Abstract
The human Wilms’ tumor gene (WT1) was originally isolated in a Wilms’ tumor of the kidney as a tumor suppressor gene. Numerous isoforms of WT1, by combination of alternative translational start sites, alternative RNA splicing and RNA editing, have been well documented. During human ontogenesis, according to the antibodies used, anti-C or N-terminus WT1 protein, nuclear expression can be frequently obtained in numerous tissues, including metanephric and mesonephric glomeruli, and mesothelial and sub-mesothelial cells, while cytoplasmic staining is usually found in developing smooth and skeletal cells, myocardium, glial cells, neuroblasts, adrenal cortical cells and the endothelial cells of blood vessels. WT1 has been originally described as a tumor suppressor gene in renal Wilms’ tumor, but more recent studies emphasized its potential oncogenic role in several neoplasia with a variable immunostaining pattern that can be exclusively nuclear, cytoplasmic or both, according to the antibodies used (anti-C or N-terminus WT1 protein). With the present review we focus on the immunohistochemical expression of WT1 in some tumors, emphasizing its potential diagnostic role and usefulness in differential diagnosis. In addition, we analyze the WT1 protein expression profile in human embryonal/fetal tissues in order to suggest a possible role in the development of organs and tissues and to establish whether expression in some tumors replicates that observed during the development of tissues from which these tumors arise.
1. Introduction
The human Wilms’ tumor gene (WT1), first isolated as a tumor suppressor gene and involved in the development of Wilms’ tumor of the kidney [1], was among the principal tumor suppressor genes to be cloned [2]. The WT1 gene maps to chromosomal band 11p13, and encodes a transcription factor of the zinc finger type family, containing four zinc finger motifs at the C-terminus and a proline/glutamine-rich DNA-binding domain at the N-terminus. It spans approximatively 50 kb of genomic sequence and comprises 10 exons that produce a 3 kb mRNA [3,4].
Numerous isoforms, developing from a combination of alternative translational start sites, alternative RNA splicing, and RNA editing, have been well-documented [5,6,7,8]. The most studied are the exon 5 variants and the KST (lysine–threonine–serine) isoforms, and, in some cases, these can have specific functions. Although WT1 was originally discovered as a tumor suppressor gene involved in the pathogenesis of renal Wilms’ tumor, subsequent studies emphasized its potential oncogenic role in hematologic malignancies [9] and in different malignant solid tumors [10], including lung cancer [11], colorectal cancer [12], pancreatic cancer [13], breast cancer [14], desmoid tumors [15], ovarian cancer [16,17], brain tumors [18,19] and soft tissue sarcomas [20]. This hypothesis is supported by several functional studies showing that WT1 inhibition, by using antisense oligonucleotides, reduces cell proliferation, migration and endothelial tube formation [21]. WT1 could play an important role in angiogenesis, the onset of metastases and the inhibition of the immune response. In fact the endothelial cells, haematopoietic progenitor and myeloid-derived suppressor cells express high levels of WT1 inducing the control of the expression of CD31 and CD117. Therefore, the inactivation of WT1 in the aforementioned cells can reduce angiogenesis, the development of metastases and promote the immune response [22].
Although the first function defined for WT1 was that of a transcriptional regulator, in the last 10 years several studies have emerged indicating its function as an activator, a repressor or a coactivator [23,24]. Nevertheless, it is known that the role of WT1 is more complicated than once believed, and it could be involved in at least two distinct cellular processes: transcription control and RNA metabolism. All these roles of WT1 are supported by alternative splicing of the WT1 RNA to generate two main isoforms that differ by the insertion of three amino acids, KTS (lysine–threonine–serine), inside the zinc finger region of the protein [25,26,27]. In this regard, several studies have demonstrated that the isoform of WT1 that lacks the KTS insertion, –KTS WT1, binds to DNA with higher affinity and functions as a transcriptional regulator. On the other hand, the form having the KTS insertion, +KTS WT1, plays a role in mRNA splicing rather than transcriptional control [28,29]. Larsson et al. found the first correlation between the WT1 protein and RNA metabolism, showing that the +KTS WT1 isoform colocalized specially with splicing factors within nuclear speckles. In the last few years it has been demonstrated that a considerable amount of transcription and/or splicing factors shuttle to the cytoplasm, acquiring a new function. The cytoplasmic role attributed to shuttling proteins is predominantly the nucleocytoplasmic transport of mRNA or RNA. Niksic et al. [27], emphasized not only that WT1 is a shuttling protein, but also that a substantial part of endogenous WT1 protein is located in the cytoplasm. Furthermore, they showed that both WT1 isoforms shuttle between the nucleus and the cytoplasm [30].
WT1 is not only differentially spliced, but in turn alters the splicing and function of other genes, such as the vascular endothelial growth factor (VEGF), through the activation of Serine/arginine-rich, protein-specific splicing factor kinase (SRPK1), and indirectly Serine/arginine-rich splicing factor 1 (Srsf1). Comparing healthy tissues with neoplastic tissues, there is a greater expression of Wt1, Srpk1, Srsf1 and the pro-angiogenic VEGF isoforms. WT1 inhibition regulates negatively the expression of Srpk1 and Srsf1 in endothelial cells, inducing the development of the antiangiogenic VEGF isoform, associated with apoptotic cell death [31,32].
In the past many authors believed that WT1 immunohistochemical expression, using antibodies directed against the C-terminal portion of the protein [33,34], was exclusively limited to the nucleus, and that the occasional cytoplasmic staining obtained in some tumors was the result of an artifact (non-specific staining). The variability of staining with anti-WT1 C-terminus antibodies could be due to the use of different clones of the same antibody. Subsequently, with the advent of newly available antibodies against the N-terminal portion of WT1 (clone WT6F-H2), it has been demonstrated that WT1 expression can be found in the nucleus or in the cytoplasm, or concurrently in both the nucleus and cytoplasm [35,36,37,38,39,40,41,42].
During human ontogenesis, according to the antibodies used, anti-C or N-terminus WT1 protein, [36,39,40,41,42,43,44,45,46,47], nuclear expression can be frequently obtained in metanephric and mesonephric glomeruli, primary sex cords, gonadic stroma and mesothelial and sub-mesothelial cells, while cytoplasmic staining is usually found in developing skeletal muscles, myocardium, radial glia of the spinal cord and cerebral cortex, sympathetic neuroblasts, adrenal cortical cells and the endothelial cells of blood vessels [33,34,37,38,48,49,50,51,52]. With regard to neoplastic tissues, WT1 protein has been found in several tumors with variable immunostaining patterns, i.e., exclusively nuclear, cytoplasmic or both, according to the antibodies used (anti-C or N-terminus WT1 protein) [35,36,39,40,41,42,43,44,45,46,47]. The variable nuclear and cytoplasmic WT1 staining may be explained by assuming that the expression of this transcription factor in some neoplastic tissues (Wilms tumor, ovarian, mesothelial neoplasms, Sertoli cell tumor and rhabdomyosarcoma) mirrors its normal developmental regulation [37,38,41]. In addition, our research group have recently demonstrated that WT1 shows an oncofetal expression pattern, being abundantly detected in developing and neoplastic skeletal muscle tissues, while its expression is down-regulated in adult normal skeletal muscle tissues [41,46].
The present review focuses on the immunohistochemical expression of WT1 in some common and less common tumors (Table S1), emphasizing its potential diagnostic role and usefulness in differential diagnosis. In addition, we analyze WT1 protein expression profiles during human ontogenesis to provide suggestions about its role in the development of organs and tissues, and to establish if its expression in some tumors replicates that observed during the development of tissues from which these tumors arise.
2. WT1 Immunohistochemical Expression in Human Embryonal/Fetal and Neoplastic Tissues
2.1. WT1 Immunohistochemical Expression in Human Embryonal/Fetal Tissues
From gestational weeks 7 to 24, WT1 expression has been found in several tissues, in a nuclear or a cytoplasmic localization. WT1 nuclear staining can be observed in different structures of metanephros and mesonephros [37,51,52], including glomeruli and sub-capsular blastema of nephrogenic zone (Figure 1). WT1 expression in round, undifferentiated mesenchymal cells (blastematous component) undergoing epithelial differentiation is a model of controlled epithelial–mesenchymal transition resulting in nephrogenesis [53,54,55,56]. WT1 is expressed in mesothelial cells (excoelomic epithelium) that are found above both ovaries and testes, in the developing sex cords and in the gonadal mesenchyme (Figure 1). In the later phases of development, nuclear staining is manteined in the secondary sex cords of both testes and ovaries, while it gradually disappears from their surrounding mesenchyme, to be restricted to epithelial cells surrounding the oocytes [51]. Between the seventh and the tenth week of gestational age, WT1 nuclear expression can be found in the mesothelial cells of serosal surfaces covering the abdominal and pelvic visceral organs (uterus and ovaries, bladder, stomach, small and large intestine, pancreas) pleura and peritoneum. Notably, a conspicuous amount of mesenchymal sub-mesothelial cells along mesothelial cells displays WT1 nuclear expression [37].
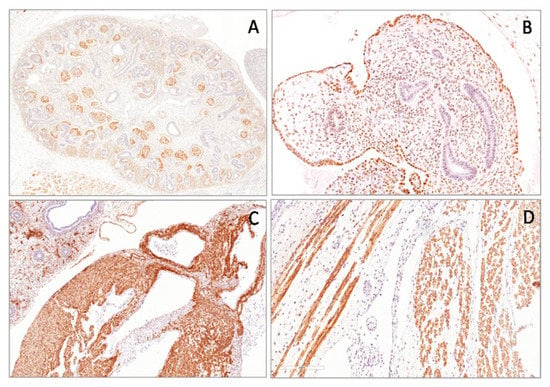
Figure 1.
Nuclear expression of the Wilms’ tumor gene (WT1) (N-terminus) in human metanephros (A), in gonadic stromal cells (B) and in the mesothelial cells covering the surface of the ovary (B) of the fetus of 11 weeks of gestational age. Cytoplasmic expression of WT1 (N-terminus) in myocardium (C) and in the skeletal cells (D) in a fetus of 22 weeks of gestational age.
During human developmental phases (from 8 to 28 wGA) small clusters of neuroblasts are located in the paravertebral regions, in the cortex of adrenal glands and within the muscle wall of the developing stomach and small/large intestine. WT1 cytoplasmic expression in neuroblasts is strong and diffuse during the different phases of development, while no nuclear immunoreactivity can be demonstrated. These cells gradually differentiate into ganglion cells, at first as immature ganglion cells and finally as mature ganglion cells. In immature ganglion cells, unlike neuroblasts, WT1 cytoplasmic immunoexpression is weak/focal and sometimes absent, while ganglion cells of adult sympathetic ganglia, adrenal glands and myoenteric nervous plexuses, are completely negative. Similar to the adult adrenal medulla and paraganglia, extra- and intra-adrenal differentiating chromaffin cells are not WT1 stained. The expression of WT1 is strongly localized in the cytoplasm of radial glia cells of both the developing spinal cord and cerebral cortex along the entire thickness of the neural tube.WT1 expression in the undifferentiated radial glial cells seems to support the theory that it is necessary to maintain cells in an undifferentiated state [30,57,58,59].
During the early phases of development (from 6 wGA) embryonic myoblasts forming myotomes exhibit strong cytoplasmic staining for WT1. Moreover, in both primary and secondary myotubes of the developing muscles, WT1 is strongly and diffusely expressed in the cytoplasm of these cells. From 20 wGA, WT1 cytoplasmic expression becomes more heterogeneous, ranging from focally strong to weak or absent within the same muscle fiber (mosaic-type expression) (Figure 1). WT1 nuclear immunoreactivity is lacking in skeletal muscle during all phases of myogenesis [41]. Several studies have shown that WT1 is crucial for heart development [60,61], and it may also be involved in the proliferation of cardiac myocytes. In addition, WT1 plays a key role in the conversion of epicardial cells to mesenchymal cells. This is demonstrated by strong WT1 cytoplasmic expression in cardiomyocytes of both atria and ventricles during the different phases of heart development, as in somatic skeletal muscle cells. In addition, WT1 is expressed in the cytoplasm of the endothelial cells of blood vessels (aorta, arteries, veins, capillaries) in all developing tissues (Figure 1) [62]. These findings are also maintained in the endothelial cells of both adult tissues and benign and malignant tumors. In developing human lungs, from 8 to 14 wGA, WT1 nuclear staining is limited to the mesothelial cells of visceral pleura, with a weak cytoplasmic expression in some mesenchymal cells surrounding branching epithelial structures. Between 7 and 14 wGA, WT1 staining is missing in the human fetal epidermis, except for an intense cytoplasmic expression in the progenitor cells of the developing dermis [41].
WT1 is expressed in the myoenteric plexus of the developing gastroenteric system [38], while a weak cytoplasmic staining is observed in the cells of the muscularis propria, especially of the small intestine [52]. A strong WT1 nuclear staining is found in the mesothelial cells of serosal surfaces covering the stomach, small and large intestine, pancreas and liver [52].
