Abstract
Syndepositional diagenesis is a complicating factor when interpreting geochemical proxies in carbonate sedimentary environments. Previous studies have suggested that carbonate deposits may preserve the geochemical and isotopic signatures of seawater that can be used for paleo-redox reconstructions. However, more work is necessary to understand how these trace metals are preserved. The present study examines shallow marine carbonate sediments from the Bahamas to better understand diagenetic effects on trace metal uptake and sequestration. Analysis of diagenetic effects and trace metal uptake follows a multi-method approach, combining sequential extraction, stable isotope analyses, and rare earth elemental analysis. Stable isotopes track bacterial sulfate reduction, denitrification, and organic matter source and provide insight into thresholds and processes for the authigenic trace metal uptake. Importantly, exchangeable phases exhibit authigenic accumulation of molybdenum, uranium and vanadium, and intensified bacterial sulfate reduction is evidenced by most depleted sulfur isotope signatures. In addition, rare earth element values are very indicative proxies that suggest altered primary seawater trace element in carbonates (no cerium or lanthanum anomaly, moderate heavy rare earth element enrichment, decreased y/ho ratios and positive correlations between aluminum, manganese, and iron). Taken together, these results allow the development of a framework to better understand how to apply sedimentary geochemistry of carbonate rocks to paleo-environments as this study shows significant authigenic accumulation of redox-sensitive trace metals by exchangeable phases.
1. Introduction
The geochemistry of carbonate rocks has long been a useful tool for investigating global ocean conditions, such as global oxygen and carbon cycles, over vast periods of time [1,2,3,4]. More recently, novel metal isotope proxies (e.g., U and Mo isotopes) have been developed which utilize carbonate rocks for reconstruction of global environmental changes [5,6,7]. Ultimately, however, it is important to reconstruct the chemical composition of seawater as recorded in the rock record without post-depositional alterations which may obscure the recording of global signatures in seawater paleo-redox proxies [8,9,10,11,12,13,14]. For example, recent studies have shown authigenic accumulations of U and Mo in shallow Bahamian carbonates associated with shifts in the isotopic composition of bulk sediments. However, the mode of uptake and diagenetic controls require further investigation [8,9,10].
The goal of the present study is to examine syndepositional diagenetic environments possessing varying degrees of organic matter content and composition, terrestrial mineral input, meteoric input, and restriction from the open sea, to systematically investigate the effects of variable early diagenetic conditions on the alteration of the geochemical signatures of modern carbonates. The present study employs sequential extractions and stable isotope analyses to investigate how REY and trace metals are incorporated into a variety of early diagenetically altered carbonate sediments. The results reveal both the chemical phases in which these elements are hosted and the driving factors of their uptake.
2. Materials and Methods
2.1. Site Location
The Bahamian carbonate platform is among the largest and best studied carbonate platforms, e.g., [8,9,11,12,13]. As such, it is an ideal natural location to study early diagenesis of shallow carbonates. The geographic isolation of the platform far from any continental masses provides a unique environment for the study of primary and authigenic geochemical signatures, which are mostly isolated from terrestrial siliciclastic influence from nearby continents. However, windblown dust may provide some siliciclastic material [14]. Carbonate sediment samples were collected from different locations around Eleuthera, Water Cay in the Schooner Cays, and from Darby Island in the Exuma Cays (Figure 1).
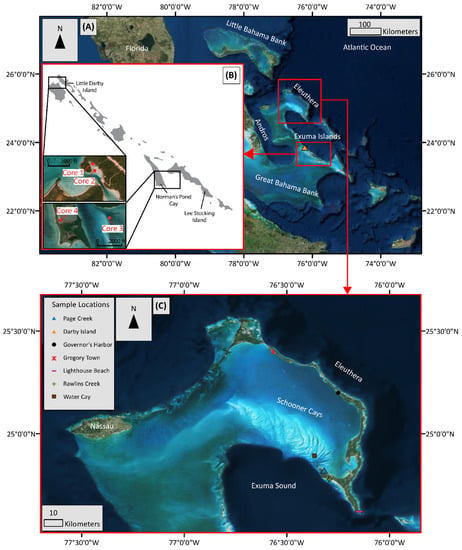
Figure 1.
Map of sampling locations on Eleuthera and Exuma Islands, Bahamas (A). Sediment cores 1, 2, 3, and 4 were collected on the Exuma Island Chain of the Greater Bahamas near and around Little Darby Island (B). Near-surface sediment grab samples were collected mainly around Eleuthera and the Schooner Cays (C), with one sample from Darby Island.
2.1.1. Exuma Islands—Darby Island
Darby Island is a vegetated cay located in the Exuma Cays island chain. The Exuma Cays are located along the southwestern margin of the Exuma Sound (Figure 1). Carbonate sediment core samples in the present study were collected along the Exuma Islands, centered around Little Darby (Figure 1). Geochemical work on these samples has been carried out previously [8,9]. Samples from four cores from a variety of environments and depths were resampled and analyzed. Cores 1 (42 cm long) and 2 (24 cm long) were collected in the area around the Little Darby research station while cores 3 (22 cm long) and 4 (30 cm long) were collected near Lee Stocking island. Core 1 was collected in an area dominated by calcareous green algae and turtle grass and the core location was chosen to avoid adjacent shrimp (Callianassa sp.) mounds and burrows. Core 2 was collected on a tidal flat approximately 100 m to the southeast of core 1. The tidal flat was largely free of any vegetation (turtle grass or green algae). Core 3 was collected in an area similar to core 1, with dense turtle grass and calcareous green algae, but in a deeper water (4 m) environment. Core 4 was collected from a saltwater pond which is tidally connected to the open ocean (salinity = 39‰). For detailed core description, see [8].
2.1.2. Eleuthera Island
Holocene and Pleistocene sea-level fluctuations are the main influences for the construction of Eleuthera during the Quaternary [15]. Periods of growth mainly occurred when sediments that accumulated during interglacial periods were transported to the island during glacial periods and lithified [15]. The sedimentary response to Quaternary sea-level fluctuations is seen in the variety of carbonate deposits forming the island which include beach, tidal, and aeolianite deposits along with in situ reefs and well developed paleosols [15,16,17]. Additionally, with Eleuthera being exposed to the Atlantic on its Easterly side, storms and high winds transport sediments to produce washover lobes on the leeward side [15].
The five sampling locations on Eleuthera are Gregory Town, Governor′s Harbor, Page Creek, Rawlins Creek, and Lighthouse Beach (Figure 1). Gregory Town, Governor′s Harbor, and Lighthouse Beach are high-energy nearshore beach sand samples. Page Creek and Rawlins Creek samples are collected from tide-dominated areas in southern Eleuthera which contain tidal channels connected to calm shallow water environments (approximately <1 m water depth) dominated by mangrove vegetation. Sediments below the SWI at Page Creek and Rawlins Creek display a dark gray color and produce a strong sulfide odor, indicating a sulfidic environment.
In total, eight samples were collected from Page Creek at different locations and sediment depths: Page Creek Fine Mud 1 (PCFM1), Page Creek Mud 2 (PCM2), Page Creek Fine Mud 3 1/2 (PCFM 3 1/2), Page Creek Fine Mud 3 2/2 (PCFM 3 2/2), Page Creek Mud 3 Anoxic (PCM3A), Page Creek Mud 3 (PCM3), Page Creek Mud 4 (PCM4), and Page Creek Black Mud (PCBM). Two samples were collected from Rawlins Creek: Rawlins Creek (RC) and Lower Creek (LC; oolitic tidal channel sample). One sample each was collected from Gregory Town (GT), Governor′s Harbor (GH), and Lighthouse Beach (EL).
2.1.3. Schooner Cays
The Schooner Cays is a 16 km × 60 km ooid shoal complex located off the coast of Cape Eleuthera. The geological setting and depositional system of the Schooner Cays have been well described [18] and is summarized here. The morphology of the ooid shoal complex is mainly influenced by tidal cycles with flood tides coming in from, and ebb tides going out to, the Exuma Sound to the south-west. General morphologic features of the Schooner Cays include sand ridges, parabolic bars, and flow-oblique shoulder bars along with tidal channels. Within the Schooner Cays there are six small vegetated cays, four of which are located approximately 6.5 km to 9.5 km off the northern point of Cape Eleuthera. Of these four, Schooner Cays samples in the present study were collected from the eastern most cay named Water Cay. It is located approximately 6.5 km north of Cape Eleuthera with coordinates of 24°54′16.1″ N, 76°21′12.5″ W. Water Cay is approximately 100 m × 200 m to 150 m × 200 m, displaying a rough circular shape. The rim of the island is composed of ooid sand with crusts of oolitic limestone, most likely derived from the local ooid sand. The interior of the island is covered by moderate to thick vegetation.
