Repurposing Established Compounds to Target Pancreatic Cancer Stem Cells (CSCs)

 , and
, and
Abstract
:1. Introduction
2. CSC-Specific Treatment Approaches: Preclinical Setting
2.1. Influence on the Metabolic Profile of CSCs
2.1.1. Ionophores
2.1.2. Antibiotics with Inhibitory Effects on the Mitochondria
2.1.3. Modulators of Metabolic Functions
2.2. CSC Pathway Inhibition
2.2.1. CSC Pathway Modulation
2.2.2. Inhibiting CSC Plasticity by Modulation of EMT
3. Future Directions
4. Conclusions
5. Outstanding Questions
- ∗
- Are authentic mini-organs or organoids derived from primary tumor specimens valuable for preclinical testing of the outlined strategies?
- ∗
- Can we identify markers as predictors for the antineoplastic efficacy of any of the above-mentioned drugs?
- ∗
- Is successful translation of the outlined approaches into beneficial clinical applications with CSC-specific potential feasible in PDAC?
Acknowledgments
Conflicts of Interest
References
- Quante, A.S.; Ming, C.; Rottmann, M.; Engel, J.; Boeck, S.; Heinemann, V.; Westphalen, C.B.; Strauch, K. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med. 2016, 5, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Rosso, T.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2016 with focus on leukaemias. Ann. Oncol. 2016, 27, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, W.; Werner, J.; Jäger, D.; Debus, J.; Büchler, M.W. Improvement of surgical results for pancreatic cancer. Lancet Oncol. 2013, 14, e476–e485. [Google Scholar] [CrossRef]
- Renz, B.W.; Boeck, S.; Roeder, F.; Trumm, C. Oligometastatic Disease in Pancreatic Cancer-How to Proceed. Visc. Med. 2017, 33, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tiede, B.; Massagué, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007, 17, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Lorenzo, I.; Dorado, J.; Lonardo, E.; Alcala, S.; Serrano, A.G.; Clausell-Tormos, J.; Cioffi, M.; Megias, D.; Zagorac, S.; Balic, A.; et al. Intracellular autofluorescence: A biomarker forepithelial cancer stem cells. Nat. Methods 2014, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; Baldwin, S.A.; Mackey, J.; Sawyer, M.B.; Cass, C.E. Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [Google Scholar] [CrossRef] [PubMed]
- Munakata, K.; Uemura, M.; Tanaka, S.; Kawai, K.; Kitahara, T.; Miyo, M.; Kano, Y.; Nishikawa, S.; Fukusumi, T.; Takahashi, Y.; et al. Cancer Stem-like Properties in Colorectal Cancer Cells with Low Proteasome Activity. Clin. Cancer Res. 2016, 22, 5277–5286. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [PubMed]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Trabulo, S.M.; Sanchez-Ripoll, Y.; Miranda-Lorenzo, I.; Lonardo, E.; Dorado, J.; Reis Vieira, C.; Ramirez, J.C.; Hidalgo, M.; Aicher, A.; et al. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut 2015, 64, 1936–1948. [Google Scholar] [CrossRef] [PubMed]