2.2. Wilms’ Tumor
Wilms’ tumor is a malignant embryonal neoplasm, also known as a nephroblastoma of pediatric age, resulting from a disturbance of the differentiation of nephrogenic blastematous cells, which replicates various stages of the developing kidneys [53,54,63,64]. Microscopically, Wilms’ tumor is typically composed of three components: blastematous, mesenchymal (stromal) and epithelial [65,66,67]. Most Wilms’ tumors show all three components, but their proportions vary widely. Some tumors are biphasic, while others are monophasic. The blastematous component is extremely cellular and composed of small round-to-oval cells with scanty cytoplasm, dark nuclei and frequent mitotic figures, arranged in diffuse, nodular, cord-like (serpentine), or basaloid (with peripheral palisading) patterns. The stromal component is composed of spindle-shaped cells set in a myxoid stroma.
In most tumors, stromal differentiation, including smooth and skeletal muscle, bone and chondroid tissues, adipocytic tissue, or more rarely, neuroglia and mature ganglion cells, may be observed. Skeletal muscle in various stages of differentiation, including rhabdomyoblasts, represents the most common heterologous component [68,69]. The epithelial component is composed of tubules, papillary and glomerular-like structures, which are closely reminiscent of normal nephrogenesis.
There is not a single marker or panel of immunostains that is diagnostic of Wilms’ tumor, but the immunohistochemical profile depends on the different tumor components examined. WT1 is certainly the most sensitive and specific marker for the diagnosis of Wilms’ tumor, with positivity in more than 90% of cases (Figure 2). It is expressed in the nuclei of primitive blastemal cells using antibodies directed both to the C-terminal or N-terminal portions of the protein, while it is absent in the differentiated epithelial and stromal elements. When a Wilms’ tumor is composed predominantly/exclusively of the blastematous component, it may be diagnostically challenging, especially when pathologists are dealing with small biopsies, and the differential diagnosis includes other small, round cell tumors (differential diagnosis with other small, round blue cell tumors-PNETs/Ewing sarcoma, neuroblastomas, rhabdomyosarcomas, clear cell sarcomas, synovial sarcomas and lymphomas). In this context, the demonstration of WT1 nuclear expression with antibodies directed against the C-terminal portion of the protein (clone WT1C19) is helpful in confirming the diagnosis.
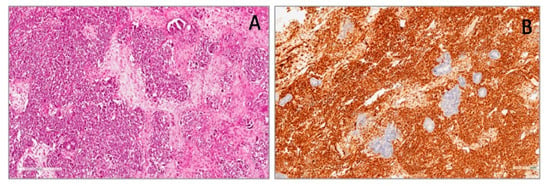
Figure 2.
Wilms’ tumor showing blastematous, epithelial and stromal components (A). Blastematous component showing diffuse and strong nuclear WT1 (N-terminus) expression (B).
2.3. Malignant Mesothelialioma
Malignant mesothelioma is a primary tumor of serosal membranes, including pleura, peritoneum, pericardium and tunica vaginalis of the testis. The majority of these tumors arise from pleura followed by peritoneum, and are related to asbestos exposure. Histologically, mesotheliomas are classified in three forms: epithelioid, sarcomatoid, or mixed (biphasic). The majority of these tumors are predominantly epithelioid, with tumor cells forming papillae, pseudoacini or solid epithelial nests [70]. These tumor cells show abundant and acidophilic cytoplasm with round nuclei and occasionally prominent nucleoli. Early forms can be difficult to distinguish from reactive mesothelial hyperplasia. Infiltration of deep tissues, obvious cytologic atypia, prominent cell groupings and necrosis favor the diagnosis of malignant mesothelioma [71]. Sarcomatoid mesothelioma [72] is composed of spindle cells with oval nuclei, scant amphophilic cytoplasm and occasionally prominent nucleoli, arranged in a fascicular pattern, sometimes with a fibrosarcoma-like appearance.
Epithelioid mesotheliomas may show a wide variety of morphologies, which can mimic numerous metastatic carcinomas, especially adenocarcinoma of the lung. The correct diagnosis of mesothelioma is usually achieved by the application of a panel of immunohistochemistry, including WT1 antibodies. Early studies on WT1 expression in mesotheliomas were published in 2000, reporting a positivity of 45–75% of the cases using a polyclonal antibody [73,74]. With the advent of new, commercially-available antibodies (6F-H2), a higher percentage of expression has been reported. Currently, WT1 is considered a highly sensitive and relatively specific marker for distinguishing mesothelioma from lung adenocarcinoma [75]. However, in daily diagnostic practice, WT1 must be used in combination with other, usually positive, markers for mesothelioma (calretinin, Keratin5/6).
In mesotheliomas, WT1 protein expression is restricted to the nucleus (Figure 3), even if it can be focally observed in the cytoplasm of the cells of sarcomatoid mesotheliomas [74,75,76,77,78]. As normal/hyperplastic mesothelial cells lining pleura show nuclear staining for WT1 [37], this marker is not helpful in distinguishing benign from malignant lesions. Nuclear WT1 expression in mesothelioma is not surprising if we consider that a similar expression has been documented in embryonal/fetal mesothelial cells covering cavities. The diagnosis of mesothelioma continues to rely on a multimodal approach that incorporates clinical features, gross, microscopic and immunohistochemical features [79].
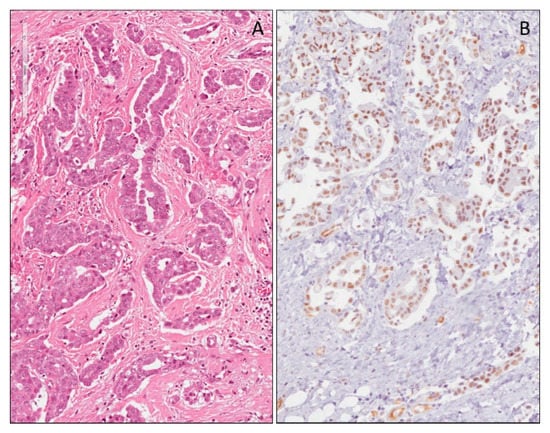
Figure 3.
Epithelioid mesothelioma of the pleura (A). Diffuse nuclear expression of WT1 (N-terminus) in the neoplastic cells of mesothelioma (B).
2.4. WT1 Expression in Epithelial Tumors of Ovary
Epithelial tumors are the most common ovarian tumors, comprising about 60% of all ovarian tumors. Serous tumors constitute approximately 30% of all ovarian tumors. About half of the cases are benign, approximately 35% are malignant and the remaining cases are classified as borderline. They usually show positive staining for CK7, and they do not stain for CK20 or CDX-2. Parenti et al. [37] reported WT1 nuclear expression in mesothelial cells lining the fetal ovaries, thus it is not surprising if the tumors that originate from the epithelium covering the ovary express WT1. In fact, several studies show that the WT1 nuclear expression in serous ovarian tumors is around 90–95%, and that this marker is also helpful in differentiating ovarian serous carcinoma from endometrial serous carcinoma, which has a similar microscopic appearance, but is less likely to stain for WT1 [16,80,81,82,83,84,85,86,87,88,89]. WT1 shows a different expression in the different histological subtypes of ovarian carcinomas. Serous ovarian carcinomas that originate in the fallopian tubes, in the peritoneum and in the ovarian cortical inclusion cysts, are always WT1 positive. Recent studies demonstrated not only the diagnostic utility, but also the possible prognostic role of WT1. High grade serous ovarian carcinomas positive to WT1 have a better prognosis, especially when the neoplastic cells co-express WT1 and the estrogen receptor [90]. Mucinous and clear-cell carcinomas are negative, according to Waldstrøm and Grove [89], Goldstein et al. [80] and Hashi et al. [82]. In contrast, another study carried out by Shimizu et al. [16] demonstrated the immunohistochemical expression of WT1 in both mucinous and clear-cell carcinomas, with a higher expression in serous carcinomas than in clear-cell carcinomas, while not a significantly higher expression than mucinous carcinomas. It is likely that these conflicting results may be due to the use of different primary antibodies. Indeed, Shimizu et al. [16], used the C19 clone, whereas in other studies the 6F-H2 clone was the antibody used against WT1.
Numerous reports show that WT1 is not expressed in endometrioid carcinomas [82,86,87], with only a few studies showing focal WT1 positivity [81,83,84]. Recently, immunohistochemical studies indicated a significant difference in WT1 expression between highly-differentiated, endometrioid carcinomas, compared with tumors of lower grade [89]. As suggested by Gilks [88], low-grade endometrioid carcinomas differ from high-grade endometrioid carcinomas in biological behavior and gene expression profile, and this theory may explain the different expression of WT1.
2.5. WT1 Expression in Granulosa Cell Tumor
Granulosa cell tumors are sex cord–stromal ovarian neoplasms showing differentiation toward follicular granulosa cells. The two types of granulosa cell tumors are known as adult and juvenile types. Macroscopically, adult granulosa cell tumors appear as unilateral, usually solid or solid-cystic masses, with an area of hemorrhage or necrosis following torsion. In the adult type granulosa cell tumors, the tumor cells resemble normal granulosa cells. They are small and round, cuboidal, or spindle shaped, with pale cytoplasm and ill-defined cell borders. The nuclei are round or oval, with fine chromatin and a single small nucleolus. An important diagnostic feature is the presence of longitudinal folds or grooves in the nuclei with a ‘coffee-bean’ appearance [91]. Occasionally, mitotic figures, nuclear pleomorphism and bizarre nuclei are seen, but do not appear to affect the prognosis adversely [92,93,94]. The tumor cells are rarely extensively (>50% of cells) luteinized with abundant eosinophilic cytoplasm, well-defined cell borders and central nuclei resembling the luteinized granulosa cells of the corpus luteum. Cells are arranged in different patterns, including diffuse, trabecular, micro-follicular, macro-follicular, insular and pseudopapillary. Macroscopically juvenile granulosa cell tumors are solid-cystic masses, rarely exclusively solid. They are variably composed of a double component, granulosa and theca component. The former shows polygonal cells with abundant cytoplasm, from eosinophilic to vacuolated, and clear and hyperchromatic nuclei without grooves. The second is composed of oval- to spindle-shaped cells with pale cytoplasm. Several growth patterns are observed, from solid to follicular or pseudo-papillary. In the atypical forms, brisk mitotic activity is seen. Bizarre nuclei may occur. WT1 is a marker that has not been widely studied in granulosa cell tumors. It is expressed in 65% to 88% of adult granulosa cell tumors (Figure 4) [95,96,97,98]. Although several different non-sex cord-stromal tumors can express WT1 [99], most of them are not in the differential diagnosis of ovarian sex cord-stromal tumors, and tumors that are in the differential diagnosis are typically negative or limited for WT1 expression.
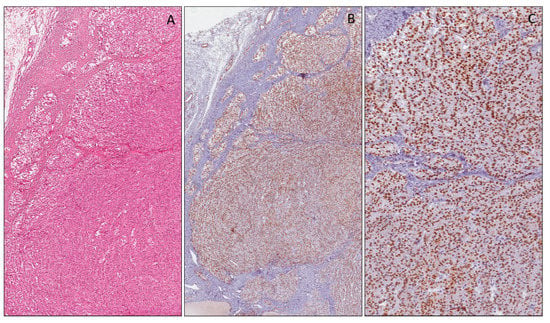
Figure 4.
Ovarian granulosa cell tumor, adult type (A). Nuclear WT1 (N-terminus) expression in neoplastic cells at low (B) and high (C) magnification.
2.6. WT1 Expression in Sertoli Cell Tumors
Sertoli cell tumors are composed of Sertoli cells with rare Leydig cells. They are typically a solid mass with a tan to yellow cut surface. The tumor cells are cuboidal or columnar with pale cytoplasm, and less frequently, deeply eosinophilic or vacuolated. The nuclei are round–oval in shape and uniform with inconspicuous nucleoli. Atypical and bizarre nuclei are rarely observed. Mitotic activity is variable, but not > 5/10 HPF. The lesion is well limited by the surrounding ovarian parenchyma.
The cells are typically arranged in a diffuse or nodular pattern, but they can be observed as a mixture of different patterns (tubules, cord or trabeculae, sheets, pseudo-papillae and more rarely, retiform, islands or spindly patterns). The main differential diagnoses are the Sertoli–Leydig cell tumor, endometrioid carcinoma, carcinoid tumor and female adnexa tumor.
Several studies have investigated the expression of WT1 in Sertoli cell tumors in order to identify new diagnostic markers. Zhao et al. [99] suggested including WT1 protein (6F-H2 antibody) in the immunohistochemical panel, together with inhibin, to distinguish Sertoli cell tumors from endometrioid and neuroendocrine tumors. In addition, the authors stated that the diagnostic utility of WT1 in Sertoli cell tumors is similar to inhibin, and better than that of calretinin [99]. Another study evaluated WT1 expression in ovarian stroma and its tumors. The results showed that ovarian stromal cells, ovarian fibroma, cellular fibroma, fibrothecoma and ovarian leiomyoma expressed WT1, whereas non-gynecologic smooth muscle tumors and other spindle cell tumors were usually negative for WT1. These findings suggest that WT1 may be used as an immunohistochemical marker for spindle cell tumors derived from the ovary and uterus, including Sertoli cell tumors (Figure 5). WT1, SF-1 and inhibin are the most informative sex cord-stromal markers to be used for the distinction of non-sex cord-stromal tumors; however, the usefulness of immunohistochemistry for the diagnosis of fibroma/fibrothecoma is limited [100].