Two samples were collected from Water Cay in the Schooner Cays. One sample was collected near the coastline (Schooner Cays Near Shore; SCNS) and the other (Schooner Cays Off Shore; SCOS) was collected further off shore within approximately 50 m of the coastline of Water Cay. Sediments from the Schooner Cays were also hand-picked under a microscope in order to produce pure ooid and non-ooid samples labeled SCPO and SCNO, respectively.
2.2. Inorganic Oxygen and Carbon Isotope Analysis
All stable isotope analyses were performed at the Oxy-Anion Stable Isotope Consortium (OASIC) at Louisiana State University. Isotopic measurements are reported in delta notation as per mil (‰) deviation relative to VPDB standard.
Approximately 200 μg of sample was dried at 75 °C for 12 h after being loaded in 12 mL borosilicate glass Exetainer vials (Labco Ltd., Lampeter, UK), then sealed and flushed with 99.999% He for 2 min. After flushing, sample vials were loaded into a 96-position Thermo Finigan Gas Bench II kept at 72 °C. Samples were then reacted for 3 h with 100 μL phosphoric acid (density = 1.92 g/mL) kept at 75 °C and added by manual injection. After 3 h, CO2 produced from the reaction was passed through a Nafion water trap and GC in Gas Bench. Analysis was performed on a Delta V Advantage mass spectrometer. One in-house standard CMA (δ13C = 2.57‰ and δ18O = −4.31‰) was used as check standards. External precision (1σ) is less than 0.1‰ for δ13C and 0.2‰ for δ18O.
2.3. Organic Nitrogen, Organic Carbon, and Sulfur Isotopes and Elemental Analysis
Sediment core and grab samples were decalcified in 1 M HCl. Samples were then washed with DI, dried, and weighed into combustible tin containers. Weights were determined by targeting ~400 μg of carbon and ~100 μg of N, approximately 2–20 mg of bulk decalcified sample. CO2, N2, and SO2 gas produced from the samples by flash combusting the tin capsules (at 950 °C for CO2 and N2, 1150 °C for SO2) in a furnace with an elemental analyzer (Micro Vario Cube, Isoprime Ltd., Cheadle, UK) was analyzed by continuous flow with an Isoprime100 (Isoprime 100, Cheadle, UK) gas source mass spectrometer.
Nitrogen and carbon isotopic composition was calibrated using one in-house standard Acetanilide-OASIC (δ15N = + 1.61‰ and δ13C = −27.63‰). Analytical precision of Nitrogen and carbon isotopic composition was <0.3‰ and 0.1‰, respectively. δ15N and δ13Corg is reported in ‰ air and VPDB variation. Mass calibrations were performed to allow adjustment of carbon content based on TOC%. TOC is calculated based on C concentration of organics and mass calculations based on weight of the original samples. Sulfur isotope compositions are reported in ‰ deviations from VCDT. Two in-house standards are used for calibration (LSU-Ag2S-1: −4.3‰; LSU-Ag2S-2: +20.2‰;). Analytical precision is <0.3‰.
2.4. Sequential Extractions and ICP–MS Analyses
2.4.1. ICP–MS Elemental Analysis of Sediment Grab Samples
Elemental concentrations of minor and trace elements in the sediment grab samples were measured using a Thermo iCap Qc quadrupole ICP–MS at Louisiana State University. Samples were prepared by dissolving approximately 1 g of sediment in 1M HCl to dissolve the carbonate mineral phases, although dissolution of amorphous iron oxides and desorption of metals adsorbed onto inorganic and organic material may also occur [19,20,21]. The residual organic and non-carbonate material was then removed by filtering the HCl leachate through Whatman GF/C glass microfiber filters (pore size = 1.2 μm). The filtered leachate was diluted with 2% HNO3 prior to analysis. The instrument was externally calibrated using a serial dilution of a solution containing QCP-QCS-3 multi-element standard, REE standard, and single-element standards of Ca, Mg, Rb, and Sr. Check standards and internal standards were used to ensure minimal instrument drift and sample suppression.
2.4.2. ICP–MS Analysis of Sequentially Extracted Trace Elements
A subset of sediment core samples were sequentially extracted following a modified Tessier extraction [21,22] method. The extraction method utilizes different reagents to chemically separate sedimentary trace metals into separate partitions: exchangeable, carbonates, oxyhydroxides, organics, and residual (Table 1). Modifications from the Tessier method include modifying the exchangeable reagents to extract non-crystalline uranium [23]. Bicarbonate effectively leeches U from natural samples and is held in solution as a U(VI)–carbonate complex [24]. Recent work has shown that sodium bicarbonate extraction is an effective wet chemistry technique to separate non-crystalline U(IV) (reductively precipitated U(IV) from U(VI), termed mononuclear U(IV)) from uraninite [25]. Other studies have shown the relative importance of non-crystalline U(IV) as a microbial reduction product in a variety of sediments [23,26,27,28]. Sodium bicarbonate dissociates in solution, supplying HCO3− and Na+ ions. The use of bicarbonate over other anions, such as CO32−, provides a mild ionic strength and is stable at pH values near neutral and allows for the dissociation of loose ionic bonds while not affecting the pH and associated reactions. HCO3− likely has the greatest leeching effect on positively charged elements, such as U. The difference in ionic character between elements adds complications when directly comparing concentrations of elements in the exchangeable phase.

Table 1.
The list of parameters, reagents, and chemical phases used during the sequential extraction. Extracts were performed using increasingly aggressive reagents, progressing down the table. Smp:Sol is abbreviated for sample to solution ratio.
The chemical partitions were separated using progressively more aggressive chemical leaches (Table 1). After each chemical leaching, samples were centrifuged at 5000 rpm for 5 min. The effluent was then carefully pipetted off of the remaining solids. The solids were rinsed twice with DI before the next reagent was added. Total digests were performed using a combination of H2O2 and concentrated nitric acid at 80 °C.
Samples were analyzed using an iCap Qc quadrupole ICP–MS at Louisiana State University using the same calibration procedure as the sediment grab samples. REY calculations were also performed in the same manner as grab samples using Equations (1)–(4). In preparation for analysis, all samples were dried and prepared in a 2% HNO3 solution. Individual chemical phases from the sequential extraction were run separately in order to reduce variations in matrix effects due to the variety of reagents used.
2.5. Normalization of Rare Earth Elements
REY were normalized to the Post-Archean Australian Shale (PAAS) [29]. Ce anomalies (CeSN/CeSN*), Y/Ho ratios, and HREE enrichments were calculated using the formulas below. Subscript “SN” indicates shale-normalized values were used. Ce anomaly calculations follow the formula of [30].
Total REY = Σ[REY]
CeSN/CeSN* = ([Ce]SN)/([Pr]2 SN/[Nd] SN)
Y/Ho = Y⁄Ho
HREE enrichment = [Yb]SN/[Nd]SN
3. Results
3.1. Inorganic Carbon and Oxygen Isotopes
The δ13Ccarb and δ18Ocarb values from cores 1, 2, and 3 do not vary significantly (Figure 2), plot near the equilibrium range for low-magnesium calcite (LMC), and are in the range of expected values for marine carbonates [11]. Modern sediment grab samples have similar δ13Ccarb to the samples of cores 1, 2, and 3. Grab samples likewise have similar δ13Ccarb and δ18Ocarb values to modern sediment collected to the west of Andros on the GBB (δ13Ccarb = 4.8 ± 0.3‰, δ18Ocarb = 0.4 ± 0.4‰).
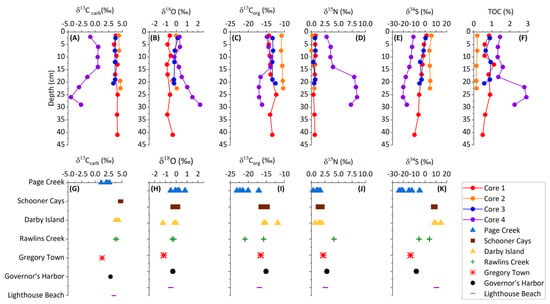
Figure 2.
Light stable isotope and TOC core profiles for sediment cores and isotopic data for sediment grab samples. (A–F) are depth profiles of the four different cores and (G–K) are individual grab samples from different locations.