- Buczacki, S.; Davies, R.J.; Winton, D.J. Stem cells, quiescence and rectal carcinoma: An unexplored relationship and potential therapeutic target. Br. J. Cancer 2011, 105, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, L.; Xia, Y.; Chen, X.; Chang, A.E.; Hollingsworth, R.E.; Hurt, E.; Owen, J.; Moyer, J.S.; Prince, M.E.P.; et al. Therapeutic Efficacy of Cancer Stem Cell Vaccines in the Adjuvant Setting. Cancer Res. 2016, 76, 4661–4672. [Google Scholar] [CrossRef] [PubMed]
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial–Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T. To differentiate or not—routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Hausen, A.Z.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. DCLK1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016, 18, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.-Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014, 146, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Ilmer, M.; Boiles, A.R.; Regel, I.; Yokoi, K.; Michalski, C.W.; Wistuba, I.I.; Rodriguez, J.; Alt, E.; Vykoukal, J. RSPO2 Enhances Canonical Wnt Signaling to Confer Stemness-Associated Traits to Susceptible Pancreatic Cancer Cells. Cancer Res. 2015, 75, 1883–1896. [Google Scholar] [CrossRef] [PubMed]
- Recio Boiles, A.; Ilmer, M.; Rhea, P.R.; Kettlun, C.; Heinemann, M.L.; Ruetering, J.; Vykoukal, J.; Alt, E. JNK pathway inhibition selectively primes pancreatic cancer stem cells to TRAIL-induced apoptosis without affecting the physiology of normal tissue resident stem cells. Oncotarget 2016, 7, 9890–9906. [Google Scholar] [PubMed]
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin Signaling Drives Self-Renewal and Tumorigenicity of Pancreatic Cancer Stem Cells and Provides a Target for Combined Drug Therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the Notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2014, 11, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Masamune, A.; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M.; Hamada, H.; Kobune, M.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS ONE 2011, 6, e20636. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, J.-J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218:e5–2227:e5. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-M.; Dong, T.-T.; Wang, L.-L.; Feng, B.; Zhao, H.-C.; Fan, X.-K.; Zheng, M.-H. Suppression of colorectal cancer metastasis by nigericin through inhibition of epithelial-mesenchymal transition. World J. Gastroenterol. 2012, 18, 2640–2648. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Choi, M.Y.; Yu, J.; Castro, J.E.; Kipps, T.J.; Carson, D.A. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13253–13257. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Najumudeen, A.K.; Jaiswal, A.; Lectez, B.; Oetken-Lindholm, C.; Guzmán, C.; Siljamäki, E.; Posada, I.M.D.; Lacey, E.; Aittokallio, T.; Abankwa, D. Cancer stem cell drugs target K-ras signaling in a stemness context. Oncogene 2016, 35, 5248–5262. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, E.; Cagnol, S.; Beaudry, K.; Carrier, J.; Rivard, N. Oncogenic KRAS signalling promotes the Wnt/β-catenin pathway through LRP6 in colorectal cancer. Oncogene 2015, 34, 4914–4927. [Google Scholar] [CrossRef] [PubMed]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.J.; Yao, D.E.; Zhuang, Y.F.; Hong, Y.; Zhu, X.C.; Fang, Z.R.; Yu, J.; Yu, Z.Y. Azithromycin enhances the favorable results of paclitaxel and cisplatin in patients with advanced non-small cell lung cancer. Genet. Mol. Res. 2014, 13, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, 536. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2015, 22, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Yang, C.S.; DiPaola, R.S.; Tan, X.-L. Repurposing of Metformin and Aspirin by Targeting AMPK-mTOR and Inflammation for Pancreatic Cancer Prevention and Treatment. Cancer Prev. Res. 2014, 7, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef] [PubMed]
- Balic, A.; Sørensen, M.D.; Trabulo, S.M.; Sainz, B. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and Hedgehog signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.; Kloog, Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014, 5, e1065. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Neth, O.; Ilmer, M.; Garnier, A.; Salinas-Martín, M.V.; de Agustín Asencio, J.C.; Schweinitz, von, D.; Kappler, R.; Muñoz, M. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J. Hepatol. 2014, 60, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.S.; Vykoukal, J.; Hubertus, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M.; Ilmer, M. Targeting the neurokinin-1 receptor inhibits growth of human colon cancer cells. Int. J. Oncol. 2015, 47, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Ilmer, M.; Garnier, A.; Vykoukal, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M. Targeting the Neurokinin-1 Receptor Compromises Canonical Wnt Signaling in Hepatoblastoma. Mol. Cancer Ther. 2015, 14, 2712–2721. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, P.; Subramaniam, D.; Paul, S.; Kwatra, D.; Palaniyandi, K.; Islam, S.; Harihar, S.; Ramalinagam, S.; Gutheil, W.; Putty, S.; et al. Crocetinic acid inhibits Hedgehog signaling to inhibit pancreatic cancer stem cells. Oncotarget 2015, 6, 27661–27673. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Kim, H.; Lee, D.-H.; Kim, T.