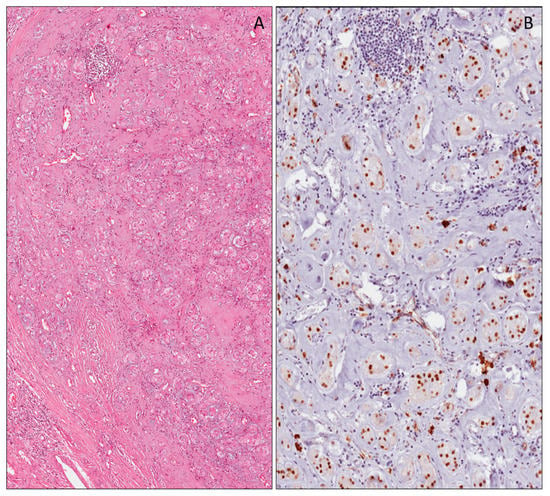
Figure 5.
Sertoli cell tumor (A). Neoplastic cells showing nuclear WT1 (N-terminus) expression (B).
2.7. WT1 Expression in Breast Carcinoma
Several studies have demonstrated WT1 expression in breast carcinomas, with low expression in adjacent normal breast tissues. Nasomyon et al. [101], confirmed WT1 protein expression by using Western blotting. These results suggested that WT1 (17AA+) might be a crucial isoform in cancer progression and development, and might work together with WT1 (17AA−) as a protein partner. In the same study, the authors showed that WT1 plays an oncogenic role in ERα and HER2 protein regulation.
In addition, they observed that ER-α and HER2 proteins were highly expressed in breast cancer, but expressed at low levels in adjacent normal breast tissues, while WT1 (17AA+) was strongly expressed in breast cancer and slightly in adjacent normal breast tissues. The result of WT1 (17AA+) mRNA expression was confirmed at the protein level.
Several studies focused on immunohistochemical nuclear expression of WT1 in breast cancer, predominantly in the mucinous histotype [43,102,103,104], while it is occasionally co-expressed in non-mucinous carcinoma components of mixed mucinous carcinoma [102]. In addition, WT1 expression was observed, not only in mucinous carcinoma, but also in associated solid papillary carcinoma. A good correlation of WT1 immunohistochemical expression between solid papillary carcinoma and associated mucinous carcinomas does exist, indicating that the former could be the precursor of the latter [105]. With the advent of immunotherapy in malignancies, expression of WT1in mucinous breast carcinomas provides a molecular target in these relatively indolent breast tumors [102]. Conflicting results are reported regarding WT1 expression and the prognosis of breast carcinoma. A study demonstrated that WT1 expression in breast cancer is correlated to poor prognosis, due to cancer-related epithelial-to-mesenchymal transition (EMT) and poor chemotherapy response [106]. Conversely, another study displayed that the immunoreactivity for WT1, together with RSPO1 and P16, was significantly associated with a more favorable disease-free survival [107]. Furthermore, high levels of Wilms’ Tumor 1 (WT1) protein and mRNA had been associated with aggressive phenotypes of breast tumors, because HER2/neuoncogene increases WT1 expression. Increased levels of WT1 are due to the engagement of Akt, resulting in HER2/neu overexpression [108].
2.8. WT1 Expression in Lung Carcinomas
WT1 expression was analyzed, by using immunohistochemical and quantitative real-time (qRT-PCR) analyses, in several types of lung carcinoma (41 adenocarcinomas, 13 squamous cell carcinomas, two large cell carcinomas and six small cell carcinomas). Data obtained from immunohistochemical analyses showed cytoplasmic WT1 immunoreactivity in 5/6 small cell carcinomas, 1/2 large cell carcinomas, 1/1 squamous cell carcinomas and in 4/5 adenocarcinomas. (In normal lung tissues, no immunoreactivity was observed). In the same study WT1 genomic DNA obtained from seven lung cancer tissues was PCR-amplified and examined for mutations by direct sequencing. The absence of mutations in all of the 10 exons of the WT1 gene was demonstrated, suggesting that the non-mutated, wild-type WT1 gene plays an important role in the tumorigenesis of de novo lung cancers, and may provide the rationale for new therapeutic strategies for lung cancer targeting the WT1 gene and its products [11].
2.9. WT1 Expression in Pancreatic Ductal Adenocarcinomas
WT1 expression was also reported in a series of pancreatic ductal adenocarcinomas. Oji et al. [13] showed that WT1 was expressed at the cytoplasmic level in 30/40 cases of pancreatic ductal carcinoma, while normal pancreatic cells adjacent to carcinoma cells were negative. No significant correlation was observed between WT1 expression and age, sex, T or N stage, tumor site and differentiation. These results suggest that the WT1 gene plays an important role in the tumorigenesis of pancreatic ductal adenocarcinoma [13]. Conversely, more recent studies have shown that the nuclear expression of WT1 in pancreatic ductal adenocarcinoma correlates with gender and tumor stage, while cytoplasmic staining correlates with gender, histological grade and perineural invasion. In addition, the same authors reported that a high nuclear expression of WT1 in pancreatic tumor tissues was significantly associated with poor overall survival, suggesting a possible role for it as a molecular biomarker of a poor prognosis among patients with pancreatic ductal adenocarcinoma [109].
2.10. WT1 Expression in Melanocytic Lesions
Several studies have reported the usefulness of WT1 in the differential diagnosis between nevi and melanomas. The first study on WT1 expression in melanocytic lesions dates back to 1997 [110], when it was observed that WT1 in melanoma, in the context of absent or mutated p53, acted as a transcriptional activator, whereas in the presence of wild-type p53, it acted as a repressor. Later, Wilsher and Cheerala [111] showed that WT1 (6F-H2) was a complementary marker of malignant melanoma. Indeed, by using immunohistochemistry, they demonstrated cytoplasmic WT1 expression in most invasive primary cutaneous melanomas, including spindle cell and desmoplastic melanomas, and in metastatic melanomas. However, its usefulness was limited by the fact that most Spitz nevi and a minority of dysplastic nevi expressed WT1. In another study, apart from demonstrating cytoplasmic, and more rarely, nuclear WT1 expression in melanoma, it was proven that the silencing of WT1 inhibited melanoma cell proliferation, supporting the role of WT1 cell proliferation in melanoma [21,112,113]. In contrast, Garrido-Ruiz et al. [114] reported a higher rate of WT1 staining in melanocytic nevi against melanomas, and an increased expression in advanced stages of melanoma progression. In addition, they supported an association of WT1 protein expression with a shorter overall survival. A significant expression of WT1 in desmoplastic (71%), compared with non-desmoplastic melanoma (47%), has also been recently observed. The same study reported that vertical growth phase melanomas exhibited an expression of WT1 more frequently than radial growth phase melanomas (46.5% vs. 16.0%) [115]. Finally, a study on 40 cases of desmoplastic melanomas showed a strong and diffuse cytoplasmic expression of WT1 (6F-H2 antibody) in all cases examined, suggesting the use of WT1 along with S-100p, SOX10, p75 and nestin as an optimal panel for the diagnosis of desmoplastic melanoma [116].
2.11. WT1 Expression in Colorectal Carcinoma
Colorectal cancer is one of the most commonly diagnosed cancers worldwide, accounting for an estimated 9.4% of all malignancies [117]. The treatment is surgery, but approximately 60% of patients experience local recurrence and/or distant metastases (Andre and Schmiegel, 2005). Despite advances in surgical techniques and in chemotherapy, around 20% of patients with colorectal carcinoma die from disease recurrence [117]. Thus, new diagnostic tools and therapeutic approaches are required. A previous study showed immunoreactivity in 89% of the cases examined [118]. In addition, in the same study, a quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) was performed that showed that WT1 mRNA was expressed in all (100%) of the 28 cases of colorectal adenocarcinoma. Moreover, WT1 mRNA expression levels were higher in 71% of the cases when compared to those of normal-appearing mucosal tissues. These results suggested an important role of WT1 in the tumorigenesis of primary colorectal adenocarcinoma, and that WT1 could be a new molecular target for the treatment of colorectal adenocarcinoma expressing WT1. However, no significant correlations were observed between WT1 mRNA expression levels and age, gender, site of tumors, T stage, N stage and M stage [118]. Miyata et al. recently evaluated the correlation between WT1 mRNA levels and other tumor-associated antigens with clinicopathological factors [119]. Notably, WT1 expression in colorectal carcinoma is significantly correlated with tumor progression, lymph node and distant metastasis, and clinical stage. These results suggest that WT1 could be an important novel independent marker for prognosis and tumor progression in colorectal adenocarcinoma [120].
2.12. WT1 Expression in Cerebral Tumors
Several authors have studied WT1 expression in cerebral neoplasms, including glial tumors and medulloblastomas [121]. The WT1 cytoplasmic expression using the anti-WT1 C-19 and anti-WT1 6F-H2 antibodies, in high-grade astrocytic tumors (glioblastoma and anaplastic astrocytoma), was significantly higher than that in low-grade ones (pilocytic astrocytoma and diffuse astrocytoma) (Figure 6) [122]. WT1 expression was also examined in anaplastic ependymomas, and anaplastic oligodendrogliomas, which showed diffuse cytoplasmic staining.
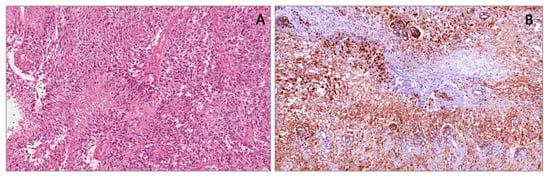
Figure 6.
Glioblastoma multiforme showing pseudopalisading necrosis (A). Neoplastic cells showing strong and diffuse cytoplasmic expression of WT1 (N-terminus) (B).
Some authors correlated the expression of WT1 with the MIB-1 staining index because histological examination areas with features of anaplasia and high perivascular proliferation and cellularity show a strong WT1 protein expression [123]. A more recent article reported that in diffuse astrocytic tumors, high levels of WT1 expression are related to a higher World Health Organization (WHO) tumor grade, absence of IDH1 mutation, older age, but not related to O(6)- methyl guanine methyl transferase (MGMT) promoter methylation status. In addition, it would seem that WT1 expression is associated with a worse outcome in patients with diffuse astrocytoma, but not glioblastoma [124].
2.13. WT1 Gene Expression in Soft Tissue Sarcomas
Sotobori et al. [125] showed that WT1 mRNA expression levels in adult cases of soft tissue sarcomas were significantly higher than in normal soft tissues, and that the disease-specific survival rate for patients with elevated WT1 mRNA expression levels was found to be significantly worse in patients with low levels. Furthermore, their results demonstrated that the WT1 mRNA expression level is a significant prognostic indicator in patients with soft tissue sarcoma. On the contrary, other authors [126] evaluated WT1 protein and mRNA expression levels in various pediatric tumors, and showed that while WT1 protein was widely detected in these malignancies, WT1 mRNA expression varied widely in the different types of pediatric cancers. However, no significant relationship was evaluated between WT1 mRNA expression and clinical factors.
2.14. WT1 Expression in Malignant Peripheral Nerve Sheath Tumors
Malignant peripheral nerve sheath tumors (MPNSTs) are an aggressive and rare type of sarcoma, usually arising from peripheral nerves. They can occur sporadically or more frequently (up to 50% of cases) from pre-existing neurofibromas in the context of neurofibromatosis type 1 (NF1) [127]. Most MPNSTs are deeply localized, and often they are greater than 10 cm in maximum diameter by the time of presentation. Histologically, they show a spindle-cell fascicular appearance with abrupt alternations between cellular and more myxoid areas, suggesting neural differentiation. Some cases have a uniformly cellular and fascicular pattern reminiscent of a monophasic synovial sarcoma. In other cases, an abundant myxoid stroma can be observed, configuring the diagnosis of myxoid MPNST. Cells have a pale, poorly defined cytoplasm and narrow nuclei, often with a wavy or buckled configuration. In addition, the nuclei tend to be hyperchromatic, and at least focally pleomorphic with inconspicuous nucleoli. Mitoses are generally frequent. In about 10–15% of MPNSTs, especially in those arising in patients with NF1, heterologous differentiation can be present [128]. The most frequent divergent differentiation is the rhabdomyosarcomatous component, configuring the so-called malignant Triton tumor, which is associated with a poor prognosis. Less frequently, osteosarcomatous or chondrosarcomatous differentiation can be observed. Immunohistochemically, the S-100 protein is positive in about 50% of cases of MPNST [129,130].
Early studies on WT1 expression reported the positivity in Schwann cells of normal nerves. In peripheral nerve sheath tumors, including MPNSTs, neurofibromas and schwannomas, neoplastic cells expressed WT1 protein at the cytoplasmic level (Figure 7) [44]. A recent study by Kim et al. [131] evaluated WT1 expression in 87 cases of soft tissue sarcoma, including liposarcoma, MFH, rhabdomyosarcoma, leiomyosarcoma, MPNST, synovial sarcoma, fibrosarcoma and others. The authors observed cytoplasmic expression of WT1 (6F-H2) in 71% of the cases of malignant peripheral nerve sheath tumors, revealing no association between WT1 expression and overall survival or disease-free survival. Conversely, Ueda et al. [132] reported that the WT1 gene is frequently overexpressed in various types of soft tissue sarcoma [132], and that WT1 mRNA overexpression is significantly associated with a poor prognosis. This result was proved only in 4 out of 52 and 3 out of 36 samples, by immunoblotting [121] and immunohistochemistry. Therefore, further studies on the correlation between the protein and mRNA of the WT1 gene in larger cohorts are required, together with survival analysis, to validate the WT1 expression level as a prognostic factor. Parenti et al. [133] investigated WT1 expression in the MPNST sNF96.2 cell line, showing a strong WT1 staining in the nuclear and perinuclear areas of neoplastic cells. In addition, they studied the effects of silencing WT1 by RNA interference through Western Blot analysis and proliferation assays. The result was a reduction of cell growth in a time- and dose-dependent manner, suggesting that WT1 is involved in the development and progression of MPNSTs, and that it could be a potential therapeutic target for MPNSTs.