Core 4, collected from a restricted saltwater pond, exhibits depleted δ13Ccarb and enriched δ18Ocarb values which change with depth (Figure 2) and are negatively correlated (Figure 3). Core 4 contains isotopic values representative of two distinct environments. The first environment is represented in the upper 14 cm of the core with δ13Ccarb and δ18Ocarb near 0. The second environment is represented in the lower 16 cm and is identified by decreasing δ13Ccarb to ~−5‰ and increasing δ18Ocarb to ~2‰. The lower 16 cm of core 4 additionally corresponds with elevated TOC up to 3%.
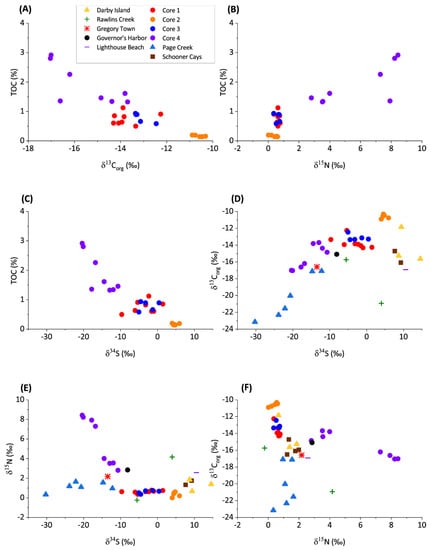
Figure 3.
Cross plots of stable isotopes of the de-calcified organic residual material, indicating various attributes of the diagenetic circumstances during deposition. (A) TOC versus δ13Corg. (B) TOC versus δ15Norg. (C) TOC versus δ34S. (D) δ13Corg versus δ34S. (E) δ15Norg versus δ34S. (F) δ13Corg versus δ15Norg.
3.2. Organic Carbon, Nitrogen, and Sulfur Isotopes
The δ13Corg values in samples from cores 1, 2, and 3 do not vary significantly (Figure 2). In core 4, there is a shift to more negative δ13Corg at approximately 15 cm depth. δ13Corg from the grab samples varies between −11.86‰ and −23.15‰. The isotopically lightest values are observed in Page Creek and Rawlins Creek. A negative covariation between TOC and δ13Corg is evident from the core samples (Figure 3). TOC data are not available for the grab samples; however, highly negative δ13Corg and other similarities between Page Creek samples and core 4 (Figure 3, Figure 4 and Figure 5) suggest similar conditions.
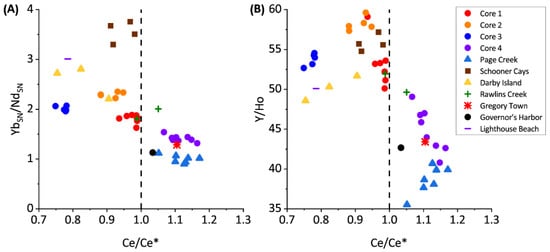
Figure 4.
Rare earth geochemistry of the investigated samples.
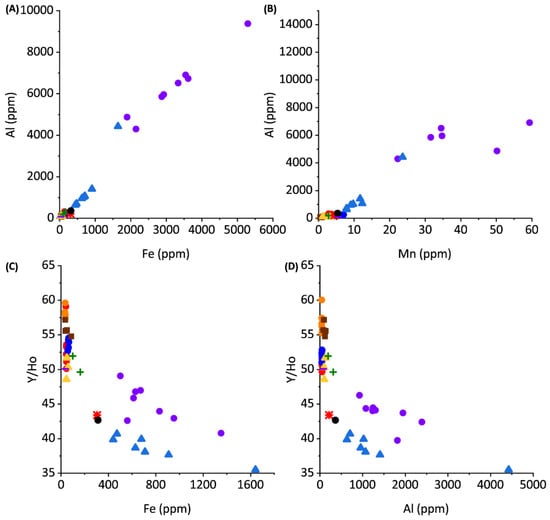
Figure 5.
Proxies for terrestrial and siliciclastic contribution to rare earth geochemistry of sediments.
δ15N values in cores 1, 2, and 3 do not vary significantly (Figure 2). The δ15N values vary between 0.13‰ and 0.66‰, with an average of 0.55‰. In core 4, δ15N varies between 2.82‰ and 8.43‰, with an average of 5.71‰. There is a shift in δ15N at approximately 15 cm depth similar to the shift in δ13Corg, although the shift in δ15N is to isotopically heavier values. The δ15N of grab samples varies between −0.23‰ and 4.17‰, with the majority of samples being comparable to the cores 1, 2, and 3. The isotopically heaviest values are measured in Rawlins Creek. Except for core 4, which possesses a positive correlation between TOC and δ15N, there is no correlation between TOC and δ15N (Figure 3).
δ34S values in all core samples show similar trends where δ34S decreases with depth (Figure 2). Despite having the same trend, average δ34S values between the cores are offset from each other. δ34S values in the cores vary between 5.94‰ and −20.40‰ with isotopically lightest values in core 4, heaviest values in core 2, and intermediate values in cores 1 and 3. δ34S measured in grab samples are also variable between sample locations and fall within a similar range of values as core samples. Schooner Cays, Lighthouse Beach, and Darby Island grab sample δ34S values are most similar to those measure in core 2. δ34S in the remaining grab samples are isotopically lighter and most similar to cores 1, 3, and 4. Sediment core δ34S values have a negative correlation with TOC and a positive correlation with δ13Corg (Figure 3). There appears to be no correlation between δ34S and δ15N (Figure 3)
3.3. Rare Earth Elements and Yttrium (REY) and Terrigenous Input Proxies (Al, Mn, Fe) in Bahamian Cores and Grab Samples
The PAAS-normalized REYs in cores 1 through 3 and most grab samples from Darby Island, Lighthouse Beach, Schooner Cays, and Rawlins Creek express slightly negative Ce anomalies and Y/Ho ratios of approximately 50–60 (Figure 4). NdSN/YbSN ratios of near 2 in the previously listed core and grab samples indicates no significant LREE enrichment (Figure 4) and these samples have low Al, Mn, and Fe concentrations (Figure 5).
3.4. Uranium Concentrations in Bahamian Cores and its Distribution in Different Sediment Phases
Sequential extractions performed in the present study indicate all chemical phases remain essentially constant downcore with respect to U (±0.5 ppm), with the exception of the exchangeable fraction (Figure 6). While exchangeable U remains relatively constant in core 2, the exchangeable fraction increases in concentration from ~1.3 to ~4.5 ppm in core 1, ~1.0 to ~1.9 ppm in core 3, and ~1.5 to ~4.5 ppm in core 4 (Figure 6). This suggests that the exchangeable phases host the majority of authigenically accumulated U. The oxide and residual phases both contain <1 ppm U and the organic phase contains <0.15 ppm U.
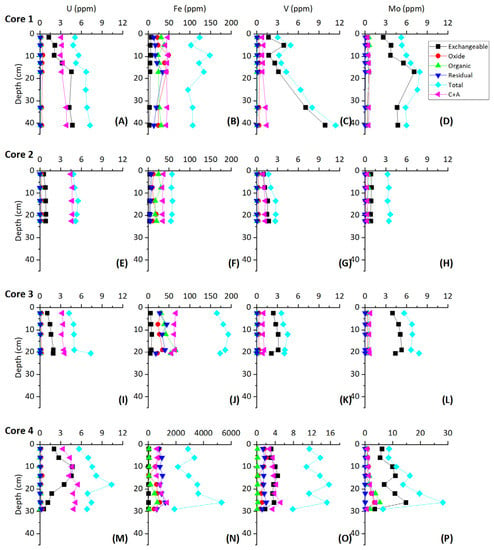
Figure 6.
Sequential extraction results for the shallow sediment core samples.
3.5. Molybdenum Concentrations in Bahamian Cores and its Distribution in Different Sediment Phases
Similar to U, sequential extractions show that all chemical phases remain essentially constant downcore with respect to Mo, with the exception of the exchangeable fraction (Figure 6). The exchangeable fraction contains the largest fraction of Mo in the sediment (Figure 6), suggesting the exchangeable phases host the majority of authigenically accumulated Mo.
3.6. Vanadium Concentrations in Bahamian Cores and its Distribution in Different Sediment Phases
4. Discussion
4.1. Multi-Proxy Organic Geochemistry of Bahamian Bulk and Core Sediments
Surface and shallow surface grab samples were collected from a variety of depositional environments in the Bahamas to constrain the variability of primary precipitates in non-buried near-surface sediments that have not yet undergone extensive early diagenesis in contact with porewater. In shallow water carbonates, the majority of the organic matter is derived from shallow water carbonate organisms such as calcareous green algae or sea grass, or from terrestrial input [11]. Marine organisms utilize C from dissolved inorganic carbon (DIC), while terrestrial organisms sequester C from atmospheric CO2 [31]. Terrestrial organic matter may be ~12–15‰ depleted in δ13Corg with respect to organic matter produced in marine environments [12,32]. In the samples studied, a negative covariation between TOC and δ13Corg suggests that the addition of terrestrial organics drives the changes in organic C isotopes (Figure 3).
