
Genome-Wide Association Study for Weight-Related Traits in Scylla paramamosain Using Whole-Genome Resequencing



Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample and Phenotype Collection
2.2. DNA Extraction, Sequencing, and Variant Calling
2.3. Phenotypic Heritability and Correlations
2.4. Population Structure Analysis
2.5. Genome-Wide Association Study
2.6. Gene Annotation and Functional Enrichment Analysis
3. Results
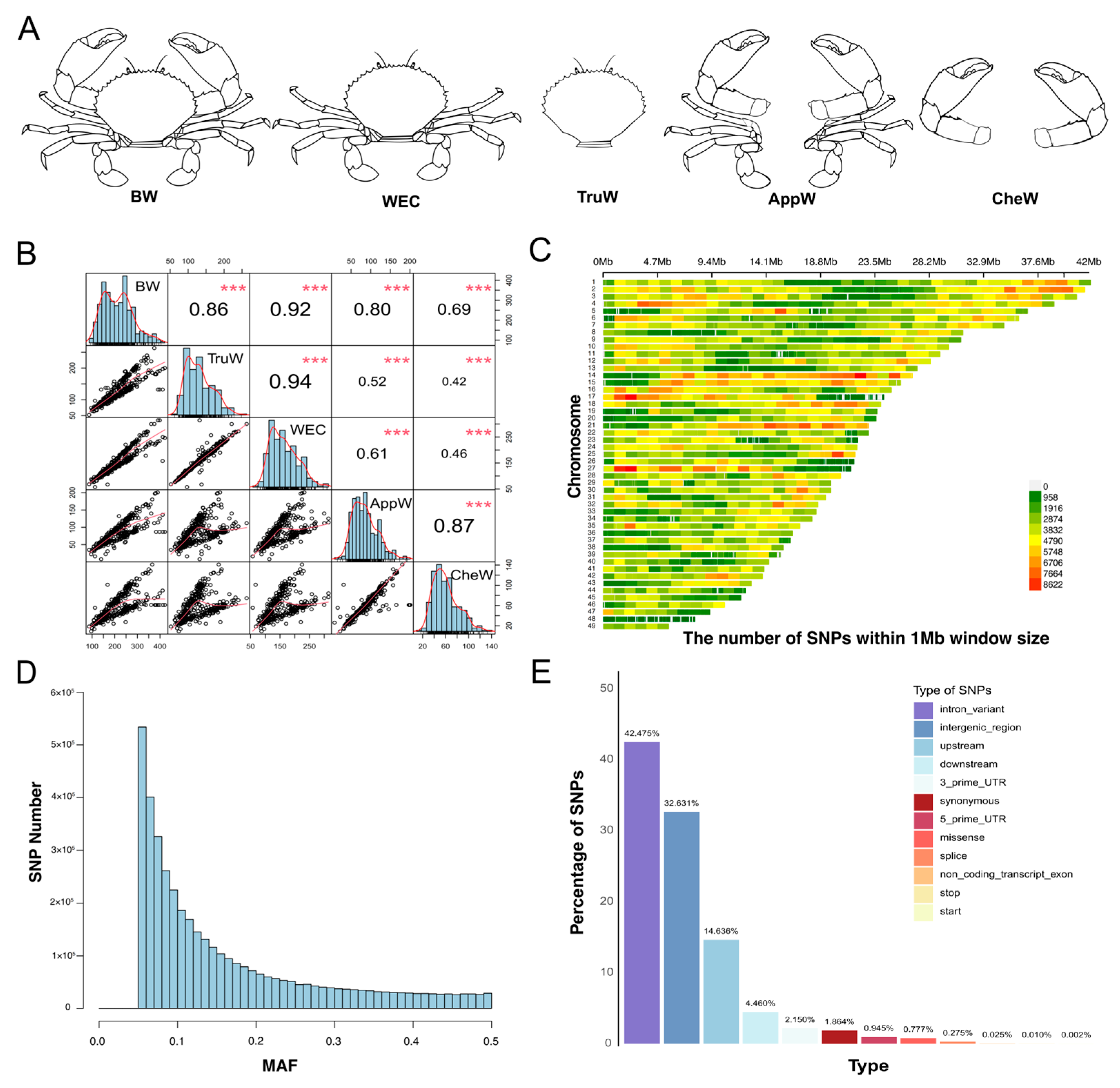
3.1. Measurement of Phenotypic and Genomic Data
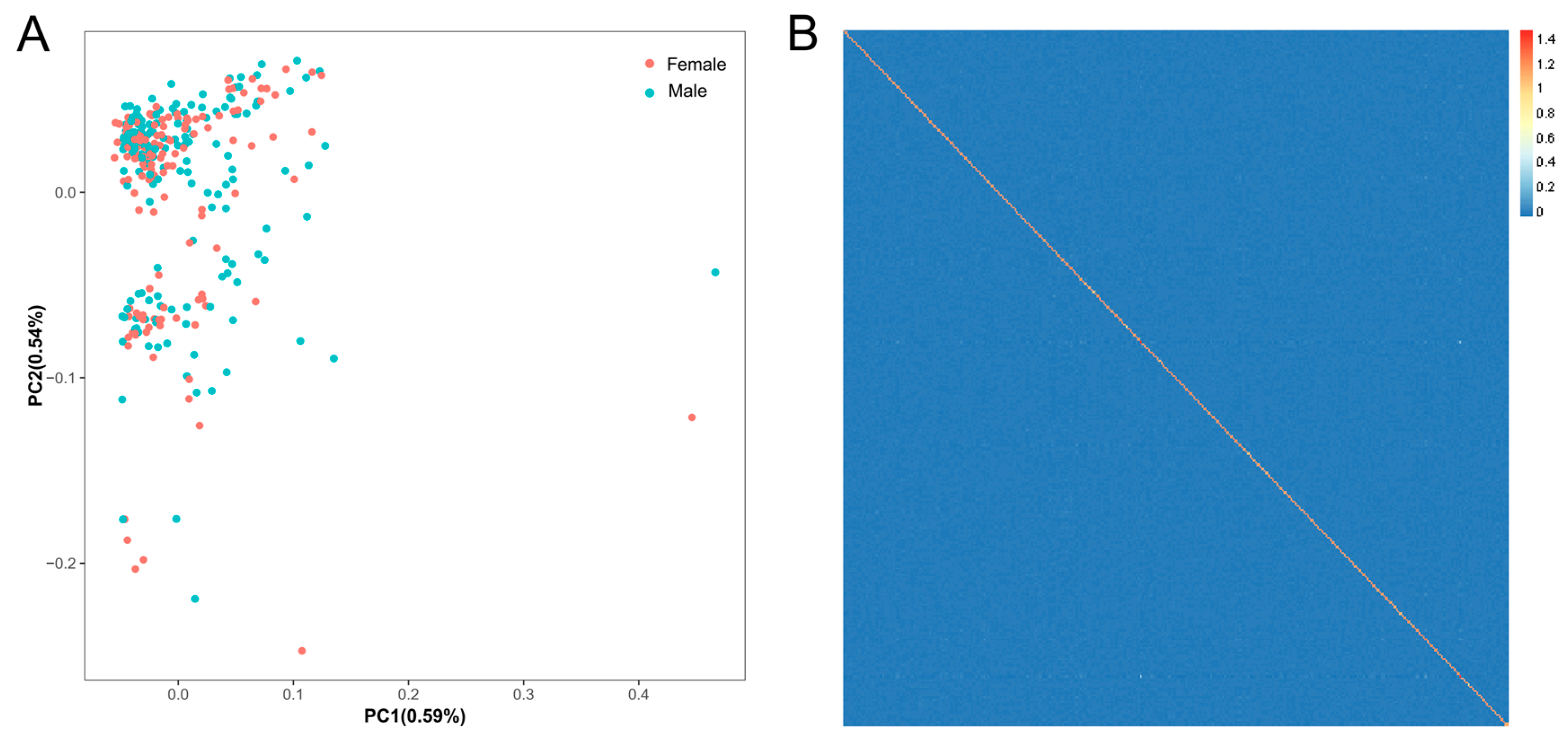
3.2. Population Structure
