierroz, V.; Rubbiani, R.; Gentili, C.; Patra, M.; Mari, C.; Gasser, G.; Ferrari, S. Dual mode of cell death upon the photo-irradiation of a RuII-polypyridyl complex in interphase or mitosis. Chem. Sci. 2016, 7, 6115–6124. [Google Scholar] [CrossRef]
- Hess, J.; Huang, H.; Kaiser, A.; Pierroz, V.; Blacque, O.; Chao, H.; Gasser, G. Evaluation of the Medicinal Potential of Two Ruthenium(II) Polypyridine Complexes as One-and Two-Photon Photodynamic Therapy Photosensitizers. Chem. Eur. J. 2017, 23, 9888–9896. [Google Scholar] [CrossRef]
- Gill, M.; Garcia-Lara, J.; Foster, S.; Smythe, C.; Battaglia, G.; Thomas, J.A. A ruthenium(II) polypyridyl complex for direct imaging of DNA structure in living cells. Nat. Chem. 2009, 1, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Xu, G.; Zhao, J.; Wang, Z.; Lu, J.; Sun, W.; Gou, S. DNA-Targeting RuII-Polypyridyl Complex with a Long-Lived Intraligand Excited State as a Potential Photodynamic Therapy Agent. Chem. Eur. J. 2020, 26, 17495–17503. [Google Scholar] [CrossRef]
- Mulcany, S.P.; Li, S.; Korn, R.; Xie, X.; Meggers, E. Solid-phase synthesis of tris-heteroleptic ruthenium(II) complexes and application to acetylcholinesterase inhibition. Inorg. Chem. 2008, 47, 5030–5032. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Golde, T.E.; Ghiso, J.; Cheung, T.T.; Estus, S.; Shaffer, L.M.; Cai, X.D.; McKay, D.M.; Tintner, R.; Frangione, B.; et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 1992, 258, 126–129. [Google Scholar] [CrossRef]
- Cook, N.P.; Torres, V.; Jain, D.; Martí, A.A. Sensing Amyloid-β Aggregation Using Luminescent Dipyridophenazine Ruthenium(II) Complexes. J. Am. Chem. Soc. 2011, 133, 11121. [Google Scholar] [CrossRef]
- Jiang, Y.; Fang, X.; Bai, C. Signaling Aptamer/Protein Binding by a Molecular Light Switch Complex. Anal. Chem. 2004, 76, 5230–5235. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yu, T.; Guo, L.; Xie, J.; Shao, N.; He, Z. In vitro selection of DNA aptamer against abrin toxin and aptamer-based abrin direct detection. Biosens. Bioelectron. 2007, 22, 2456–2463. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, Y.; Zhou, C.; Fang, X. Aptamer-Based ATP Assay Using a Luminescent Light Switching Complex. Anal. Chem. 2005, 77, 3542–3546. [Google Scholar] [CrossRef]
- Hu, L.; Bian, Z.; Li, H.; Han, S.; Yuan, Y.; Gao, L.; Xu, G. [Ru(bpy)2dppz]2+ electrochemiluminescence switch and its applications for DNA interaction study and label-free ATP aptasensor. Anal. Chem. 2009, 81, 9807–9811. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | MeCN | H2O | Ref | ||||||
|---|---|---|---|---|---|---|---|---|---|
| λabs/nm (ε/104 M−1 cm−1) | λem/nm | Φ/% | τair/ns (τAr/ns) | λabs/nm (ε/104 M−1 cm−1) | λemi/nm | Φ/% | τair/ns (τAr/ns) | ||
| [Ru(bpy)2(dppz)]2+ | - | 615 | - | - | 263 (11.7) 372 (2.18) 439 (2.0) | - | - | - | [70,71] |
| [Os(tpy)(pydppz)]2+ | 325 (5.05) 375 (2.21) 475 (1.46) | 800 | 0.8 | 50 | - | - | - | - | [91] |