δ15Norg of cores 1, 2, and 3, show small variations (Figure 2) and poor correlations with both TOC and δ13Corg (Figure 3), indicating little fractionation associated with denitrification is taking place. The lack of fractionation in nitrogen isotopes is indicative of primary organic matter. Covariation between δ15Norg and TOC and a negative covariation between δ15Norg and δ13Corg in core 4, however, indicates a relative increase in input of terrestrial organic matter in the lower core as C3 and C4 saltmarsh organic matter possesses δ15Norg of ~6–14‰, [33]. Grab samples show no correlations between δ15Norg and δ13Corg and similar δ15Norg values to cores 1, 2, and 3 (Figure 2 and Figure 3). Terrestrial organic matter input may explain elevated δ15Norg in Rawlins Creek grab samples, as the Rawlins Creek location contains abundant mangrove vegetation.
Sulfur isotopes experience fractionation in pore waters during bacterial sulfate reduction (BSR), in which pore waters become isotopically heavy and preserved organics become isotopically light [12,34]. Sulfide oxidation and sediment reworking keep levels of free dissolved sulfide relatively low. Cores 1 and 3 have depleted δ34S values with respect to core 2, indicating that BSR in cores 1 and 3 is preferentially releasing 34S to the pore water and preserving 33S in organic material in agreement with previously reported elevated sulfide (500–1500 µM) in pore waters [9]. The lack of covariation between TOC and δ34S in cores 1 and 3 suggests that BSR affected a small percentage of TOC available for oxidation [35] (Figure 3). Core 2 has a positive and invariant δ34S signature due to decreased BSR, consistent with decreased porewater sulfide [9]. In core 4, δ34S decreases with depth and negatively covaries with TOC (Figure 2 and Figure 3).
Grab samples from the Schooner Cays, Darby Island, and Lighthouse beach have similar δ34S to core 2 samples (Figure 2 and Figure 3) and, likewise, indicate low levels of BSR. Similar δ34S values to cores 1 and 3 in Rawlins Creek and Governor′s Harbor samples suggests a greater influence of BSR in Rawlins Creek and Governor′s Harbor compared to core 2, Schooner Cays, Darby Island, and Lighthouse beach samples. Core 4, Page Creek, and Gregory Town samples have the most negative δ34S values, suggesting that BSR is an important process in these sediments. In core 4, highly negatively fractionated δ34S may be related to increased TOC which can facilitate a greater prevalence of anaerobic remineralization pathways such as BSR [36]. While high TOC also likely drives BSR and negative fractionation of δ34S in Page Creek and Gregory Town samples, a lack of grab sample TOC data makes comparison somewhat difficult. However, the strong positive linear correlation between δ34S and δ13Corg in core 4 and Page creek samples (Figure 3) suggests that input of terrestrial organic matter, in addition to marine organic matter, is an important driver of anaerobic processes and early diagenetic intensity in these environments.
4.2. Rare Earth and Yttrium Geochemistry of Bahamian Bulk and Core Sediments
Redox-sensitive proxies REY, U, Mo, and V have different mechanisms which affect their accumulation and distribution in sediments. In terms of REY, rare earth elements consist of fourteen elements forming the Lanthanide series. Y is often included with REEs (hence REY) due to its similar chemical properties to Ho. The oxidation state of REY (typically +3), ionic radius, and other chemical properties, result in predictable distributions in oceans and sediment [37,38]. REEs are supplied to the ocean by riverine input, hydrothermal vents, or through eolian dust, and removed through particle scavenging to the sediments [38,39]. Hydrothermal vents and riverine input carry flat shale-normalized patterns. Once freshwater interacts with saline waters; however, patterns quickly acquire seawater characteristics with negative Ce anomalies and HREE enrichments (low Ce/Ce* and NdSN/YbSN ratios) [38]. Ce is unlike other REEs in that it occurs naturally in a 3+ or 4+ valance state, and can be readily removed to the sediment in the oxidized Ce(IV) state [37,39]. This results in a negative Ce anomaly in oxidized oceans and can be used to track redox conditions.
The PAAS-normalized REE’s express slightly negative Ce anomalies in cores 1 through 3 and grab samples (Figure 4) consistent with seawater values [37,40,41,42]. Y/Ho ratios are ~50–60 for these samples signifying little to no detrital input, as expected in Bahamian samples [40].
Previous work has shown Light REE’s (LREEs) are enriched in Bahamian soils due to aeolian dust input; however, there is no significant LREE enrichment, as reflected in NdSN/YbSN ratios of ~2 in cores 1 through 3 and grab samples, suggesting that the REE contribution from terrestrial soils are minimal in these samples [14]. The seawater-type distribution of REYs and lack of covariation among diagenetic-sensitive elements suggests that seawater REY values are preserved in cores 1, 2, and 3 (Figure 4 and Figure 5). This indicates that REY values are not significantly impacted, even during diagenesis under reducing pore water conditions, consistent with previous studies of REE incorporation into carbonate sediments [43,44].
Core 4 and Page Creek have REY distributions unlike those of the remaining samples. Core 4 and Page Creek samples show no Ce anomaly (Figure 4), moderate HREE enrichment, and decreased Y/Ho ratios. The decreased Y/Ho ratios suggest increased terrestrial input which is consistent with stable isotope data, positive correlations between Al, Mn, and Fe, and high Al, Mn, and Fe concentrations (Figure 5). Thus, there is evidence for clay leaching in core 4 and Page creek data which may impart increased ƩREY, lower Y/Ho, and flatter REY profiles in carbonate leaches [39].
4.3. Paleoredox Applications
4.3.1. Uranium Sequestration
Authigenic accumulation of U can be partly controlled through BSR reactions as sulfate-reducing organisms can gain energy for growth by electron transport to U(VI) [45]. This process for authigenic U accumulation is confirmed to be a driving factor for authigenic U accumulation in the exchangeable phases, where authigenic U accumulates, through negative covariation of U and δ34S in cores 1, 2, and 3 (Figure 7). Core 4 shows no covariation between U and δ34S, as a result of the meteoric induced dolomitization in which oxic waters flushed through the sediment, re-oxidizing and mobilizing authigenic U [46].
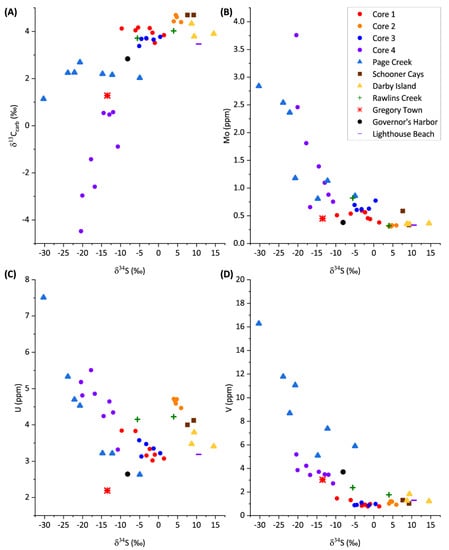
Figure 7.
Trace metal and stable isotope geochemistry of bulk sediment samples.
The accumulation of U downcore in cores 1 and 3 in the exchangeable phases, along with reducing pore water conditions conducive to precipitation of mononuclear U(IV), suggests an authigenic accumulation of mononuclear U(IV). The remaining chemical phases sum to a concentration of U between ~3 and 5 ppm, similar to primary precipitates [8], indicating authigenically accumulated U is quantitatively removed in the first sequential extraction phase.
Reduction of U(VI) to U(IV) provides the largest known natural fractionation of 238U/235U, making it a potentially valuable proxy for ocean redox conditions [5,6,8]. Due to the long ocean residence time of U (~350 ka) compared to ocean mixing timescales (~1.5 ka), the composition of open seawater should theoretically reflect the globally uniform 238U/235U values of a given time and give a global view of redox conditions [47]. While primary precipitates incorporate the 238U/235U values of seawater, early diagenesis imparts an authigenic signature creating a bulk heavier 238U/235U value of 0.2–0.4‰ with depth and suggests an authigenic accumulation of isotopically heavy U downcore [10]. Until recently, authigenic accumulations of U were generally thought to be the product of microbial or chemical reduction and take the form of uraninite, though uraninite is rarely found in nature [8,25,26]. Others have also demonstrated little isotope fractionation of U isotopes in primary carbonate precipitates [48,49,50].