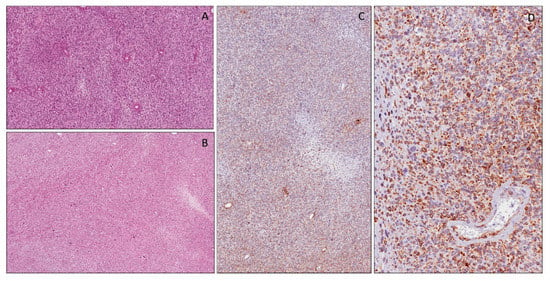
Figure 7.
Peripheral nerve sheath tumor consisting of spindle cells arranged in a fascicular pattern (A,B). Immunohistochemical cytoplasmic expression of WT1 (N-terminus) in neoplastic cells at low (C) and high (D) magnification.
2.15. WT1 Expression in Desmoplastic Small Round Cell Tumors (DSRCTs)
DSRCTs are highly aggressive tumors, typically with intra-abdominal localization (pelvic peritoneum, mesentery, surface of the liver and omentum), but can arise in many other sites, including meninges, scalp, pleura, paranasal sinuses, parotid gland, pancreas, scrotum, ovary, kidney and bone [134]. These tumors are typical of pediatric and adolescent age, with a male predilection. Histologically, they are composed of round cells with scant cytoplasm, indistinct cell borders and hyperchromatic round to oval, or slightly angulated, nuclei that have finely granular chromatin and small nucleoli. The cells are arranged in variably-sized nests, trabeculae, or lobules, usually separated by a prominent fibro-sclerotic stroma. In some cases, necrosis in the central portion and calcification may be seen. Mitoses are usually observed. In rare cases neoplastic cells with glandular or pseudo-rosettes formation and rhabdoid or signet ring appearance have been reported [134]. Immunohistochemically, the neoplastic cells show a polyphenotypic profile, including desmin, vimentin and epithelial markers, such as epithelial membrane antigen and cytokeratin and WT1 (Figure 8) [134].
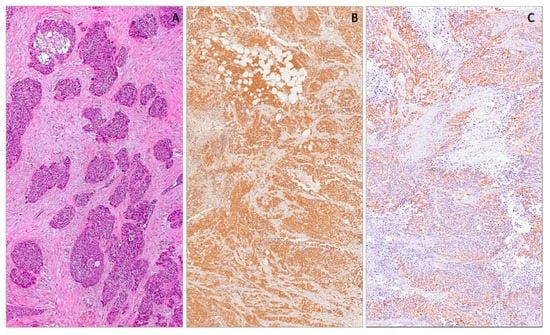
Figure 8.
Desmoplastic small round cell tumor showing neoplastic cells arranged in variably-sized nests, separated by a prominent fibro-sclerotic stroma (A). Neoplastic cells showing diffuse nuclear expression with WT1 (C-terminus) (B) and cytoplasmic expression with WT1 (N-terminus) (C).
In most cases (>90%) of desmoplastic small round cell tumors, strong nuclear staining with antibodies directed against the C-terminal portion of WT1 protein (clone WT1 C19), due to a recurrent chromosomal translocation (11;22)(p13;q12) resulting in the EWSR1-WT1 fusion transcript, is observed [134,135,136]. In rare cases, unusual nuclear WT1 staining with N-terminus antibodies was described, and was due, probably, to novel fusion transcripts [35,43,137].
2.16. WT1 Expression in Malignant Rhabdoid Tumors
Malignant rhabdoid tumors are highly aggressive tumors that usually occur in the kidneys of children. Other sites are rarely affected, such as somatic soft tissues, liver, gastrointestinal tract, pelvis, retroperitoneum, abdomen, heart and central nervous system [138]. Interestingly, soft tissue localization prevails in fetuses, newborns and young children, while in adolescents, renal and nervous system sites, they are more frequent [138]. In some cases, tumors are metastatic at presentation and have a fatal course [138]. Histologically, they show solid or trabecular growth patterns composed of round/epithelioid to polygonal cells. Typically, neoplastic cells have abundant, deeply eosinophilic cytoplasm with a paranuclear eosinophilic, PAS-positive inclusion and large, round, vescicular nuclei with finely dispersed chromatin, containing a prominent eosinophilic nucleolus. Mitoses and necrosis are commonly seen. In some cases, minor tumor components consisting of smaller, round, undifferentiated cells with a scant cytoplasmic rim may be present or even prominent. Similarly to desmoplastic small round cell tumors, malignant rhabdoid tumors exhibit polyphenotipic profiles, with variable co-expression of different markers, including vimentin, cytokeratins, epithelial membrane antigen (EMA) and CD99 [138]. However, the complete absence of nuclear immunoreactivity for INI1 protein is the most useful diagnostic clue [139,140,141,142]. Tumors can express additional markers, such as muscle specific actin, alpha-smooth muscle actin, S100 protein, synapthophisin, and CD56. Occasionally, WT1 (clone WT-C19) can be expressed at nuclear or nucleo-cytoplasmic levels (Figure 9) [33,34,138]. We have experience of similar results also by using antibodies against the N-terminal portion (clone WT 6F-H2) [143].
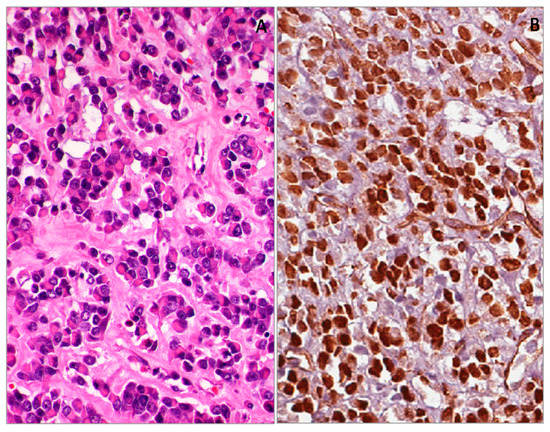
Figure 9.
Malignant rhabdoid tumor of the kidney (A). Diffuse and strong nuclear WT1 (N-terminus) expression of neoplastic cell (B).
2.17. WT1 Expression in Rhabdomyosarcomas
Rhabdomyosarcomas (RMSs) are malignant tumors composed of cells that show evidence of skeletal muscle differentiation. Based on morphological, immunohistochemical and molecular features, at least four major subtypes can be recognized: (i) embryonal; (ii) alveolar; (iii) spindle cell/sclerosing; and (iv) pleomorphic. Embryonal, alveolar and spindle cell/sclerosing rhabdomyosarcomas are predominantly tumors of children and adolescents, while the pleomorphic subtype tends to occur in adults. Histologically, the typical pattern of embryonal rhabdomyosarcoma consists of small, round or spindle-shaped cells, admixed with variable numbers of round, strap-, or tadpole-shaped eosinophilic rhabdomyoblasts set in a myxoid stroma. In 20–30% of cases, cytoplasmic cross striations are present. Spindle cell/sclerosing rhabdomyosarcoma most commonly arises in the head and neck or paratesticular soft tissues, and shows a striking male predilection. Alveolar rhabdomyosarcoma is a distinct subtype of rhabdomyosarcoma usually associated with an aggressive behavior. It consists of larger, more rounded, undifferentiated cells with larger nuclei than those in the embryonal variant, admixed with variable numbers of eosinophilic rhabdomyoblasts and multinucleate giant cells with peripheral nuclei. These cells are most often arranged in an alveolar pattern. Spindle cell rhabdomyosarcomas are mainly composed of primitive-looking spindle, ovoid, or round cells, often associated with pseudovascular clefts, embedded in a prominent hyalinized collagenous stroma. Small foci of obvious rhabdomyoblastic differentiation may be evident. Immunohistochemically, regardless of histological type (embryonal, alveolar, spindle/sclerosing), albeit with a variable extension, desmin, myogenin and MyoD1 are considered the most sensitive and specific markers of skeletal muscle differentiation [144]. Several studies have reported a diffuse and strong cytoplasmic expression of WT1 N-terminus antibodies (cloneWT 6F-H2) in all variants of rhabdomyosarcomas, including embryonal, sclerosing/spindle cell and alveolar (Figure 10) [35,36,41,45,143,145]. These results are consistent with those reported in studies on WT1 expression in the cytoplasm of human developing myoblasts and myotubes during the early phases of skeletal myogenesis [37,41,42,46,52,146].
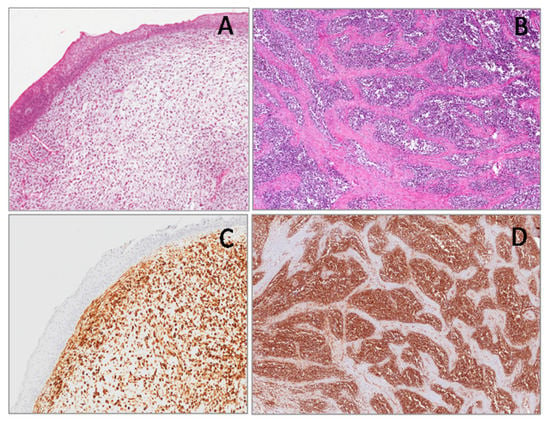
Figure 10.
Embryonal rhabdomyosarcoma, botryoid type (A) and alveolar rhabdomyosarcoma (B). Neoplastic cells showing strong and diffuse cytoplasmic WT1 (N-terminus) staining (C,D).
2.18. WT1 Expression in Neuroblastic Tumors
Neuroblastic tumors arise in children and adolescents. They occur especially in the adrenal gland, retroperitoneum or, and more rarely, in the posterior mediastinum. On the basis of a different degree of differentiation of immature neuroblasts in mature ganglionic cells, three variants of neuroblastic tumors are identified: (i) neuroblastoma in which the schwannian stroma component is poor, including undifferentiated, poorly differentiated and differentiating neuroblastomas; (ii) ganglioneuroblastoma in which the schwannian stroma component is dominant, but foci of neuroblasts are still present, including intermixed and nodular ganglioneuroblastomas; (iii) ganglioneuroma in which the schwannian stroma is predominant and neuroblasts are absent, including maturing and mature ganglioneuromas [147]. Neuroblastoma consist of small, round cells, with scant cytoplasm and round, hyperchromatic nuclei, arranged around a fibrillar area (neuropil). The neuroblastoma is classified as poorly differentiated if ≤5% of the tumor cells show ganglionic differentiation, while, if the neuroblastic component shows ganglionic differentiation in more than 5% of the tumor cells, the neuroblastoma is classified as differentiating; the neuroblastoma is classified as undifferentiated when ganglionic differentiation is almost or completely absent [147]. Ganglioneuroblastoma is a neuroblastic tumor with intermediate features of differentiation between neuroblastoma and ganglioneuroma. Histologically, two variants are recognized, intermixed and nodular. In the former, the ganglioneuromatous component is associated with a collection of immature ganglion cells that are interspersed among the schwannian stroma, while in the latter, in addition to the ganglioneuromatous component, there is a well-circumscribed area of neuroblastoma [147]. When the tumor is composed of mature or maturing ganglion cells surrounded by fascicles of Schwann cells, it is called ganglioneuroma [147]. This tumor model perfectly summarizes what happens during developmental stages of a normal peripheral sympathetic nervous system [148,149,150,151,152,153]. In some studies, a focal and weak nuclear WT1 staining in neuroblastoma was reported [35,135,145]. Wang et al. showed WT1 expression preferentially in ganglioneuroblastoma and ganglioneuroma [154].
In our experience, by using antibodies against the N-terminal portion of the WT1 protein (clone WT6F-H2), a variable cytoplasmic WT1 staining was found only in the ganglion cell component of both ganglioneuroblastoma and ganglioneuroma, while the neuroblastic component was negative (Figure 11) [143].
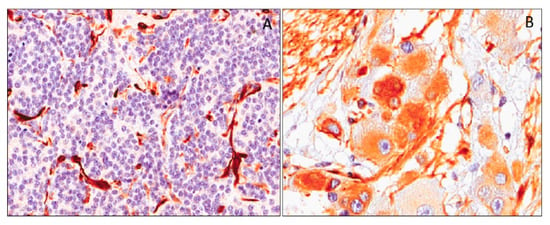
Figure 11.
Neoplastic cells of poorly-differentiated neuroblastoma showing no immunohistochemical expression of WT1 (N-terminus) (A). Ganglion cell of a maturing ganglioneuroma showing heterogeneous cytoplasmic WT1 (N-terminus) expression (B).