-S.; Kim, T.; Chung, C.; Koh, G.Y.; Kim, H.; Lim, D.-S. Reversing the intractable nature of pancreatic cancer by selectively targeting ALDH-high, therapy-resistant cancer cells. PLoS ONE 2013, 8, e78130. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wu, G.; Chang, C.; Zhu, F.; Xiao, Y.; Li, Q.; Zhang, T.; Zhang, L. Disulfiram inhibits TGF-β-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-κB/Snail pathway. Oncotarget 2015, 6, 40907–40919. [Google Scholar] [PubMed]
- Blaj, C.; Bringmann, A.; Schmidt, E.M.; Urbischek, M.; Lamprecht, S.; Fröhlich, T.; Arnold, G.J.; Krebs, S.; Blum, H.; Hermeking, H.; et al. ADNP is a therapeutically inducible repressor of WNT signaling in colorectal cancer. Clin. Cancer Res. 2017, 23, 2769–2780. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schuler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Ponnurangam, S.; Dandawate, P.R.; Dhar, A.; Tawfik, O.W.; Parab, R.R.; Mishra, P.D.; Ranadive, P.; Sharma, R.; Mahajan, G.; Umar, S.; et al. Quinomycin A targets Notch signaling pathway in pancreatic cancer stem cells. Oncotarget 2016, 7, 3217–3232. [Google Scholar] [PubMed]
- Huang, S.; Ren, X.; Wang, L.; Zhang, L.; Wu, X. Lung-cancer chemoprevention by induction of synthetic lethality in mutant KRAS premalignant cells in vitro and in vivo. Cancer Prev. Res. 2011, 4, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Ilmer, M.; Horst, D. Pancreatic CSCs and microenvironment. Genes Cancer 2015, 6, 365–366. [Google Scholar] [PubMed]
- Guinot, A.; Oeztuerk-Winder, F.; Ventura, J.-J. miR-17-92/p38α dysregulation enhances Wnt signal and selects Lgr6+ cancer stem cell like cells during human lung adenocarcinoma progression. Cancer Res. 2016, 76, 2015–4022. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Chen, J.; Morikawa, T.; Ogino, S.; Kirchner, T.; Shivdasani, R.A. Differential WNT Activity in Colorectal Cancer Confers Limited Tumorigenic Potential and Is Regulated by MAPK Signaling. Cancer Res. 2012, 72, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Cairo, S.; Armengol, C.; De Reyničs, A.; Wei, Y.; Thomas, E.; Renard, C.A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L. Hepatic Stem-like Phenotype and Interplay of Wnt/[beta]-Catenin and Myc Signaling in Aggressive Childhood Liver Cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.-T.; Lee, H.-T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, S.; Alcala, S.; Bayon, G.F.; Kheir, T.B.; Schoenhals, M.; Gonzalez-Neira, A.; Fraga, M.F.; Aicher, A.; Sainz, B.; Heeschen, C. DNMT1 inhibition reprograms pancreatic cancer cells via upregulation of the miR-17-92 cluster. Cancer Res. 2016, 76, 4546–4558. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
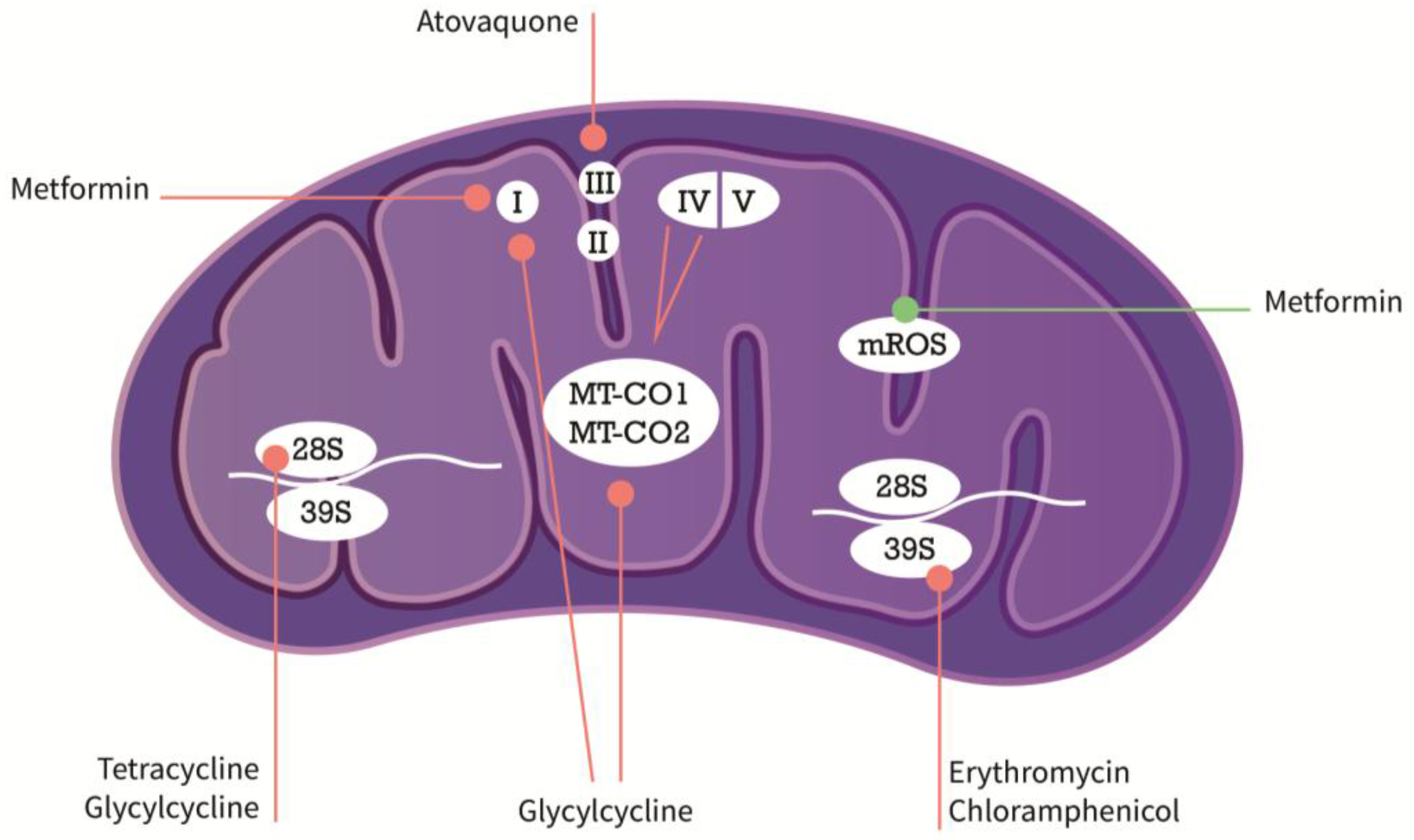