Isotopically heavy U may precipitate under reducing conditions [5,51,52]. An authigenic accumulation of non-crystalline U(IV) in reduced pore waters could contribute a heavier isotopic signature to bulk carbonate values, consistent with previous observation [8]. Thus, instead of applying a correction factor to δ238U values of carbonates [8,53,54], it may be more effective to chemically remove mononuclear U(IV) using sodium bicarbonate and authigenically precipitated carbonate in order to isotopically analyze the remaining sediment.
Estimates for δ238U values of the isotopically heavier mononuclear U(IV) are possible. Assuming that non-authigenic U records δ238U values of seawater and any increase above that is due to authigenic enrichment in the exchangeable phases, a two end-member mixing equation can be set up.
where:
δ238Ubulk is the isotopic composition of the bulk sediment, including all phases. δ238Ubulk-exchange is the isotopic composition of the non-authigenic material, presumably representing the seawater composition. Using δ238Ubulk values from [8] and setting δ238Ubulk-exchange to that of seawater (−0.4‰), δ238Uexchange ranges from 0.03–0.63‰ (average 0.31‰, standard deviation 0.15 ‰) in cores 1 and 3 where authigenic enrichment is observed. Slightly higher δ238Uexchange values of 0.89–1.86‰ (average 1.36‰, standard deviation 0.36‰) would be required in core 2 where very little U is measured. This range is similar to that proposed by [8] (0.6–1.2‰) and consistent with studies of δ238U fractionation in reducing environments [5,8,51,52,55], suggesting that it may be possible to remove the authigenic diagenetic overprint through sequential extraction yielding a more reliable estimate of the uranium isotopic composition of seawater. The above calculations are based on samples from the original U isotope work in The Bahamas [8], ensuring a direct comparison. However, more recent work in that area are consistent with these values and magnitude of isotope offsets [56,57].
4.3.2. Molybdenum Sequestration
Mo, as with U, has a long residence time in the open ocean (~700 ka) with respect to ocean mixing time, making it additionally an attractive proxy for potentially recording a globally integrated redox state [58]. Many studies have demonstrated molybdenum’s utility in black shales in which Mo may be quantitatively removed [59,60,61]. However, black shales do not constitute all of geologic history and may be biased towards restrictive epicontinental seas [62]. Furthermore, it is difficult to prove quantitative removal of Mo which is necessary for obtaining reliable seawater value estimates [60,63,64]. Mo isotopic signatures may be otherwise preserved in carbonate sediments [7,65,66], with little Mo isotope fractionation, during Mo incorporation into calcite [67]. However, authigenic Mo accumulations may be isotopically light and result in bulk values shifting away from seawater [9].
Mo concentrations, as with U, are higher in shallow Bahamian sediment than the primary precipitates that make up the sediment based on the results of the grab sample—core comparison. Mo concentrations are variable in the sediment cores (<0.1–30 ppm), while modern primary skeletal and non-skeletal precipitates are generally lower (0.005–0.17 ppm) [7,9]. Although a large percentage of the total concentration of Mo is soluble in the exchangeable phases of the sequential extraction (Figure 6), carbonate phase Mo concentrations in cores 1, 2, and 3 are similar to those of primary precipitates (~0.1 ppm) and is generally constant downcore. The similarities in Mo concentrations between the carbonate phase of cores 1, 2, and 3 and primary precipitates, in addition to consistent downcore carbonate phase Mo concentrations, suggests that the carbonate phase may retain Mo which coprecipitated with primary precipitates.
Core 4 incorporates progressively more Mo with depth, primarily into the exchangeable phases, but also into carbonate, oxide, and organic phases. As stable isotopes and REY patterns have shown, core 4 experiences a high terrestrial influx, effecting the preservation and uptake of trace metals. While much of the total Mo abundance occurs in the exchangeable phases, it is unlikely that remaining phases of core 4 consist only of Mo from primary precipitates as the concentration is much higher (~3–5 ppm) than those measured in primary precipitates.
Authigenic enrichment of Mo is mediated by the amount of free H2S in the pore waters produced through BSR [68]. As such, excluding an anomalously low Mo value in core 4 (3.3 ppm Mo at depth 28 cm), Mo and δ34S negatively correlate with an R2 of 0.47. Similarly, using H2S data from [8], Mo and H2S correlate slightly better with an R2 of 0.61, suggesting H2S accumulation is driving authigenic Mo accumulation in core 4. The remaining cores generally have poor correlations within themselves. However, when considering all the cores together in a net capacity, exchangeable Mo and δ34S negatively correlate (excluding the anomalous core 4 value). The net correlation of Mo and δ34S between all cores suggests that more intense BSR enhances authigenic Mo accumulation across the different depositional environments in the present study.
A “switching” point of 11 µM porewater H2S, after which MoO42− is rapidly converted to MoS42−has been previously proposed [69]. Further studies and modelling have suggested a step-wise conversion of MoO42− to MoS42−, with a series of intermediates, MoOxS4−x2−, and associated isotopic fractionations [9,60,70]. Sediments in which there is a quantitative conversion of MoO42− to MoS42− are thought to capture seawater Mo isotopic compositions, though it is unclear if shallow sediments (such as those found in The Bahamas) are capable of such conditions [9,60,63,70,71]. Alternatively, non-skeletal carbonate primary precipitates preserve seawater Mo isotopic values [7] with little Mo isotope fractionation during Mo incorporation into calcite [67]. By chemically removing the authigenic Mo fraction in bulk sediment through sequential extractions, it may be possible to mitigate the complexities of stepwise Mo fractionation occurring in pore waters and capture original seawater isotopic values in the non-exchangeable phases. Since the majority of bulk sediment Mo is found in the exchangeable phases (~50–85%), a mass balance of non-exchangeable phases could result in a wide range of isotopic values and requires further study to determine the potential for directly recording seawater values.
4.3.3. Vanadium Sequestration
The large fraction of V in the exchangeable phases suggests that free H2S in pore waters drives the uptake of V, similarly to Mo. V may be taken up in organic complexes as well as hydroxides, thus the presence of organics in these cores may aid in V uptake. In core 1, V concentrations increase sharply with depth below ~15 cm in the exchangeable phases while Mo concentrations remain relatively constant. The differing behavior between exchangeable phase Mo and V concentrations indicate that Mo may have been quantitatively removed from pore waters. Quantitative removal of Mo from pore waters is additionally evidenced by decreasing δ98Mo in the lower 20 cm of core 1 [9]. In core 4, a decrease in V in the bottom 16 cm can be explained by removal by flushing of oxic waters similar to the removal of U. While V concentrations are low in all cores (up to 12 ppm compared to average carbonates of 20 ppm) the increase with depth in the exchangeable phases and covariation with Mo suggests a redox-controlled uptake in The Bahamas sections [72]. Similar to the net correlation between exchangeable Mo and δ34S across all cores, exchangeable V and δ34S also show a negative correlation across all cores with an R2 of 0.68 (excluding two anomalous high V values from core 1 and V values from the bottom 16 cm of core 4). This likewise suggests a large control of BSR on authigenic V accumulation across different carbonate depositional environments.
4.4. U, Mo, and V Accumulation in Authigenic Carbonates
Results reported in the above sections indicate authigenic carbonate precipitation, associated with increased BSR in some sediment samples partially evidenced by decreasing δ13Ccarb beginning when δ34S decreases below 0 (Figure 7). Similar trends are seen between the carbonate phase of redox-sensitive trace elements (U, Mo, and V) and δ34S where U, Mo, and V concentrations increase when δ34S decreases below 0. Increased U, Mo, and V concentrations when δ34S < 0 indicates euxinic environments appear to precipitate authigenic carbonate which incorporates these elements and is indicative of a switch point threshold for the beginning of intense trace metal sequestration resulting from intense sulfate reduction. This seems most significant for Mo and V, which show concentrations 6x larger in sample where δ34S < 0 than in samples where δ34S > 0. Therefore, measuring δ34S concurrently with trace metal abundances is a potential screening tool for identifying the presence of secondary trace metals that were sequestered during syndepositional diagenesis which may obscure the primary seawater signature of the carbonate sediments.
4.5. Screening for Primary Geochemical Signals
Tidal flat sediments measured in core 2 lacked overlying vegetation and have low TOC (~0.2%), which indicates little organic matter input. Any organic matter that is input into the environment is quickly remineralized due to repeated reoxygenation that allows for more sustained aerobic respiration which is thermodynamically more favorable and kinetically faster than anaerobic respiration. BSR occurs further below the sediment-water interface (depths > ~10 cm as indicated by measured pore water H2S; [10]), though at a lower rate (relatively low H2S and high δ34S) than other depositional environments in this study supposedly due to lower TOC content. Periodic flushing of oxidized water and a low influence of BSR prevent accumulations of high U, Mo, and V concentrations in the exchangeable phases. Low amounts of BSR also prevent significant incorporation of U, Mo, and V in authigenic carbonates. Periodic flushing also does not alter the REY signature of the carbonates and suggests that the combined isotope and organic geochemistry indicate primary signatures.