2.19. Infantile-Type Fibromatoses
Young-type fibromatoses are a group of fibroblastic and myofibroblastic tumor and tumor-like lesions occurring in the soft tissues of children and adolescents. This group includes the fibrous hamartoma of infancy, myofibroma/myofibromatosis and lipofibromatosis. The biological behavior of these lesions is variable, with lesions showing a benign course such as fibrous hamartomatous of infancy, and lesions with a tendency to local recurrence such as myofibroma/myofibromatosis and lipofibromatosis. Although each of these entities exhibits characteristic morphological features, differential diagnostic problems may arise especially from small biopsy specimens. Interesting results have emerged from the study of WT1 in these lesions. All cases of young-type fibromatoses, including fibrous hamartoma of infancy, myofibroma/myofibromatosis and lipofibromatosis, exhibited a diffuse WT1 cytoplasmic staining. On the contrary, all cases of adult-type fibromatoses and nodular fasciitis, from which young-type fibromatoses are to be distinguished, are not immunoreactive for WT1 [39]. Amini Nik et al. [15], investigated the expression of WT1 mRNA and protein in desmoid-type fibromatoses. They found that the levels of WT1 mRNA, revealed by TaqMan quantitative PCR, in all examined miofibroblastic tumor cells, were from medium to high, while contiguous normal fibroblasts exhibited a lower expression. WT1 protein overexpression was confirmed by Western blot and immunohistochemistry analyses. These data suggest that WT1 may play a role in the tumorigenesis of desmoid-type fibromatoses.
2.20. Congenital/Infantile Fibrosarcoma
This is a malignant tumor currently classified in the category of intermediate neoplasms. It occurs in the first two years of life, with a significant number of cases diagnosed at birth or antenatally [155]. It occurs in the soft tissues of the trunk and distal extremities, with only rare cases reported in the retroperitoneum. Distant metastases rarely occur and when they do, prognosis is favorable [155]. Histologically, it shows a fascicular pattern, focally with a herring-bone configuration; it consists predominantly of spindle cells showing only a mild to focally moderate degree of nuclear atypia. Mitoses are usually numerous. The diagnosis of fibrosarcoma is frequently of exclusion, being mainly based on negative results for specific lineage markers such as desmin, myogenin, CD34, S-100, HMB-45, and cytokeratins. Fibrosarcoma may variably express, even if with focal extension, alpha-smooth muscle actin and/or desmin. We recently reported a series of congenital/infantile fibrosarcoma with a strong and diffuse cytoplasmic expression of WT1 by using antibodies against the N-terminal portion of the WT1 protein (cloneWT6F-H2) (Figure 12) [39,143]. Then the WT1 protein could be useful as an immunomarker to support a diagnosis of fibrosarcoma, mostly in daily practice, in distinguishing congenital/infantile fibrosarcoma from desmoid-type fibromatosis.
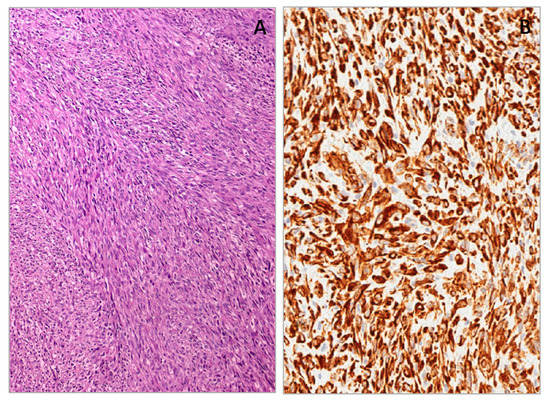
Figure 12.
Congenital/infantile fibrosarcoma with herring-bone configuration (A). Neoplastic cells showing diffuse cytoplasmic WT1 (N-terminus) expression (B).
Although both tumors may show overlapping morphological and immunohistochemical features (expression of alpha-smooth muscle actin), desmoid-type fibromatosis is typically WT1-negative. However, the diagnosis of congenital/infantile fibrosarcoma is usually confirmed by the identification of the recurrent translocation t (12;15)(p13;q25) with an ETV6-NTRK3 gene fusion [156,157].
2.21. WT1 Expression in Vascular Tumors
Vascular lesions are a heterogeneous group of tumors, including benign and malignant tumors, as well as malformations. Vascular tumors and malformations can be diagnostically challenging. Although they may initially appear very similar, they have distinct clinical courses and management. To provide a correct diagnosis, several studies have been conducted to identify immunohistochemical markers. Among the antibodies studied, interesting results have emerged from the study of WT1 cytoplasmic expression. Based on the studies available in the literature that compare vascular lesions, both benign and malignant, with malformations, it emerged that almost all benign lesions, such as capillary hemangioma, pyogenic granulomas, cherry angiomas and tufted angiomas, are WT1-positive, while lymphangioma and cavernous hemangioma are negative. Malignant tumors, such as angiosarcomas, hemangioendotheliomas and Kaposi’s sarcomas, are strongly WT1-positive (Figure 13). WT-1 expression in vascular malformations (angiokeratoma/verrucous hemangioma, combined vascular malformations, venous malformations, glomuvenous malformations, lymphatic malformations/lymphangioma, telangiectasia and targetoid hemosiderotic hemangioma) is generally completely negative or weak and focal positive. Interestingly, a strong and diffuse WT-1 staining was reported in a case of thrombosed vascular malformation with prominent endothelial hyperplasia [158,159,160]. WT1 expression in vascular tumors was also analyzed in fetal tissues, where a moderate to strong staining intensity in the cytoplasm of the endothelial cells of blood vessels was reported [37,52]. As WT1 staining has been obtained in the cytoplasm of embryonal/fetal endothelial cells, the reported cytoplasmic expression of this marker in many vascular tumors is not at all surprising.
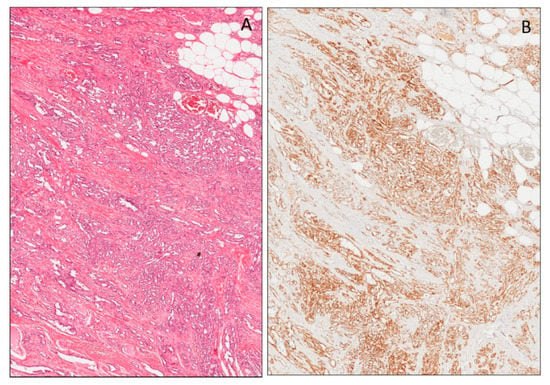
Figure 13.
Angiosarcoma (A). Diffuse nuclear expression of WT1 (N-terminus) in neoplastic cells (B).
2.22. Mammary Myofibroblastoma, Epithelioid Cell Variant
Myofibroblastomais a rare, benign tumor composed of both fibroblastic and myofibroblastic cells [161,162,163], which localizes typically in the breast [163], vulvovaginal region [164,165] and soft tissues. It is composed of a proliferation of bland-looking spindle-shaped cells to epithelioid cells set in a predominant fibrous stroma. When the epithelioid cell component predominates, the myofibroblastoma is referred to as “epithelioid”. It was observed that WT1 expression is limited to epithelioid-type myofibroblastoma, while all other variants (classic-type, collagenized/fibrotic-type, myxoid-type, lipomatous-type and palisaded/Schwannian-like) are completely negative [40]. Indeed, all cases reported of epithelioid-type myofibroblastomas exhibited a diffuse and strong WT1 cytoplasmic expression. In the evaluation of small breast biopsies, WT1 is an important marker, together with desmin, alpha-smooth muscle actin, Myogenin, MyoD1, h-caldesmon, S-100 protein, HMB45, EMA, Pancytokeratins and CD34, which are helpful in the differential diagnosis of lesions with epithelioid morphology.
Supplementary Materials
The supplementary materials are available online at https://www.mdpi.com/2076-3417/10/1/40/s1.
Author Contributions
Conceptualization, L.S. and G.M. (Gaetano Magro); Data curation, L.S., G.C., R.P., G.M.V., L.P., R.C., G.M. (Giuseppe Musumeci), G.M. (Gaetano Magro); Methodology, L.S., G.M.V. and G.M. (Gaetano Magro); Resources, G.M. (Gaetano Magro); Writing—original draft, L.S.; Writing—review & editing, L.S. and G.M. (Gaetano Magro). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Call, K.M.; Glaser, T.; Ito, C.Y.; Buckler, A.J.; Pelletier, J.; Haber, D.A.; Rose, E.A.; Kral, A.; Yeger, H.; Lewis, W.H. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990, 60, 509–520. [Google Scholar] [CrossRef]
- Haber, D.A.; Buckler, A.J.; Glaser, T.; Call, K.M.; Pelletier, J.; Sohn, R.L.; Douglass, E.C.; Housman, D.E. An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms’ tumor. Cell 1990, 61, 1257–1269. [Google Scholar] [CrossRef]
- Gessler, M.; Poustka, A.; Cavenee, W.; Neve, R.L.; Orkin, S.H.; Bruns, G.A. Homozygous deletion in Wilms’ tumours of a zinc-finger gene identified by chromosome jumping. Nature 1990, 343, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Mrowka, C.; Schedl, A. Wilms’ tumor suppressor gene WT1: From structure to renal pathophysiologic features. J. Am. Soc. Nephrol. 2000, 11 (Suppl. S16), S106–S115. [Google Scholar] [PubMed]
- Bruening, W.; Pelletier, J. A non-AUG translational initiation event generates novel WT1 isoforms. J. Biol. Chem. 1996, 271, 8646–8654. [Google Scholar] [CrossRef] [PubMed]
- Scharnhorst, V.; Dekker, P.; van der Eb, A.J.; Jochemsen, A.G. Internal translation initiation generates novel WT1 protein isoforms with distinct biological properties. J. Biol. Chem. 1999, 274, 23456–23462. [Google Scholar] [CrossRef]
- Haber, D.A.; Sohn, R.L.; Buckler, A.J.; Pelletier, J.; Call, K.M.; Housman, D.E. Alternative splicing and genomic structure of the Wilms’ tumor gene WT1. Proc. Natl. Acad. Sci. USA 1991, 88, 9618–9622. [Google Scholar] [CrossRef]
- Sharma, P.M.; Bowman, M.; Madden, S.L.; Rauscher, F.J., 3rd; Sukumar, S. RNA editing in the Wilms’ tumor susceptibility gene, WT1. Genes. Dev. 1994, 8, 720–731. [Google Scholar] [CrossRef]
- Miwa, H.; Beran, M.; Saunders, G.F. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 1992, 6, 405–409. [Google Scholar]
- Oji, Y.; Ogawa, H.; Tamaki, H.; Oka, Y.; Tsuboi, A.; Kim, E.H.; Soma, T.; Tatekawa, T.; Kawakami, M.; Asada, M.; et al. Expression of the Wilms’ tumor gene WT1 in solid tumors and its involvement in tumor cell growth. Jpn. J. Cancer Res. 1999, 90, 194–204. [Google Scholar] [CrossRef]
- Oji, Y.; Miyoshi, S.; Maeda, H.; Hayashi, S.; Tamaki, H.; Nakatsuka, S.; Yao, M.; Takahashi, E.; Nakano, Y.; Hirabayashi, H.; et al. Overexpression of the Wilms’ tumor gene WT1 in de novo lung cancers. Int. J. Cancer 2002, 100, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Koesters, R.; Linnebacher, M.; Coy, J.F.; Germann, A.; Schwitalle, Y.; Findeisen, P.; von Knebel Doeberitz, M. WT1 is a tumor-associated antigen in colon cancer that can be recognized by in vitro stimulated cytotoxic T cells. Int. J. Cancer 2004, 109, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Oji, Y.; Nakamori, S.; Fujikawa, M.; Nakatsuka, S.; Yokota, A.; Tatsumi, N.; Abeno, S.; Ikeba, A.; Takashima, S.; Tsujie, M.; et al. Overexpression of the Wilms’ tumor gene WT1 in pancreatic ductal adenocarcinoma. Cancer Sci. 2004, 95, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Loeb, D.M.; Evron, E.; Patel, C.B.; Sharma, P.M.; Niranjan, B.; Buluwela, L.; Weitzman, S.A.; Korz, D.; Sukumar, S. Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001, 61, 921–925. [Google Scholar]
- Amini Nik, S.; Hohenstein, P.; Jadidizadeh, A.; Van Dam, K.; Bastidas, A.; Berry, R.L.; Patek, C.E.; Van der Schueren, B.; Cassiman, J.J.; Tejpar, S. Upregulation of Wilms’ tumor gene 1 (WT1) in desmoid tumors. Int. J. Cancer 2005, 114, 202–208. [Google Scholar] [CrossRef]
- Shimizu, M.; Toki, T.; Takagi, Y.; Konishi, I.; Fujii, S. Immunohistochemical detection of the Wilms’ tumor gene (WT1) in epithelial ovarian tumors. Int. J. Gynecol. Pathol. 2000, 19, 158–163. [Google Scholar] [CrossRef]
- Andersson, C.; Oji, Y.; Ohlson, N.; Wang, S.; Li, X.; Ottander, U.; Lundin, E.; Sugiyama, H.; Li, A. Prognostic significance of specific anti-WT1 IgG antibody level in plasma in patients with ovarian carcinoma. Cancer Med. 2014, 3, 909–918. [Google Scholar] [CrossRef]