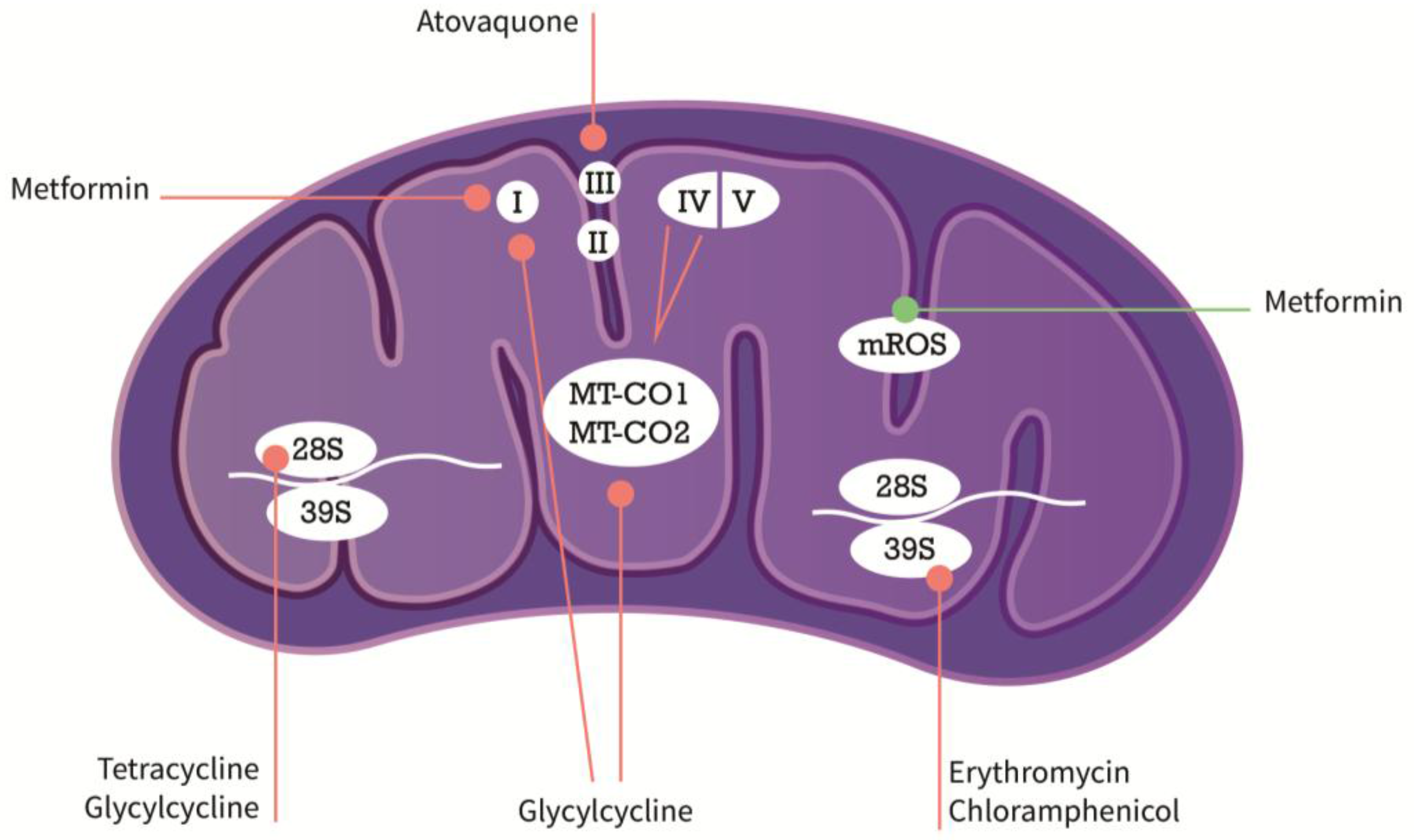{kind=link}
| Marker | Reference |
|---|---|
| Activin | [29] |
| Aldefluor activity | [35] |
| Autofluorescence | [10] |
| c-MET | [36] |
| CD133 | [25] |
| CD24 | [24] |
| CD44 | [24] |
| CXCR4 | [25] |
| DCLK1 | [23,26] |
| ESA/EpCAM | [24] |
| Nodal | [29] |
| Wnt susceptibility | [27] |
| Molecule | Function | Pathway | Cancer Type | Phase/Clinical Trial | NCT Number | Reference |
|---|---|---|---|---|---|---|
| Aprepitant | Anti-emetic | AKT/mTOR, Wnt | Hepatoblastoma, Colorectal cancer, PDAC | N/A | N/A | [54,55,56] |
| Atovaquone | Anti-malaria | Mitochondrial inhibition | Breast cancer | Phase I | NCT02628080 | [50,51] |
| Azithromycin | Macrolide | Unknown | various: e.g., NSCLC, PDAC | N/A | N/A | [44,45] |
| Chloroquine | Anti-malaria | CXCL12/CXCR4; Autophagy | PDAC | Phase I | NCT01777477 | [52] |
| Crocetinic acid | Safron compound | Hedgehog inhibition | PDAC | N/A | N/A | [57] |
| Disulfiram | ALDH inhibitor | ERK/NFκB/EMT | Breast, Colorectal, Melanoma, PDAC | Phase I | NCT02671890 | [58,59] |
| Ketamine | NMDA receptor antagonist | Wnt inhibition | Colorectal cancer | N/A | N/A | [60] |
| Metformin | Biguanide | Mitochondrial inhibition | PDAC, colorectal | Phase II | multiple: e.g., NCT01210911, NCT01971034 | [48] |
| Mocetinostat | Class I HDAC inhibitor | EMT inhibition | PDAC, prostate cancer | Phase I/II | multiple: e.g., NCT02805660, NCT00372437 | [61] |
| Nigericin | Ionophore | EMT inhibition | Colorectal cancer | N/A | N/A | [38] |
| Quinomycin A | Quinoxaline antibiotic | Notch inhibition | PDAC | N/A | N/A | [62] |
| Salinomycin | Ionophore | Wnt inhibition, K-ras | Breast cancer; CLL | N/A | N/A | [41,63] |
| Tigecycline | Glycylcyclin | Mitochondrial inhibition | AML; Breast, Lung, Prostate, PDAC | Phase I | NCT01332786 | [43,44] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renz, B.W.; D’Haese, J.G.; Werner, J.; Westphalen, C.B.; Ilmer, M. Repurposing Established Compounds to Target Pancreatic Cancer Stem Cells (CSCs). Med. Sci. 2017, 5, 14. https://doi.org/10.3390/medsci5020014
Renz BW, D’Haese JG, Werner J, Westphalen CB, Ilmer M. Repurposing Established Compounds to Target Pancreatic Cancer Stem Cells (CSCs). Medical Sciences. 2017; 5(2):14. https://doi.org/10.3390/medsci5020014
Chicago/Turabian StyleRenz, Bernhard W., Jan G. D’Haese, Jens Werner, C. Benedikt Westphalen, and Matthias Ilmer. 2017. "Repurposing Established Compounds to Target Pancreatic Cancer Stem Cells (CSCs)" Medical Sciences 5, no. 2: 14. https://doi.org/10.3390/medsci5020014
APA StyleRenz, B. W., D’Haese, J. G., Werner, J., Westphalen, C. B., & Ilmer, M. (2017). Repurposing Established Compounds to Target Pancreatic Cancer Stem Cells (CSCs). Medical Sciences, 5(2), 14. https://doi.org/10.3390/medsci5020014