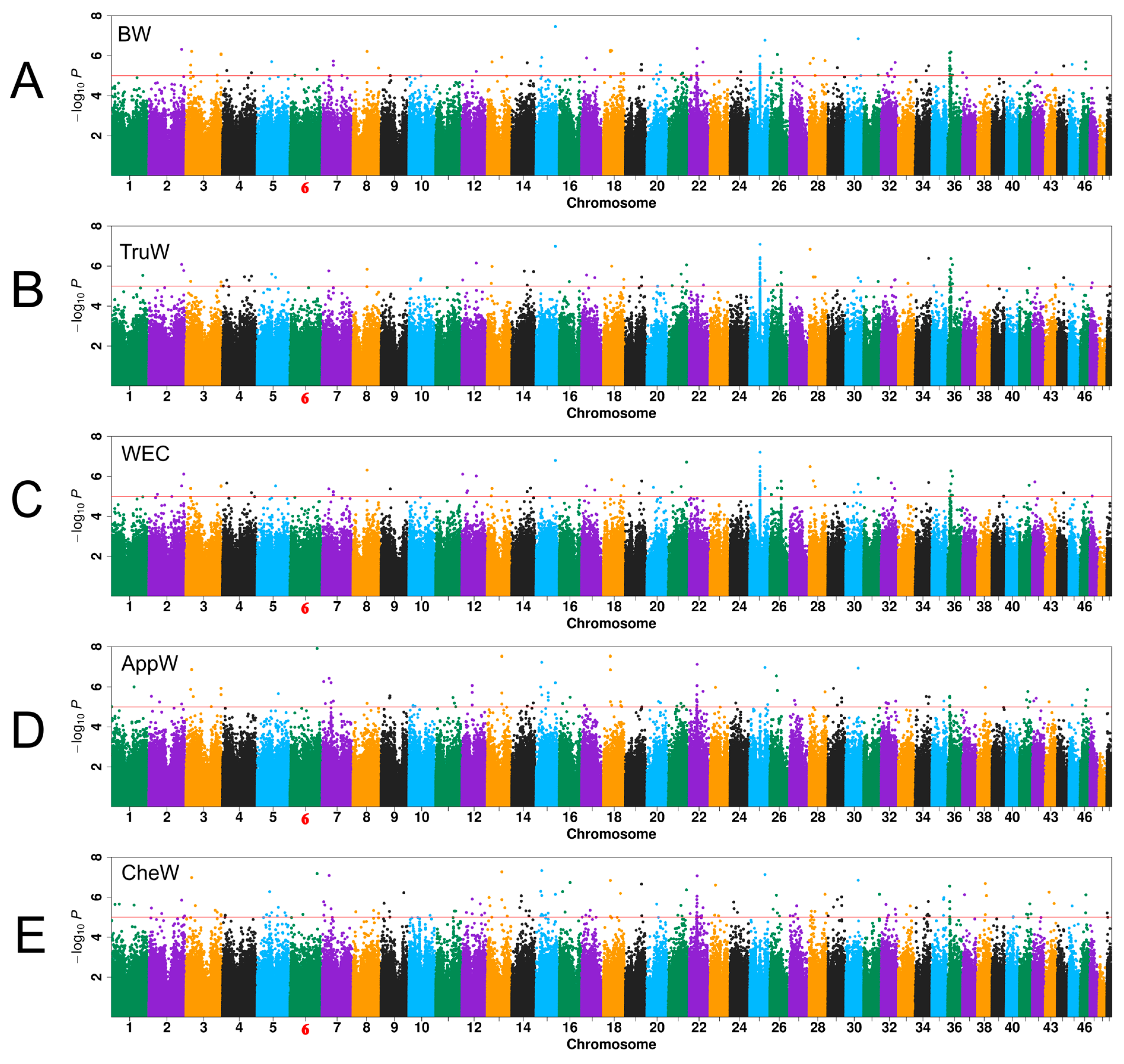
3.3. GWAS Results
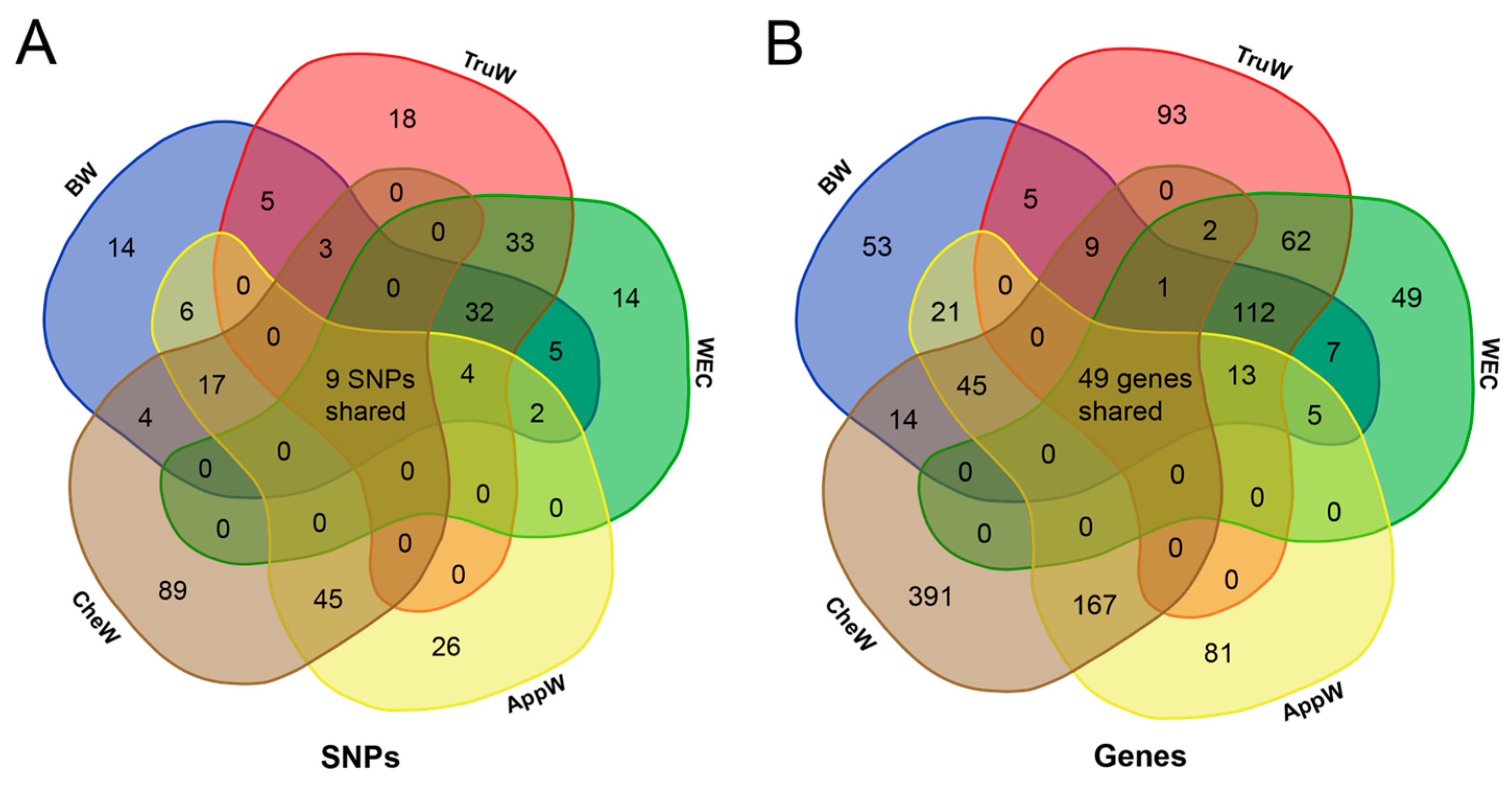
3.4. Functional Annotation
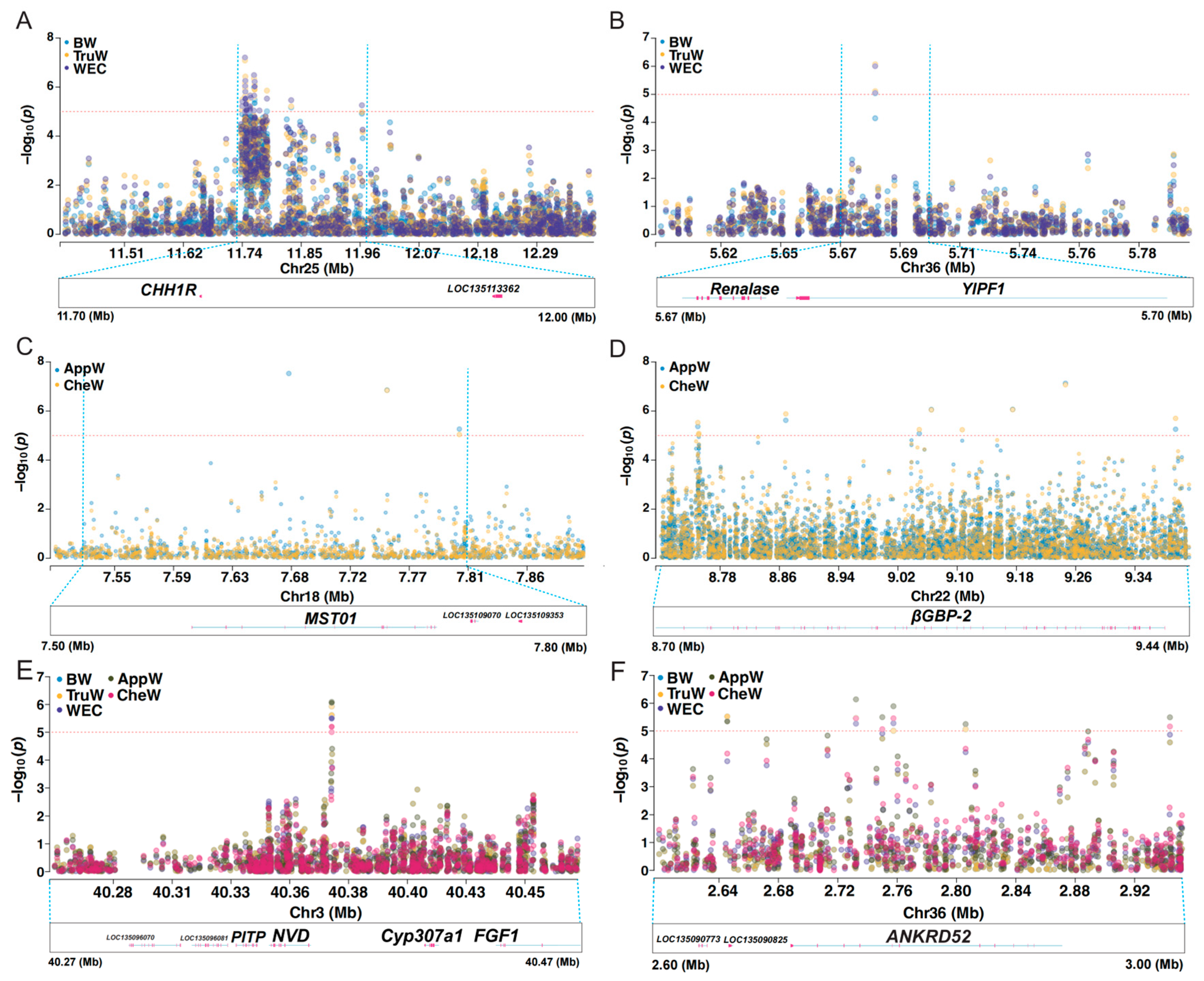
3.5. Key Candidate Area
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, M.; Wang, W.; Zhang, F.; Ma, C.; Liu, Z.; Yang, M.H.; Chen, W.; Li, Q.; Cui, M.; Jiang, K.; et al. A chromosome-level genome of the mud crab (Scylla paramamosain estampador) provides insights into the evolution of chemical and light perception in this crustacean. Mol. Ecol. Resour. 2021, 21, 1299–1317. [Google Scholar] [CrossRef] [PubMed]