- Oji, Y.; Suzuki, T.; Nakano, Y.; Maruno, M.; Nakatsuka, S.; Jomgeow, T.; Abeno, S.; Tatsumi, N.; Yokota, A.; Aoyagi, S.; et al. Overexpression of the Wilms’ tumor gene WT1 in primary astrocytic tumors. Cancer Sci. 2004, 95, 822–827. [Google Scholar] [CrossRef]
- Menssen, H.D.; Bertelmann, E.; Bartelt, S.; Schmidt, R.A.; Pecher, G.; Schramm, K.; Thiel, E. Wilms’ tumor gene (WT1) expression in lung cancer, colon cancer and glioblastoma cell lines compared to freshly isolated tumor specimens. J. Cancer Res. Clin. Oncol. 2000, 126, 226–232. [Google Scholar] [CrossRef]
- Athale, U.H.; Shurtleff, S.A.; Jenkins, J.J.; Poquette, C.A.; Tan, M.; Downing, J.R.; Pappo, A.S. Use of reverse transcriptase polymerase chain reaction for diagnosis and staging of alveolar rhabdomyosarcoma, Ewing sarcoma family of tumors, and desmoplastic small round cell tumor. J. Pediatr. Hematol. Oncol. 2001, 23, 99–104. [Google Scholar] [CrossRef]
- Wagner, N.; Michiels, J.F.; Schedl, A.; Wagner, K.D. The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: Expression in tumour vessels in vivo. Oncogene 2008, 27, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Cherfils-Vicini, J.; Hosen, N.; Hohenstein, P.; Gilson, E.; Hastie, N.D.; Michiels, J.F.; Wagner, N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 2014, 5, 5852. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Haber, D.A. Wilms tumor and the WT1 gene. Exp. Cell. Res. 2001, 264, 74–99. [Google Scholar] [CrossRef] [PubMed]
- Little, M.; Holmes, G.; Walsh, P. WT1: What has the last decade told us? Bioessays 1999, 21, 191–202. [Google Scholar] [CrossRef]
- Davies, R.; Moore, A.; Schedl, A.; Bratt, E.; Miyahawa, K.; Ladomery, M.; Miles, C.; Menke, A.; van Heyningen, V.; Hastie, N. Multiple roles for the Wilms’ tumor suppressor, WT1. Cancer Res. 1999, 59, 1747s–1750s. [Google Scholar]
- Hastie, N.D. Life, sex, and WT1 isoforms--three amino acids can make all the difference. Cell 2001, 106, 391–394. [Google Scholar] [CrossRef]
- Niksic, M.; Slight, J.; Sanford, J.R.; Caceres, J.F.; Hastie, N.D. The Wilms’ tumour protein (WT1) shuttles between nucleus and cytoplasm and is present in functional polysomes. Hum. Mol. Genet. 2004, 13, 463–471. [Google Scholar] [CrossRef]
- Scholz, H.; Kirschner, K.M. A role for the Wilms’ tumor protein WT1 in organ development. Physiology 2005, 20, 54–59. [Google Scholar] [CrossRef]
- Roberts, S.G. Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 2005, 15, 542–547. [Google Scholar] [CrossRef]
- Hohenstein, P.; Hastie, N.D. The many facets of the Wilms’ tumour gene, WT1. Hum. Mol. Genet. 2006, 15, 196–201. [Google Scholar] [CrossRef]
- Wagner, K.D.; El Maï, M.; Ladomery, M.; Belali, T.; Leccia, N.; Michiels, J.F.; Wagner, N. Altered VEGF splicing isoform balance in tumor endothelium involves activation of splicing factors Srpk1 and Srsf1 by the Wilms’ tumor suppressor Wt1. Cells 2019, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Ramani, P.; Cowell, J.K. The expression pattern of Wilms’ tumour gene (WT1) product in normal tissues and paediatric renal tumours. J. Pathol. 1996, 179, 162–168. [Google Scholar] [CrossRef]
- Charles, A.K.; Mall, S.; Watson, J.; Berry, P.J. Expression of the Wilms’ tumour gene WT1 in the developing human and in paediatric renal tumours: An immunohistochemical study. Mol. Pathol. 1997, 50, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Carpentieri, D.F.; Nichols, K.; Chou, P.M.; Matthews, M.; Pawel, B.; Huff, D. The expression of WT1 in the differentiation of rhabdomyosarcoma from other pediatric small round blue cell tumors. Mod. Pathol. 2002, 15, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Bisceglia, M.; Vairo, M.; Galliani, C.; Lastilla, G.; Parafioriti, A.; De Maglio, G. Immunohistochemical investigation of WT1 expression in 117 embryonal tumors. Pathologica 2011, 103, 182–183. [Google Scholar]
- Parenti, R.; Perris, R.; Vecchio, G.M.; Salvatorelli, L.; Torrisi, A.; Gravina, L.; Magro, G. Immunohistochemical expression of Wilms’ tumor protein (WT1) in developing human epithelial and mesenchymal tissues. Acta Histochem. 2013, 115, 70–75. [Google Scholar] [CrossRef]
- Parenti, R.; Puzzo, L.; Vecchio, G.M.; Gravina, L.; Salvatorelli, L.; Musumeci, G.; Vasquez, E.; Magro, G. Immunolocalization of Wilms’ Tumor protein (WT1) in developing human peripheral sympathetic and gastroenteric nervous system. Acta Histochem. 2014, 116, 48–54. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Vecchio, G.M.; Musumeci, G.; Rita, A.; Parenti, R. Cytoplasmic expression of Wilms’ tumor transcription factor-1 (WT1): A useful immunomarker for young-type fibromatoses and infantile fibrosarcoma. Acta Histochem. 2014, 116, 1134–1140. [Google Scholar] [CrossRef]
- Magro, G.; Longo, F.; Salvatorelli, L.; Vecchio, G.M.; Parenti, R. Wilms’ tumor protein (WT1) in mammary myofibroblastoma: An immunohistochemical study. Acta Histochem. 2014, 116, 905–910. [Google Scholar] [CrossRef]
- Magro, G.; Salvatorelli, L.; Puzzo, L.; Musumeci, G.; Bisceglia, M.; Parenti, R. Oncofetal expression of Wilms’ tumor 1 (WT1) protein in human fetal, adult and neoplastic skeletal muscle tissues. Acta Histochem. 2015, 117, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Longo, F.R.; Angelico, G.; Spadola, S.; Amore, F.F.; Salvatorelli, L. Immunohistochemistry as potential diagnostic pitfall in the most common solid tumors of children and adolescents. Acta Histochem. 2015, 117, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, S.; Oji, Y.; Horiuchi, T.; Kanda, T.; Kitagawa, M.; Takeuchi, T.; Kawano, K.; Kuwae, Y.; Yamauchi, A.; Okumura, M.; et al. Immunohistochemical detection of WT1 protein in a variety of cancer cells. Mod. Pathol. 2006, 19, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, J.; Thiericke, J.; Nagel, C.; Meyermann, R.; Beschorner, R. WT1 expression in normal and neoplastic cranial and peripheral nerves is independent of grade of malignancy. Cancer Biomark. 2010, 7, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Bisceglia, M.; Magro, G.; Carosi, I.; Cannazza, V.; Ben Dor, D. Primary embryonal rhabdomyosarcoma of the prostate in adults: Report of a case and review of the literature. Int. J. Surg. Pathol. 2011, 19, 831–837. [Google Scholar] [CrossRef]
- Salvatorelli, L.; Bisceglia, M.; Vecchio, G.; Parenti, R.; Galliani, C.; Alaggio, R. A comparative immunohistochemical study of oncofetalcy-toplasmic WT1 expression in human fetal, adult and neoplasticskeletal muscle. Pathologica 2011, 103, 186. [Google Scholar]
- Singh, A.; Mishra, A.K.; Ylaya, K.; Hewitt, S.M.; Sharma, K.C.; Saxena, S. Wilms’ tumor-1, claudin-1 and ezrin are useful immunohistochemical markers that help to distinguish schwannoma from fibroblastic meningioma. Pathol. Oncol. Res. 2012, 18, 383–389. [Google Scholar] [CrossRef]
- Pritchard-Jones, K.; Fleming, S.; Davidson, D.; Bickmore, W.; Porteous, D.; Gosden, C.; Bard, J.; Buckler, A.; Pelletier, J.; Housman, D. The candidate Wilms’ tumour gene is involved in genitourinary development. Nature 1990, 346, 194–197. [Google Scholar] [CrossRef]
- Sharma, P.M.; Yang, X.; Bowman, M.; Roberts, V.; Sukumar, S. Molecular-cloning of rat Wilms’ tumor complementary DNA and a study of messenger RNA expression in the urogenital system and the brain. Cancer Res. 1992, 52, 6407–6412. [Google Scholar]
- Armstrong, J.F.; Pritchard-Jones, K.; Bickmore, W.A.; Hastie, N.D.; Bard, J.B. The expression of the Wilms’ tumour gene, WT1, in the developing mammalian embryo. Mech. Dev. 1993, 4, 85–97. [Google Scholar] [CrossRef]
- Mundlos, S.; Pelletier, J.; Darveau, A.; Bachmann, M.; Winterpacht, A.; Zabel, B. Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development 1993, 119, 1329–1341. [Google Scholar] [PubMed]
- Parenti, R.; Salvatorelli, L.; Musumeci, G.; Parenti, C.; Giorlandino, A.; Motta, F.; Magro, G. Wilms’ tumor 1 (WT1) protein expression in human developing tissues. Acta Histochem. 2015, 117, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.A.; Ladomery, M.; Hohenstein, P.; Michael, L.; Shafe, A.; Spraggon, L.; Hastie, N. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum. Mol. Genet. 2004, 13, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Miller-Hodges, E.; Hohenstein, P. WT1 in disease: Shifting the epithelial-mesenchymal balance. J. Pathol. 2012, 226, 229–240. [Google Scholar] [CrossRef]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 is required for early kidney development. Cell 1993, 74, 679–691. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D.; Scholz, H.; Kirschner, K.M.; Schedl, A. Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms’ tumor suppressor Wt1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R779–R787. [Google Scholar] [CrossRef]
- Wagner, K.D.; Wagner, N.; Vidal, V.P.; Schley, G.; Wilhelm, D.; Schedl, A.; Englert, C.; Scholz, H. The Wilms’ tumor gene Wt1 is required for normal development of the retina. EMBO J. 2002, 21, 1398–1405. [Google Scholar] [CrossRef]
- Clark, A.J.; Ware, J.L.; Chen, M.Y.; Graf, M.R.; Van Meter, T.E.; Dos Santos, W.G.; Fillmore, H.L.; Broaddus, W.C. Effect of WT1 gene silencing on the tumorigenicity of human glioblastoma multiforme cells. J. Neurosurg. 2010, 112, 18–25. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar] [CrossRef]
- Moore, A.W.; McInnes, L.; Kreidberg, J.; Hastie, N.D.; Schedl, A. YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 1999, 126, 1845–1857. [Google Scholar]
- Martínez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Wagner, K.D.; Theres, H.; Englert, C.; Schedl, A.; Scholz, H. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms’ tumor transcription factor Wt1. Genes Dev. 2005, 19, 2631–2642. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.Y.; Hastie, N.D. The role of Wt1 in regulating mesenchyme in cancer, development, and tissue homeostasis. Trends Genet. 2012, 28, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussain, T.; Ali, A.; Akhtar, M. Wilms tumor: An update. Adv. Anat. Pathol. 2014, 21, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, J.B. Wilms’ tumor and other renal tumors of childhood: A selective review from the National Wilms’ Tumor Study Pathology Center. Hum. Pathol. 1983, 14, 481–492. [Google Scholar] [CrossRef]
- Charles, A.K.; Brown, K.W.; Berry, P.J. Microdissecting the genetic events in nephrogenic rests and Wilms’ tumor development. Am. J. Pathol. 1998, 153, 991–1000. [Google Scholar] [CrossRef]
- Marsden, H.B.; Lawler, W. Primary renal tumours in the first year of life. A population based review. Virchows. Arch. A Pathol. Anat. Histopathol. 1983, 399, 1–9. [Google Scholar]
- Garvin, A.J.; Surrette, F.; Hintz, D.S.; Rudisill, M.T.; Sens, M.A.; Sens, D.A. The in vitro growth and characterization of the skeletal muscle component of Wilms’ tumor. Am. J. Pathol. 1985, 121, 298–310. [Google Scholar]
- Wigger, H.J. Fetal rhabdomyomatous nephroblastoma-a variant of Wilms’ tumor. Hum. Pathol. 1976, 7, 613–623. [Google Scholar] [CrossRef]
- Attanoos, R.L.; Gibbs, A.R. Pathology of malignant mesothelioma. Histopathology 1997, 30, 403–418. [Google Scholar] [CrossRef]
- McCaughey, W.T.; Al-Jabi, M. Differentiation of serosal hyperplasia and neoplasia in biopsies. Pathol. Annu. 1986, 21, 271–293. [Google Scholar] [PubMed]