Alternatively, cores from the slightly deeper, subtidal regime (cores 1 and 3) have elevated TOC with respect to the tidal flat (~0.8%). Pore waters are more reducing (elevated pore water H2S with respect to tidal flat; [8]) likely due to increased TOC input of mixed marine and terrestrial sources supported by higher TOC content than the tidal flat and correlation between δ13Corg and TOC. Porewaters are also more insulated from meteoric input than the tidal flat. The above support the sulfurization of Mo and V and the incorporation of reduced U into the loosely bound exchangeable phases as well as accumulation of U, Mo, and V into authigenic carbonates precipitated in these euxinic conditions. REY values of the subtidal realm, as with those of the tidal flat, reflect seawater values, suggesting that despite the conditions of this depositional environment, original seawater values are preserved in the carbonate phase.
The tidal pond deposits from core 4 have the highest concentration of TOC, the lightest δ13Corg values representing a large contribution of terrestrial organic input and supporting the most significant amount of BSR. The pond concentrates terrestrial input, unlike the other settings investigated, evidenced by elevated total Fe concentrations. Windblown dust supplies Fe to the surrounding island, as runoff carries it into and concentrates in the pond. The tidal pond study section has two distinct geochemical signatures. In the top 14 cm, bacterial respiration in the pore waters drive reducing conditions allowing for the sulfurization of Mo and V and the incorporation of reduced U into the loosely bound exchangeable phases as well as trace metal accumulation in authigenic carbonates, similar to enrichments seen in the subtidal flat. In contrast, in the lower 16 cm of core 4, flushing of oxic waters remobilized U and V, decreasing concentrations in the exchangeable phases with respect to the overlying 14 cm while Mo is still incorporated into the exchangeable phases during early diagenesis. High BSR supports a significant accumulation of U, Mo, and V into authigenic carbonates. REY distributions record an increase in terrestrial input (Y/Ho and Ce anomalies) and do not represent open marine pristine seawater conditions. Samples from Rawlins Creek and Page Creek possess similar geochemical signatures to core 4, but TOC values are missing and therefore a direct comparison is not possible. However, both of Rawlins and Page Creek originate from areas that drain enclosed shallow nearshore areas containing a high abundance of mangrove vegetation. The abundant mangrove vegetation may supply sediments in Rawlins and Page creek with substantial amounts organic carbon, relative to other sediments from intertidal flats, which would better facilitate the development of sulfate-reducing conditions. Overall, carbonate environments subject to enhanced organic matter and/or terrestrial mineral input are inadequate archives for reconstructing Earth history due to the accumulation of authigenic U, Mo, and V under BSR conditions, as well as the alteration of REY distributions by terrestrial mineral input.
5. Conclusions
In the shallow modern Bahamian cores of the present study, syndepositional diagenesis produces variable authigenic accumulation of U, Mo, and V depending on the intensity of BSR and, therefore, the depositional environment. While environments which receive little input of organic carbon are not subject to authigenic trace metal enrichments, such as the intertidal flat in the present study, environments which receive enhanced inputs of organic carbon may develop conditions which allow for accumulation of authigenic U, Mo, and V. Higher TOC in subtidal flat and tidal pond sediments allow for the development of sulfate-reducing conditions and precipitation of authigenic carbonate minerals, driving the authigenic uptake of U, Mo, and V in exchangeable and carbonate phases of the sediments. Diagenetic alteration by meteoric waters, however, may lead to removal of exchangeable phase U and V while authigenic enrichments of U and V in the carbonate phase remain.
While cores which experienced intense BSR yielded authigenic trace metal accumulation, PAAS-normalized REY values in many sediments reflect seawater characteristics, regardless of specific diagenetic circumstances. Cores with Y/Ho ratios indicative of a lack of terrestrial REY influence (~50–60) retained negative Ce anomalies (typical of oxidized seawater) and HREE enrichment. In restricted terrestrially dominated environments, REY values show evidence of terrestrial influence such as lower Y/Ho ratios (~44). Additionally, restricted terrestrially dominated environments possess more positive Ce anomalies, indicative of a near shore restricted environment or reducing waters at the time of carbonate precipitation.
Geochemical data presented from the depositional environments in the present study therefore suggest the following: (1) sulfate-reducing conditions in carbonate sediments may yield subsequent authigenic accumulations of U, Mo, and V and alter their viability as paleo-redox proxies and (2) PAAS-normalized REY values may retain seawater characteristics in varying diagenetic environments given the absence of significant terrestrial mineral input. Care should therefore be taken to ensure carbonates analyzed in future paleo-redox studies were not subjected to authigenic trace metal accumulation or authigenic accumulation should otherwise be accounted for. Additionally, future paleo-redox studies which involve analysis of carbonate REY distributions should ensure distributions are not significantly affected by terrestrial mineral input.
Author Contributions
Conceptualization, A.D.H.; methodology, A.D.H., A.T., Y.P. and E.M.; validation, A.D.H., A.T., Y.P. and E.M.; formal analysis, A.D.H., A.T., Y.P. and E.M.; investigation, A.D.H., A.T., Y.P. and E.M.; data curation, A.D.H.; writing—original draft preparation, E.M. and A.T.; writing—review and editing, A.D.H., A.T., Y.P. and E.M.; supervision, A.D.H.; project administration, A.D.H.; funding acquisition, A.D.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Petroleum Research Fund administered by the American Chemical Society, grant number ACS PRF # 55392-DNI2. The APC was funded by the journal.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Wanda Leblanc (LSU) who provided use of her lab for some portions of my sample preparations. We thank three anonymous reviewers for their constructive reviews that helped improve this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Luo, G.; Kump, L.R.; Wang, Y.; Tong, J.; Arthur, M.A.; Yang, H.; Huang, J.; Yin, H.; Xie, S. Isotopic evidence for an anomalously low oceanic sulfate concentration following end-Permian mass extinction. Earth Planet. Sci. Lett. 2010, 300, 101–111. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Planavsky, N.J.; Kendall, B.; Wang, X.; Shi, X.; Scott, C.; Anbar, A.D.; Lyons, T.W.; Jiang, G. Ocean oxygenation in the wake of the Marinoan glaciation. Nature 2012, 489, 546–549. [Google Scholar] [CrossRef]
- Hoffman, P.F.; Kaufman, A.J.; Halverson, G.P.; Schrag, D.P. A neoproterozoic snowball earth. Science 1998, 281, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Veizer, J.; Hoefs, J. The nature of O18/O16 and C13/C12 secular trends in sedimentary carbonate rocks. Geochim. Cosmochim. Acta 1976, 40, 1387–1395. [Google Scholar] [CrossRef]