| [Ru(acac)2(dppz)] | 554 (9100) | Not soluble | [109] | ||||||
| [Ru(phen)3]2+ | 262 446 | - | - | - | 421 443 (2.0) | - | - | - | [113] |
| [Ru(phen)2(dppz)]2+ | 264, 276 sh 316, 352 360, 368, 440 | 630 | 2.1 | 180 (643) | 264, 278 sh 318 sh, 358 sh 372, 440 (2.34) | - | - | - | [113] |
| [Ru(phen)2(PHEHAT)]2+ | 264, 278 sh 312 sh, 354 sh 370, 438 | 662 | 1.1 | 191 (262) | 264, 276 sh 312 sh, 356 374, 440 (2.27) | - | - | - | [113,114] |
| [Ru(phen)2(HAT)]2+ | 262 420 480 sh | 696 | 1.7 | 371 (776) | 262 430 (1.44) 494 sh | - | - | - | [115] |
| [Ru(bpy)2(HAT)]2+ | 282 420 480 sh | 703 | 1.6 a | - (620) | 277 (5.90) 432 (1.0) 484 sh | 742 | 6 × 10−3 | - (104) | [115] |
| [Ru(HAT)3]2+ | 275 410 sh 436 | 587 | 0.9 a | - (89) | 275 410 sh 440 (2.10) | 596 | 0.013 | - (145) | [115] |
| [Ru(TAP)3]2+ | 276 408 437 | 604 | 0.7 | - (68) | 276 (4.68) 408 (1.37) 437 (1.30) | 602 | 0.014 | - (223) | [116] |
| [Ru(bpy)2(TAP)]2+ | 280 428 472 | 679 | 4.2 | - (980) | 278 (7.61) 439 (1.24) 484 | 714 | 0.003 | - (145) | [115] |
| [Ru(TAP)2(dppz)]2+ | 621 | - | - | 204 (9.08) 230 (5.93) 278 (10.4) 366 (1.45) 412 (2.23) 454 (1.58) | 636 | 0.035 | 820 (1090) | [117] | |
| [Ru(NH3)4(dppz)]2+ | 550 | - | - | - | 544 (0.26) | - | - | - | [118] |
| [Ru(bpy-COOMe)2(dppz)]2+ | 471 (1.95) | 642 | - | - | 474 | 642 | - | - | [119,120] |
| [Ru(bpy-COOH)2(dppz)]2+ | - | - | - | - | 463 (1.61) | 624 | - | - | [120] |
| [Ru(bpy)2(qdpq)]2+ | 284 (7.5) 362 (1.4) 390 (1.4) 564 (0.6) | [121,122,123] | |||||||
| [Ru(bpy)2(qdppz]2+ | 285 (10.33) 378 (1.86) 395 (1.85) 442 (1.66) | [121,122,123] | |||||||
| [Ru(tpy)(pydppz)]2+ | 306 (5.72) 325 (2.96) 475 (1.48) | 698 | 0.021 a | - | - | - | - | - | [121,122,123] |
| [Ru(bpy)2(dppn)]2+ | 444 (1.35) | 617 | 0.3 | - | - | - | - | - | [124,125] |
| [(bpy)2Ru(tpphz)]2+ | 450 (19.7) 380 (33.9) 361 (24.1) 284 (133.0) 246 (76.4) | 616 | 0.010 | 634 | 0.017 | [126,127] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pietro, M.L.; La Ganga, G.; Nastasi, F.; Puntoriero, F. Ru(II)-Dppz Derivatives and Their Interactions with DNA: Thirty Years and Counting. Appl. Sci. 2021, 11, 3038. https://doi.org/10.3390/app11073038
Di Pietro ML, La Ganga G, Nastasi F, Puntoriero F. Ru(II)-Dppz Derivatives and Their Interactions with DNA: Thirty Years and Counting. Applied Sciences. 2021; 11(7):3038. https://doi.org/10.3390/app11073038
Chicago/Turabian StyleDi Pietro, Maria Letizia, Giuseppina La Ganga, Francesco Nastasi, and Fausto Puntoriero. 2021. "Ru(II)-Dppz Derivatives and Their Interactions with DNA: Thirty Years and Counting" Applied Sciences 11, no. 7: 3038. https://doi.org/10.3390/app11073038
APA StyleDi Pietro, M. L., La Ganga, G., Nastasi, F., & Puntoriero, F. (2021). Ru(II)-Dppz Derivatives and Their Interactions with DNA: Thirty Years and Counting. Applied Sciences, 11(7), 3038. https://doi.org/10.3390/app11073038