- Klebe, S.; Brownlee, S.A.; Mahar, A.; Burchette, J.L.; Sporn, T.A.; Vollmer, R.T.; Roggli, V.L. Sarcomatoid mesothelioma: A clinical-pathologic correlation of 326 cases. Mod. Pathol. 2010, 23, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Oates, J.; Edwards, C. HBME-1, MOC-31, WT1 and calretinin: An assessment of recently described markers for mesothelioma and adenocarcinoma. Histopathology 2000, 36, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, N.G. Value of thyroid transcription factor-1, E-cadherin, BG8, WT1, and CD44S immunostaining in distinguishing epithelial pleural mesothelioma from pulmonary and non pulmonary adenocarcinoma. Am. J. Surg. Pathol. 2000, 24, 598–606. [Google Scholar] [CrossRef]
- Ordóñez, N.G. The immunohistochemical diagnosis of mesothelioma: A comparative study of epithelioid mesothelioma and lung adenocarcinoma. Am. J. Surg. Pathol. 2003, 27, 1031–1051. [Google Scholar] [CrossRef]
- Ordóñez, N.G. The diagnostic utility of immunohistochemistry in distinguishing between mesothelioma and renal cell carcinoma: A comparative study. Hum. Pathol. 2004, 35, 697–710. [Google Scholar] [CrossRef]
- Kushitani, K.; Takeshima, Y.; Amatya, V.J.; Furonaka, O.; Sakatani, A.; Inai, K. Differential diagnosis of sarcomatoid mesothelioma from true sarcoma and sarcomatoid carcinoma using immunohistochemistry. Pathol. Int. 2008, 58, 75–83. [Google Scholar] [CrossRef]
- Tsuta, K.; Kato, Y.; Tochigi, N.; Hoshino, T.; Takeda, Y.; Hosako, M.; Maeshima, A.M.; Asamura, H.; Kondo, T.; Matsuno, Y. Comparison of different clones (WT49 versus 6F-H2) of WT-1 antibodies for immunohistochemical diagnosis of malignant pleural mesothelioma. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 126–130. [Google Scholar] [CrossRef]
- Harwood, T.R.; Gracey, D.R.; Yokoo, H. Pseudomesotheliomatous carcinoma of the lung. A variant of peripheral lung. Cancer Am. J. Clin. Pathol. 1976, 65, 159–167. [Google Scholar] [CrossRef]
- Goldstein, N.S.; Bassi, D.; Uzieblo, A. WT1 is an integral component of an antibody panel to distinguish pancreaticobiliary and some ovarian epithelial neoplasms. Am. J. Clin. Pathol. 2001, 116, 246–252. [Google Scholar] [CrossRef]
- Lee, B.H.; Hecht, J.L.; Pinkus, J.L.; Pinkus, G.S. WT1, estrogen receptor, and progesterone receptor as markers for breast or ovarian primary sites in metastatic adenocarcinoma to body fluids. Am. J. Clin. Pathol. 2002, 117, 745–750. [Google Scholar] [CrossRef]
- Hashi, A.; Yuminamochi, T.; Murata, S.; Iwamoto, H.; Honda, T.; Hoshi, K. Wilms’ tumor gene immunoreactivity in primary serous carcinomas of the fallopian tube, ovary, endometrium, and peritoneum. Int. J. Gynecol. Pathol. 2003, 22, 374–377. [Google Scholar] [CrossRef]
- Logani, S.; Oliva, E.; Amin, M.B.; Folpe, A.L.; Cohen, C.; Young, R.H. Immunoprofile of ovarian tumors with putative transitional cell (urothelial) differentiation using novel urothelial markers: Histogenetic and diagnostic implications. Am. J. Surg. Pathol. 2003, 27, 1434–1441. [Google Scholar] [CrossRef]
- Hecht, J.L.; Lee, B.H.; Pinkus, J.L.; Pinkus, G.S. The value of Wilms’ tumor susceptibility gene 1 in cytologic preparations as a marker for malignant mesothelioma. Cancer 2002, 96, 105–109. [Google Scholar] [CrossRef]
- Goldstein, N.S.; Uzieblo, A. WT1 immunoreactivity in uterine papillary serous carcinomas is different from ovarian serous carcinomas. Am. J. Clin. Pathol. 2002, 117, 541–545. [Google Scholar] [CrossRef]
- Al-Hussaini, M.; Stockman, A.; Foster, H.; McCluggage, W.G. WT-1 assists in distinguishing ovarian from uterine serous carcinoma and in distinguishing between serous and endometrioid ovarian carcinoma. Histopathology 2004, 44, 109–115. [Google Scholar] [CrossRef]
- Acs, G.; Pasha, T.; Zhang, P.J. WT1 is differentially expressed in serous, endometrioid, clear cell, and mucinous carcinomas of the peritoneum, fallopian tube, ovary, and endometrium. Int. J. Gynecol. Pathol. 2004, 23, 110–118. [Google Scholar] [CrossRef]
- Gilks, C.B. Subclassification of ovarian surface epithelial tumors based on correlation of histologic and molecular pathologic data. Int. J. Gynecol. Pathol. 2004, 23, 200–205. [Google Scholar] [CrossRef]
- Waldstrøm, M.; Grove, A. Immunohistochemical expression of Wilms’ tumor gene protein in different histologic subtypes of ovarian carcinomas. Arch. Pathol. Lab. Med. 2005, 129, 85–88. [Google Scholar]
- Taube, E.T.; Denkert, C.; Sehouli, J.; Kunze, C.A.; Dietel, M.; Braicu, I.; Letsch, A.; Darb-Esfahani, S. Wilms’ tumor protein 1 (WT1)—Not only a diagnostic but also a prognostic marker in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2016, 140, 494–502. [Google Scholar] [CrossRef]
- Norris, H.J.; Taylor, H.B. Prognosis of granulosa-theca tumors of the ovary. Cancer 1968, 21, 255–263. [Google Scholar] [CrossRef]
- Young, R.H.; Scully, R.E. Ovarian sex cord-stromal tumors with bizarre nuclei: A clinicopathologic analysis of 17 cases. Int. J. Gynecol. Pathol. 1983, 1, 325–335. [Google Scholar] [CrossRef]
- Gaffey, M.J.; Frierson, H.F., Jr.; Iezzoni, J.C.; Mills, S.E.; Clement, P.B.; Gersell, D.J.; Shashi, V.; von Kap-Herr, C.; Young, R.H. Ovarian granulosa cell tumors with bizarre nuclei: An immunohistochemical analysis with fluorescence in situ hybridization documenting trisomy 12 in the bizarre component [corrected]. Mod. Pathol. 1996, 9, 308–315. [Google Scholar]
- Young, R.H. Sex cord-stromal tumors of the ovary and testis: Their similarities and differences with consideration of selected problems. Mod. Pathol. 2005, 18, S81–S98. [Google Scholar] [CrossRef]
- Arora, D.S.; Cooke, I.E.; Ganesan, T.S.; Ramsdale, J.; Manek, S.; Charnock, F.M.; Groome, N.P.; Wells, M. Immunohistochemical expression of inhibin/activin subunits in epithelial and granulosa cell tumours of the ovary. J. Pathol. 1997, 181, 413–418. [Google Scholar] [CrossRef]
- Cathro, H.P.; Stoler, M.H. The utility of calretinin, inhibin, and WT1 immunohistochemical staining in the differential diagnosis of ovarian tumors. Hum. Pathol. 2005, 36, 195–201. [Google Scholar] [CrossRef]
- Deavers, M.T.; Malpica, A.; Liu, J.; Broaddus, R.; Silva, E.G. Ovarian sex cord-stromal tumors: An immunohistochemical study including a comparison of calretinin and inhibin. Mod. Pathol. 2003, 16, 584–590. [Google Scholar] [CrossRef]
- He, H.; Luthringer, D.J.; Hui, P.; Lau, S.K.; Weiss, L.M.; Chu, P.G. Expression of CD56 and WT1 in ovarian stroma and ovarian stromal tumors. Am. J. Surg. Pathol. 2008, 32, 884–890. [Google Scholar] [CrossRef]
- Zhao, C.; Bratthauer, G.L.; Barner, R.; Vang, R. Diagnostic utility of WT1 immunostaining in ovarian Sertoli cell tumor. Am. J. Surg. Pathol. 2007, 31, 1378–1386. [Google Scholar] [CrossRef]
- Zhao, C.; Vinh, T.N.; McManus, K.; Dabbs, D.; Barner, R.; Vang, R. Identification of the most sensitive and robust immunohistochemical markers in different categories of ovarian sex cord-stromal tumors. Am. J. Surg. Pathol. 2009, 33, 354–366. [Google Scholar] [CrossRef]
- Nasomyon, T.; Samphao, S.; Sangkhathat, S.; Mahattanobon, S.; Graidist, P. Correlation of Wilms’ tumor 1 isoforms with HER2 and ER-α and its oncogenic role in breast Cancer. AntiCancer Res. 2014, 34, 1333–1342. [Google Scholar]
- Domfeh, A.B.; Carley, A.L.; Striebel, J.M.; Karabakhtsian, R.G.; Florea, A.V.; McManus, K.; Beriwal, S.; Bhargava, R. WT1 immunoreactivity in breast carcinoma: Selective expression in pure and mixed mucinous subtypes. Mod. Pathol. 2008, 21, 1217–1223. [Google Scholar] [CrossRef]
- Hwang, H.; Quenneville, L.; Yaziji, H.; Gown, A.M. Wilms tumor gene product: Sensitive and contextually specific marker of serous carcinomas of ovarian surface epithelial origin. Appl. Immunohistochem. Mol. Morphol. 2004, 12, 122–126. [Google Scholar] [CrossRef]
- Lee, A.H.; Paish, E.C.; Marchio, C.; Sapino, A.; Schmitt, F.C.; Ellis, I.O.; Reis-Filho, J.S. The expression of Wilms’ tumour-1 and Ca125 in invasive micropapillary carcinoma of the breast. Histopathology 2007, 51, 824–828. [Google Scholar] [CrossRef]
- Oh, E.J.; Koo, J.S.; Kim, J.Y.; Jung, W.H. Correlation between solid papillary carcinoma and associated invasive carcinoma according to expression of WT1 and several MUCs. Pathol. Res. Pract. 2014, 210, 953–958. [Google Scholar] [CrossRef]
- Artibani, M.; Sims, A.H.; Slight, J.; Aitken, S.; Thornburn, A.; Muir, M.; Brunton, V.G.; Del-Pozo, J.; Morrison, L.R.; Katz, E.; et al. WT1 expression in breast cancer disrupts the epithelial/mesenchymal balance of tumour cells and correlates with the metabolic response to docetaxel. Sci. Rep. 2017, 7, 45255. [Google Scholar] [CrossRef]
- Choi, E.J.; Yun, J.A.; Jeon, E.K.; Won, H.S.; Ko, Y.H.; Kim, S.Y. Prognostic significance of RSPO1, WNT1, P16, WT1, and SDC1 expressions in invasive ductal carcinoma of the breast. World J. Surg. Oncol. 2013, 11, 314. [Google Scholar] [CrossRef]
- Tuna, M.; Chavez-Reyes, A.; Tari, A.M. HER2/neu increases the expression of Wilms’ Tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene 2005, 24, 1648–1652. [Google Scholar] [CrossRef]
- Li, B.Q.; Huang, S.; Shao, Q.Q.; Sun, J.; Zhou, L.; You, L.; Zhang, T.P.; Liao, Q.; Guo, J.C.; Zhao, Y.P. WT1-associated protein is a novel prognostic factor in pancreatic ductal adenocarcinoma. Oncol. Lett. 2017, 13, 2531–2538. [Google Scholar] [CrossRef]
- Rodeck, U.; Bossler, A.; Kari, C.; Humphreys, C.W.; Györfi, T.; Maurer, J.; Thiel, E.; Menssen, H.D. Expression of the WT1 Wilms’ tumor gene by normal and malignant human melanocytes. Int. J. Cancer 1994, 59, 78–82. [Google Scholar] [CrossRef]
- Wilsher, M.; Cheerala, B. WT1 as a complementary marker of malignant melanoma: An immunohistochemical study of whole sections. Histopathology 2007, 51, 605–610. [Google Scholar] [CrossRef]
- Wagner, N.; Panelos, J.; Massi, D.; Wagner, K.D. The Wilms’ tumor suppressor WT1 is associated with melanoma proliferation. Pflug. Arch. 2000, 455, 839–847. [Google Scholar] [CrossRef]
- Michiels, J.F.; Perrin, C.; Leccia, N.; Massi, D.; Grimaldi, P.; Wagner, N. PPARbeta activation inhibits melanoma cell proliferation involving repression of theWilms’ tumour suppressor WT1. Pflug. Arch. 2010, 459, 689–703. [Google Scholar] [CrossRef][Green Version]
- Garrido-Ruiz, M.C.; Rodriguez-Pinilla, S.M.; Pérez-Gómez, B.; Rodriguez-Peralto, J.L. WT1 expression in nevi and melanomas: A marker of melanocytic invasion into the dermis. J. Cutan. Pathol. 2010, 37, 542–548. [Google Scholar] [CrossRef]
- Garrido Ruiz, M.C.; Requena, L.; Kutzner, H.; Ortiz, P.; Pérez-Gómez, B.; Rodriguez-Peralto, J.L. Desmoplastic melanoma: Expression of epithelial-mesenchymal transition-related proteins. Am. J. Dermatopathol. 2014, 36, 238–242. [Google Scholar] [CrossRef]
- Plaza, J.A.; Bonneau, P.; Prieto, V.; Sangueza, M.; Mackinnon, A.; Suster, D.; Bacchi, C.; Estrozi, B.; Kazakov, D.; Kacerovska, D.; et al. Desmoplastic melanoma: An updated immunohistochemical analysis of 40 cases with a proposal for an additional panel of stains for diagnosis. J. Cutan. Pathol. 2016, 43, 313–323. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef]
- Oji, Y.; Yamamoto, H.; Nomura, M.; Nakano, Y.; Ikeba, A.; Nakatsuka, S.; Abeno, S.; Kiyotoh, E.; Jomgeow, T.; Sekimoto, M.; et al. Overexpression of the Wilms’ tumor gene WT1 in colorectal adenocarcinoma. Cancer Sci. 2003, 94, 712–717. [Google Scholar] [CrossRef]