- Weyer, S.; Anbar, A.; Gerdes, A.; Gordon, G.; Algeo, T.; Boyle, E. Natural fractionation of 238 U/235 U. Geochim. Cosmochim. Acta 2008, 72, 345–359. [Google Scholar] [CrossRef]
- Brennecka, G.A.; Herrmann, A.D.; Algeo, T.J.; Anbar, A.D. Rapid expansion of oceanic anoxia immediately before the end-Permian mass extinction. Proc. Natl. Acad. Sci. USA 2011, 108, 17631–17634. [Google Scholar] [CrossRef] [PubMed]
- Voegelin, A.R.; Nägler, T.F.; Samankassou, E.; Villa, I.M. Molybdenum isotopic composition of modern and Carboniferous carbonates. Chem. Geol. 2009, 265, 488–498. [Google Scholar] [CrossRef]
- Romaniello, S.J.; Herrmann, A.D.; Anbar, A.D. Uranium concentrations and 238 U/235 U isotope ratios in modern carbonates from the Bahamas: Assessing a novel paleoredox proxy. Chem. Geol. 2013, 362, 305–316. [Google Scholar] [CrossRef]
- Romaniello, S.J.; Herrmann, A.D.; Anbar, A.D. Syndepositional diagenetic control of molybdenum isotope variations in carbonate sediments from the Bahamas. Chem. Geol. 2016, 438, 84–90. [Google Scholar] [CrossRef]
- Andersen, M.; Romaniello, S.; Vance, D.; Little, S.; Herdman, R.; Lyons, T. A modern framework for the interpretation of 238 U/235 U in studies of ancient ocean redox. Earth Planet. Sci. Lett. 2014, 400, 184–194. [Google Scholar] [CrossRef]
- Oehlert, A.M.; Swart, P.K. Interpreting carbonate and organic carbon isotope covariance in the sedimentary record. Nat. Commun. 2014, 5, 4672. [Google Scholar] [CrossRef]
- Swart, P.K. The geochemistry of carbonate diagenesis: The past, present and future. Sedimentology 2015, 62, 1233–1304. [Google Scholar] [CrossRef]
- Baker, P.A.; Kastner, M. Constraints on the formation of sedimentary dolomite. Science 1981, 213, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Muhs, D.R.; Budahn, J.R.; Prospero, J.M.; Carey, S.N. Geochemical evidence for African dust inputs to soils of western Atlantic islands: Barbados, the Bahamas, and Florida. J. Geophys. Res. Earth Surf. 2007, 112, F02009. [Google Scholar] [CrossRef]
- Hearty, P.J. The Geology of Eleuthera Island, Bahamas: A Rosetta Stone of Quaternary Stratigraphy and Sea-Level History. Quat. Sci. Rev. 1998, 17, 333–355. [Google Scholar] [CrossRef]
- Conrad Neumann, A.; Hearty, P.J. Rapid sea-level changes at the close of the last interglacial (substage 5e) recorded in Bahamian island geology. Geology 1996, 24, 775–778. [Google Scholar] [CrossRef]
- Kindler, P.; Hearty, P.J. Carbonate petrography as an indicator of climate and sea-level changes: New data from Bahamian Quaternary units. Sedimentology 1996, 43, 381–399. [Google Scholar] [CrossRef]
- Rankey, E.C.; Reeder, S.L. Holocene oolitic marine sand complexes of the Bahamas. J. Sediment. Res. 2011, 81, 97–117. [Google Scholar] [CrossRef]
- Maynard, D.; Fletcher, W. Comparison of total and partial extractable copper in anomalous and background peat samples. J. Geochem. Explor. 1973, 2, 19–24. [Google Scholar] [CrossRef]
- Chao, T.; Zhou, L. Extraction techniques for selective dissolution of amorphous iron oxides from soils and sediments. Soil Sci. Soc. Am. J. 1983, 47, 225–232. [Google Scholar] [CrossRef]
- Tessier, A.; Campbell, P.G.; Bisson, M. Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem. 1979, 51, 844–851. [Google Scholar] [CrossRef]
- Šurija, B.; Branica, M. Distribution of Cd, Pb, Cu and Zn in carbonate sediments from the Krka river estuary obtained by sequential extraction. Sci. Total Environ. 1995, 170, 101–118. [Google Scholar] [CrossRef]
- Morin, G.; Mangeret, A.; Othmane, G.; Stetten, L.; Seder-Colomina, M.; Brest, J.; Ona-Nguema, G.; Bassot, S.; Courbet, C.; Guillevic, J. Mononuclear U (IV) complexes and ningyoite as major uranium species in lake sediments. Geochem. Perspect. Lett. 2016, 2, 95–105. [Google Scholar] [CrossRef]
- Phillips, E.J.; Landa, E.R.; Lovley, D.R. Remediation of uranium contaminated soils with bicarbonate extraction and microbial U (VI) reduction. J. Ind. Microbiol. 1995, 14, 203–207. [Google Scholar] [CrossRef]
- Alessi, D.S.; Uster, B.; Veeramani, H.; Suvorova, E.I.; Lezama-Pacheco, J.S.; Stubbs, J.E.; Bargar, J.R.; Bernier-Latmani, R. Quantitative separation of monomeric U(IV) from UO2 in products of U(VI) reduction. Environ. Sci. Technol. 2012, 46, 6150–6157. [Google Scholar] [CrossRef]
- Alessi, D.S.; Lezama-Pacheco, J.S.; Stubbs, J.E.; Janousch, M.; Bargar, J.R.; Persson, P.; Bernier-Latmani, R. The product of microbial uranium reduction includes multiple species with U (IV)–phosphate coordination. Geochim. Cosmochim. Acta 2014, 131, 115–127. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Campbell, K.M.; Kelly, S.D.; Roebbert, Y.; Weyer, S.; Bernier-Latmani, R.; Borch, T. Biogenic non-crystalline U (IV) revealed as major component in uranium ore deposits. Nat. Commun. 2017, 8, 15538. [Google Scholar] [CrossRef]
- Sharp, J.O.; Lezama-Pacheco, J.S.; Schofield, E.J.; Junier, P.; Ulrich, K.-U.; Chinni, S.; Veeramani, H.; Margot-Roquier, C.; Webb, S.M.; Tebo, B.M. Uranium speciation and stability after reductive immobilization in aquifer sediments. Geochim. Cosmochim. Acta 2011, 75, 6497–6510. [Google Scholar] [CrossRef]
- Pourmand, A.; Dauphas, N.; Ireland, T.J. A novel extraction chromatography and MC-ICP-MS technique for rapid analysis of REE, Sc and Y: Revising CI-chondrite and Post-Archean Australian Shale (PAAS) abundances. Chem. Geol. 2012, 291, 38–54. [Google Scholar] [CrossRef]
- Lawrence, M.G.; Greig, A.; Collerson, K.D.; Kamber, B.S. Rare earth element and yttrium variability in South East Queensland waterways. Aquat. Geochem. 2006, 12, 39–72. [Google Scholar] [CrossRef]
- Lajtha, K.; Marshall, J. Sources of variation in the stable isotopic composition of plants. In Stable Isotopes in Ecology and Environmental Science; Lajtha, K., Michener, R.H., Eds.; Blackwell Scientific Publications: Oxford, UK, 1994; pp. 1–21. [Google Scholar]
- Meyers, P.A. Preservation of elemental and isotopic source identification of sedimentary organic matter. Chem. Geol. 1994, 114, 289–302. [Google Scholar] [CrossRef]
- Cloern, J.E.; Canuel, E.A.; Harris, D. Stable carbon and nitrogen isotope composition of aquatic and terrestrial plants of the San Francisco Bay estuarine system. Limnol. Oceanogr. 2002, 47, 713–729. [Google Scholar] [CrossRef]
- Canfield, D.E.; Boudreau, B.P.; Mucci, A.; Gundersen, J.K. The early diagenetic formation of organic sulfur in the sediments of Mangrove Lake, Bermuda. Geochim. Et Cosmochim. Acta 1998, 62, 767–781. [Google Scholar] [CrossRef]
- Walter, L.M.; Burton, E.A. Dissolution of recent platform carbonate sediments in marine pore fluids. Am. J. Sci. 1990, 290, 601–643. [Google Scholar] [CrossRef]
- Glud, R.N. Oxygen dynamics of marine sediments. Mar. Biol. Res. 2008, 4, 243–289. [Google Scholar] [CrossRef]
- Bau, M.; Dulski, P. Distribution of yttrium and rare-earth elements in the Penge and Kuruman iron-formations, Transvaal Supergroup, South Africa. Precambrian Res. 1996, 79, 37–55. [Google Scholar] [CrossRef]
- Elderfield, H.; Greaves, M.J. The rare earth elements in seawater. Nature 1982, 296, 214–219. [Google Scholar] [CrossRef]
- Tostevin, R.; Shields, G.A.; Tarbuck, G.M.; He, T.; Clarkson, M.O.; Wood, R.A. Effective use of cerium anomalies as a redox proxy in carbonate-dominated marine settings. Chem. Geol. 2016, 438, 146–162. [Google Scholar] [CrossRef]
- Webb, G.E.; Kamber, B.S. Rare earth elements in Holocene reefal microbialites: A new shallow seawater proxy. Geochim. Cosmochim. Acta 2000, 64, 1557–1565. [Google Scholar] [CrossRef]