- Miyata, Y.; Kumagai, K.; Nagaoka, T.; Kitaura, K.; Kaneda, G.; Kanazawa, H.; Suzuki, S.; Hamada, Y.; Suzuki, R. Clinicopathological significance and prognostic value of Wilms’ tumor gene expression in colorectal Cancer. Cancer Biomark. 2015, 15, 789–797. [Google Scholar] [CrossRef]
- Aslan, A.; Erdem, H.; Celik, M.A.; Sahin, A.; Cankaya, S. Investigation of Insulin-Like Growth Factor-1 (IGF-1), P53, and Wilms’ Tumor 1 (WT1) Expression Levels in the Colon Polyp Subtypes in Colon Cancer. Med. Sci. Monit. 2019, 25, 5510–5517. [Google Scholar] [CrossRef]
- Nakahara, Y.; Okamoto, H.; Mineta, T.; Tabuchi, K. Expression of the Wilms’ tumor gene product WT1 in glioblastomas and medulloblastomas. Brain Tumor. Pathol. 2004, 21, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, J.; Mittelbronn, M.; Nguyen, T.D.; Meyermann, R.; Beschorner, R. WT1 expression distinguishes astrocytic tumor cells from normal and reactive astrocytes. Brain. Pathol. 2008, 18, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Hashiba, T.; Izumoto, S.; Kagawa, N.; Suzuki, T.; Hashimoto, N.; Maruno, M.; Yoshimine, T. Expression of WT1 protein and correlation with cellular proliferation in glial tumors. Neurol. Med. Chir. 2007, 47, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, J.; Beschorner, R.; Gierke, M.; Bisdas, S.; Braun, C.; Ebner, F.H.; Schittenhelm, J. WT1 expression increases with malignancy and indicates unfavourable outcome in astrocytoma. J. Clin. Pathol. 2014, 67, 556–561. [Google Scholar] [CrossRef]
- Sotobori, T.; Ueda, T.; Oji, Y.; Naka, N.; Araki, N.; Myoui, A.; Sugiyama, H.; Yoshikawa, H. Prognostic significance of Wilms’ tumor gene (WT1) mRNA expression in soft tissue sarcoma. Cancer 2006, 106, 2233–2240. [Google Scholar] [CrossRef]
- Oue, T.; Ueharaa, S.; Yamanakaa, H.; Takamaa, Y.; Ojib, Y.; Fukuzawa, M. Expression of Wilms’ tumor 1 gene in a variety of pediatric tumors. J. Ped. Surg. 2011, 46, 2233–2238. [Google Scholar] [CrossRef]
- Weiss, S.W.; Goldblum, J.R. Enzinger and Weiss’s Soft Tissue Tumors, 5th ed.; Mosby, J.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 903–925. [Google Scholar]
- Ducatman, B.S.; Scheithauer, B.W. Malignant peripheral nerve sheath tumors showing. Cancer 1984, 54, 1049–1057. [Google Scholar] [CrossRef]
- Wick, M.R.; Swanson, P.E.; Scheithauer, B.W.; Manivel, J.C. Malignant peripheral nerve sheath tumor. An immunohistochemical study of 62 cases. Am. J. Clin. Pathol. 1987, 87, 425–433. [Google Scholar] [CrossRef]
- Johnson, T.L.; Lee, M.W.; Meis, J.M.; Zarbo, R.J.; Crissman, J. Immunohistochemical characterization of malignant peripheral nerve sheath tumors. Surg. Pathol. 1991, 4, 121–135. [Google Scholar]
- Kim, A.; Park, E.Y.; Kim, K.; Lee, J.H.; Shin, D.H.; Kim, J.Y.; Park, D.Y.; Lee, C.H.; Sol, M.Y.; Choi, K.U.; et al. Prognostic significance of WT1 expression in soft tissue sarcoma. World J. Surg. Oncol. 2014, 12, 214. [Google Scholar] [CrossRef][Green Version]
- Ueda, T.; Oji, Y.; Naka, N.; Nakano, Y.; Takahashi, E.; Koga, S.; Asada, M.; Ikeba, A.; Nakatsuka, S.; Abeno, S.; et al. Overexpression of the Wilms’ tumor gene WT1 in human bone and soft-tissue sarcomas. Cancer Sci. 2003, 94, 271–276. [Google Scholar] [CrossRef]
- Parenti, R.; Cardile, V.; Graziano, A.C.; Parenti, C.; Venuti, A.; Bertuccio, M.P.; Furno, D.L.; Magro, G. Wilms’ tumor gene 1 (WT1) silencing inhibits proliferation of malignant peripheral nerve sheath tumor sNF96.2 cell line. PLoS ONE 2014, 9, e114333. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Ladanyi, M. Desmoplastic small round cell tumour. In WHO Classification of Tumours of Soft Tissue and Bone; Fletcher, C.D.M., Bridge, J.A., Hogendoorn, P.C.W., Mertens, F., Eds.; IARC: Lyon, France, 2013; pp. 225–227. [Google Scholar]
- Barnoud, R.; Sabourin, J.C.; Pasquier, D.; Ranchère, D.; Bailly, C.; Terrier-Lacombe, M.J.; Pasquier, B. Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: A comparative study with other small round cell tumors. Am. J. Surg. Pathol. 2000, 24, 830. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Pfeifer, J.D.; Marley, E.F.; Dehner, L.P.; Humphrey, P.A.; Zhu, X.; Swanson, P.E. WT1 staining reliably differentiates desmoplastic small round cell tumor from Ewing sarcoma/primitive neuroectodermal tumor. An immunohistochemical and molecular diagnostic study. Am. J. Clin. Pathol. 2000, 114, 345–353. [Google Scholar] [CrossRef]
- Murphy, A.J.; Bishop, K.; Pereira, C.; Chilton-MacNeill, S.; Ho, M.; Zielenska, M.; Thorner, P.S. A new molecular variant of desmoplastic small round cell tumor: Significance of WT1 immunostaining in this entity. Hum. Pathol. 2008, 39, 1763–1770. [Google Scholar] [CrossRef]
- Alaggio, R.; Coffin, C.M.; Vargas, S.O. Soft tissue tumors of uncertain origin. Pediatr. Dev. Pathol. 2012, 15 (Suppl. S1), 267–305. [Google Scholar] [CrossRef]
- Hoot, A.C.; Russo, P.; Judkins, A.R.; Perlman, E.J.; Biegel, J.A. Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am. J. Surg. Pathol. 2004, 28, 1485–1491. [Google Scholar] [CrossRef]
- Sigauke, E.; Rakheja, D.; Maddox, D.L.; Hladik, C.L.; White, C.L.; Timmons, C.F.; Raisanen, J. Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: An immunohistochemical study with implications for diagnosis. Mod. Pathol. 2006, 19, 717–725. [Google Scholar] [CrossRef]
- Judkins, A.R. Immunohistochemistry of INI1 expression: A new tool for old challenges in CNS and soft tissue pathology. Adv. Anat. Pathol. 2007, 14, 335–339. [Google Scholar] [CrossRef]
- Machado, I.; Noguera, R.; Santonja, N.; Donat, J.; Fernandez-Delgado, R.; Acevedo, A.; Baragaño, M.; Navarro, S. Immunohistochemical study as a tool in differential diagnosis of pediatric malignant rhabdoid tumor. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 150–158. [Google Scholar] [CrossRef]
- Salvatorelli, L.; Parenti, R.; Leone, G.; Musumeci, G.; Vasquez, E.; Magro, G. Wilms tumor 1 (WT1) protein: Diagnostic utility in pediatric tumors. Acta Histochem. 2015, 117, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Parham, D.M.; Alaggio, R.; Coffin, C.M. Myogenic tumors in children and adolescents. Pediatr. Dev. Pathol. 2012, 15 (Suppl. S1), 211–238. [Google Scholar] [CrossRef] [PubMed]
- Sebire, N.J.; Gibson, S.; Rampling, D.; Williams, S.; Malone, M.; Ramsay, A.D. Immunohistochemical findings in embryonal small round cell tumors with molecular diagnostic confirmation. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Castrogiovanni, P.; Coleman, R.; Szychlinska, M.A.; Salvatorelli, L.; Parenti, R.; Magro, G.; Imbesi, R. Somitogenesis: From somite to skeletal muscle. Acta Histochem. 2015, 117, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Rosai, J. Rosai and Ackerman’s Surgical Pathology, 10th ed.; Elsevier: New York, NY, USA, 2011. [Google Scholar]
- Magro, G.; Grasso, S.; Emmanuele, C. Immunohistochemical distribution of S-100 protein and type IV collagen in human embryonic and fetal sympathetic neuroblasts. Histochem. J. 1995, 27, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Ruggieri, M.; Fraggetta, F.; Grasso, S.; Viale, G. Cathepsin D is a marker of ganglion cell differentiation in the developing and neoplastic human peripheral sympathetic nervous tissues. Virchows Arch. 2000, 437, 406–412. [Google Scholar] [CrossRef]
- Magro, G.; Grasso, S. Immunohistochemical identification and comparison of glial cell lineage in fetal, neonatal, adult and neoplastichumanadrenal medulla. Histochem. J. 1997, 29, 293–299. [Google Scholar] [CrossRef]
- Magro, G.; Grasso, S. The glial cell in the ontogenesis of the human peripheral sympathetic nervous system and in neuroblastoma. Pathologica 2001, 93, 505–516. [Google Scholar]
- De Preter, K.; Vandesompele, J.; Heimann, P.; Yigit, N.; Beckman, S.; Schramm, A.; Eggert, A.; Stallings, R.L.; Benoit, Y.; Renard, M.; et al. Human fetal neuroblast and neuroblastoma transcriptome analysis confirms neuroblast origin and highlights neuroblastoma candidate genes. Genome Biol. 2007, 8, 401. [Google Scholar] [CrossRef]
- Hoehner, J.C.; Hedborg, F.; Eriksson, L.; Sandstedt, B.; Grimelius, L.; Olsen, L.; Påhlman, S. Developmental gene expression of sympathetic nervous system tumors reflects their histogenesis. Lab. Investig. 1998, 78, 29–45. [Google Scholar]
- Wang, L.L.; Perlman, E.J.; Vujanic, G.M.; Zuppan, C.; Brundler, M.A.; Cheung, C.R.; Calicchio, M.L.; Dubois, S.; Cendron, M.; Murata-Collins, J.L.; et al. Desmoplastic small round cell tumor of the kidney in childhood. Am. J. Surg. Pathol. 2007, 31, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Coffin, C.M.; Alaggio, R. Fibroblastic and myofibroblastic tumors in children and adolescents. Pediatr. Dev. Pathol. 2012, 15, 127–180. [Google Scholar] [CrossRef]
- Bourgeois, J.M.; Knezevich, S.R.; Mathers, J.A.; Sorensen, P.M. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am. J. Surg. Pathol. 2000, 24, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.Q.; Hisaoka, M.; Okamoto, S.; Tanaka, A.; Meis-Kindblom, J.M.; Kindblom, L.G.; Ishida, T.; Nojima, T.; Hashimoto, H. Congenital–infantile fibrosarcoma: A clinico-pathologic study of 10 cases and molecular detection of the ETV6-NTRK3 fusion transcripts using paraffin embedded tissues. Am. J. Clin. Pathol. 2001, 115, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Galfione, S.K.; Ro, J.Y.; Ayala, A.G.; Ge, Y. Diagnostic utility of WT-1 cytoplasmic stain in variety of vascular lesions. Int. J. Clin. Exp. Pathol. 2014, 7, 2536–2543. [Google Scholar] [PubMed]
- Timár, J.; Mészáros, L.; Orosz, Z.; Albini, A.; Rásó, E. WT1 expression in angiogenic tumours of the skin. Histopathology 2005, 47, 67–73. [Google Scholar] [CrossRef]
- Al Dhaybi, R.; Powell, J.; McCuaig, C.; Kokta, V. Differentiation of vasculartumors from vascular malformations by expression of Wilms’tumor 1 gene: Evaluation of 126 cases. J. Am. Acad. Dermatol. 2010, 63, 1052–1057. [Google Scholar] [CrossRef]
- Magro, G.; Bisceglia, M.; Michal, M.; Eusebi, V. Spindle cell lipoma-like tumor, solitary fibrous tumor and myofibroblastoma ofthe breast: A clinic pathological analysis of 13 cases in favor of a unifying histologic concept. Virchows Arch. 2002, 440, 249–260. [Google Scholar] [CrossRef]
- Magro, G.; Gurrera, A.; Bisceglia, M. H-caldesmon expression in myofibroblastoma of the breast: Evidence supporting the distinctionfrom leiomyoma. Histopathology 2003, 42, 233–238. [Google Scholar] [CrossRef]
- Magro, G.; Greco, P.; Alaggio, R.; Gangemi, P.; Ninfo, V. Polypoid angiomyofibroblastoma-like tumor of the oral cavity: A hitherto unreported soft tissue tumor mimicking embryonal rhabdomyosarcoma. Pathol. Res. Pract. 2008, 204, 837–843. [Google Scholar] [CrossRef]
- Magro, G. Stromal tumors of the lower female genital tract: Histo-genetic, morphological and immunohistochemical similaritieswith the “benign spindle cell tumors of the mammary stroma”. Pathol. Res. Pract. 2007, 203, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Caltabiano, R.; Kacerovská, D.; Vecchio, G.M.; Kazakov, D.; Michal, M. Vulvovaginal myofibroblastoma: Expanding themorphological and immunohistochemical spectrum. A clinico-pathologic study of 10 cases. Hum. Pathol. 2012, 43, 243–253. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).