- Piper, D.Z.; Bau, M. Normalized rare earth elements in water, sediments, and wine: Identifying sources and environmental redox conditions. Am. J. Anal. Chem. 2013, 4, 69. [Google Scholar] [CrossRef]
- Osborne, A.H.; Haley, B.A.; Hathorne, E.C.; Plancherel, Y.; Frank, M. Rare earth element distribution in Caribbean seawater: Continental inputs versus lateral transport of distinct REE compositions in subsurface water masses. Mar. Chem. 2015, 177, 172–183. [Google Scholar] [CrossRef]
- Webb, G.E.; Nothdurft, L.D.; Kamber, B.S.; Kloprogge, J.; ZHAO, J.X. Rare earth element geochemistry of scleractinian coral skeleton during meteoric diagenesis: A sequence through neomorphism of aragonite to calcite. Sedimentology 2009, 56, 1433–1463. [Google Scholar] [CrossRef]
- Liu, X.; Hardisty, D.; Lyons, T.W.; Swart, P. Evaluating the integrity of the cerium paleoredox tracer through analysis of a modern marine carbonate analog. Geochim. Cosmochim. Acta 2018, 248, 25–42. [Google Scholar] [CrossRef]
- Lovley, D.R.; Phillips, E.J.; Gorby, Y.A.; Landa, E.R. Microbial reduction of uranium. Nature 1991, 350, 413. [Google Scholar] [CrossRef]
- McManus, J.; Berelson, W.M.; Klinkhammer, G.P.; Hammond, D.E.; Holm, C. Authigenic uranium: Relationship to oxygen penetration depth and organic carbon rain. Geochim. Cosmochim. Acta 2005, 69, 95–108. [Google Scholar] [CrossRef]
- Dunk, R.; Mills, R.; Jenkins, W. A reevaluation of the oceanic uranium budget for the Holocene. Chem. Geol. 2002, 190, 45–67. [Google Scholar] [CrossRef]
- Chen, X.; Romaniello, S.J.; Herrmann, A.D.; Wasylenki, L.E.; Anbar, A.D. Uranium isotope fractionation during coprecipitation with aragonite and calcite. Geochim. Cosmochim. Acta 2016, 188, 189–207. [Google Scholar] [CrossRef]
- Chen, X.; Romaniello, S.J.; Herrmann, A.D.; Samankassou, E.; Anbar, A.D. Biological effects on uranium isotope fractionation (238U/235U) in primary biogenic carbonates. Geochim. Cosmochim. Acta 2018, 240, 1–10. [Google Scholar] [CrossRef]
- Livermore, B.; Dahl, T.; Bizzarro, M.; Connelly, J. Uranium isotope compositions of biogenic carbonates–Implications for U uptake in shells and the application of the paleo-ocean oxygenation proxy. Geochim. Cosmochim. Acta 2020, 287, 50–64. [Google Scholar] [CrossRef]
- Rolison, J.M.; Stirling, C.H.; Middag, R.; Rijkenberg, M.J. Uranium stable isotope fractionation in the Black Sea: Modern calibration of the 238U/235U paleo-redox proxy. Geochim. Cosmochim. Acta 2017, 203, 69–88. [Google Scholar] [CrossRef]
- Stirling, C.H.; Andersen, M.B.; Potter, E.-K.; Halliday, A.N. Low-temperature isotopic fractionation of uranium. Earth Planet. Sci. Lett. 2007, 264, 208–225. [Google Scholar] [CrossRef]
- Elrick, M.; Polyak, V.; Algeo, T.J.; Romaniello, S.; Asmerom, Y.; Herrmann, A.D.; Anbar, A.D.; Zhao, L.; Chen, Z.-Q. Global-ocean redox variation during the middle-late Permian through Early Triassic based on uranium isotope and Th/U trends of marine carbonates. Geology 2017, 45, 163–166. [Google Scholar] [CrossRef]
- Song, H.; Song, H.; Algeo, T.J.; Tong, J.; Romaniello, S.J.; Zhu, Y.; Chu, D.; Gong, Y.; Anbar, A.D. Uranium and carbon isotopes document global-ocean redox-productivity relationships linked to cooling during the Frasnian-Famennian mass extinction. Geology 2017, 45, 887–890. [Google Scholar] [CrossRef]
- Clarkson, M.O.; Stirling, C.H.; Jenkyns, H.C.; Dickson, A.J.; Porcelli, D.; Moy, C.M.; Pogge von Strandmann, P.A.E.; Cooke, I.R.; Lenton, T.M. Uranium isotope evidence for two episodes of deoxygenation during Oceanic Anoxic Event 2. Proc. Natl. Acad. Sci. USA 2018, 115, 2918–2923. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Romaniello, S.J.; Herrmann, A.D.; Hardisty, D.; Gill, B.C.; Anbar, A.D. Diagenetic effects on uranium isotope fractionation in carbonate sediments from the Bahamas. Geochim. Cosmochim. Acta 2018, 237, 294–311. [Google Scholar] [CrossRef]
- Tissot, F.L.; Chen, C.; Go, B.M.; Naziemiec, M.; Healy, G.; Bekker, A.; Swart, P.K.; Dauphas, N. Controls of eustasy and diagenesis on the 238U/235U of carbonates and evolution of the seawater (234U/238U) during the last 1.4 Myr. Geochim. Cosmochim. Acta 2018, 242, 233–265. [Google Scholar] [CrossRef]
- Anbar, A.D. Molybdenum stable isotopes: Observations, interpretations and directions. Rev. Mineral. Geochem. 2004, 55, 429–454. [Google Scholar] [CrossRef]
- Dahl, T.W.; Canfield, D.E.; Rosing, M.T.; Frei, R.E.; Gordon, G.W.; Knoll, A.H.; Anbar, A.D. Molybdenum evidence for expansive sulfidic water masses in~ 750Ma oceans. Earth Planet. Sci. Lett. 2011, 311, 264–274. [Google Scholar] [CrossRef]
- Herrmann, A.D.; Kendall, B.; Algeo, T.J.; Gordon, G.W.; Wasylenki, L.E.; Anbar, A.D. Anomalous molybdenum isotope trends in Upper Pennsylvanian euxinic facies: Significance for use of δ98Mo as a global marine redox proxy. Chem. Geol. 2012, 324, 87–98. [Google Scholar] [CrossRef]
- Siebert, C.; Kramers, J.; Meisel, T.; Morel, P.; Nägler, T.F. PGE, Re-Os, and Mo isotope systematics in Archean and early Proterozoic sedimentary systems as proxies for redox conditions of the early Earth. Geochim. Cosmochim. Acta 2005, 69, 1787–1801. [Google Scholar] [CrossRef]
- Arthur, M.A.; Sageman, B.B. Marine black shales: Depositional mechanisms and environments of ancient deposits. Annu. Rev. Earth Planet. Sci. 1994, 22, 499–551. [Google Scholar] [CrossRef]
- Gordon, G.; Lyons, T.; Arnold, G.L.; Roe, J.; Sageman, B.; Anbar, A. When do black shales tell molybdenum isotope tales? Geology 2009, 37, 535–538. [Google Scholar] [CrossRef]
- Siebert, C.; McManus, J.; Bice, A.; Poulson, R.; Berelson, W.M. Molybdenum isotope signatures in continental margin marine sediments. Earth Planet. Sci. Lett. 2006, 241, 723–733. [Google Scholar] [CrossRef]
- Bura-Nakić, E.; Andersen, M.B.; Archer, C.; de Souza, G.F.; Marguš, M.; Vance, D. Coupled Mo-U abundances and isotopes in a small marine euxinic basin: Constraints on processes in euxinic basins. Geochim. Cosmochim. Acta 2018, 222, 212–229. [Google Scholar] [CrossRef]
- Kendall, B.; Dahl, T.W.; Anbar, A.D. The stable isotope geochemistry of molybdenum. Rev. Mineral. Geochem. 2017, 82, 683–732. [Google Scholar] [CrossRef]
- Chen, X.; Romaniello, S.J.; Anbar, A.D. Preliminary exploration of molybdenum isotope fractionation during coprecipitation of molybdate with abiotic and microbial calcite. Chem. Geol. 2021, 566, 120102. [Google Scholar] [CrossRef]
- Burdige, D.J.; Zimmerman, R.C. Impact of sea grass density on carbonate dissolution in Bahamian sediments. Limnol. Oceanogr. 2002, 47, 1751–1763. [Google Scholar] [CrossRef]
- Erickson, B.E.; Helz, G.R. Molybdenum(VI) speciation in sulfidic waters. Geochim. Cosmochim. Acta 2000, 64, 1149–1158. [Google Scholar] [CrossRef]
- Dahl, T.W.; Anbar, A.D.; Gordon, G.W.; Rosing, M.T.; Frei, R.; Canfield, D.E. The behavior of molybdenum and its isotopes across the chemocline and in the sediments of sulfidic Lake Cadagno, Switzerland. Geochim. Cosmochim. Acta 2010, 74, 144–163. [Google Scholar] [CrossRef]
- Neubert, N.; Nägler, T.F.; Böttcher, M.E. Sulfidity controls molybdenum isotope fractionation into euxinic sediments: Evidence from the modern Black Sea. Geology 2008, 36, 775–778. [Google Scholar] [CrossRef]
- Turekian, K.K.; Wedepohl, K.H. Distribution of the elements in some major units of the earth’s crust. Geol. Soc. Am. Bull. 1961, 72, 175–192. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).