- Keenan, C.; Davie, P.J.F.; Mann, D.L. A revision of the genus Scylla de Haan, 1833 (Crustacea: Decapoda: Brachyura: Portunidae). Raffles Bull. Zool. 1998, 46, 217–245. [Google Scholar]
- Lovatelli, A.; Shelley, C.; Tobias-Quinitio, E.; Khor, W.; Chan, D. Status, technological innovations, and industry development needs of mud crab (Scylla spp.) aquaculture. In Proceedings of the FAO Expert Workshop, Singapore, 27–30 November 2023; FAO Fisheries and Aquaculture Proceedings, No. 73. FAO: Rome, Italy, 2023. [Google Scholar]
- Li, Y.; Ai, C.; Liu, L. Mud crab, Scylla paramamosain China’s leading maricultured crab. In Aquaculture in China: Success Stories and Modern Trends; Wiley: Hoboken, NJ, USA, 2018; pp. 226–233. [Google Scholar]
- FAO. FAO Yearbook of Fishery and Aquaculture Statistics. In Fishery and Aquaculture Statistics—Yearbook 2021; FAO: Rome, Italy, 2024. [Google Scholar] [CrossRef]
- Syafaat, M.N.; Azra, M.N.; Waiho, K.; Fazhan, H.; Abol-Munafi, A.B.; Ishak, S.D.; Syahnon, M.; Ghazali, A.; Ma, H.; Ikhwanuddin, M. A review of the nursery culture of mud crabs, genus Scylla: Current progress and future directions. Animals 2021, 11, 2034. [Google Scholar] [CrossRef] [PubMed]
- Reis Neto, R.V.; Yoshida, G.M.; Lhorente, J.P.; Yáñez, J.M. Genome-wide association analysis for body weight identifies candidate genes related to development and metabolism in rainbow trout (Oncorhynchus mykiss). Mol. Genet. Genom. 2019, 294, 563–571. [Google Scholar] [CrossRef]
- Yu, Y.; Wan, S.M.; Zhang, S.M.; Liu, J.Q.; Sun, A.L.; Wang, Y.; Zhu, Y.F.; Gu, S.X.; Gao, Z.X. Identification of SNPs and candidate genes associated with growth using GWAS and transcriptome analysis in Coilia nasus. Aquaculture 2024, 586, 740777. [Google Scholar] [CrossRef]
- Gutierrez, A.P.; Yáñez, J.M.; Fukui, S.; Swift, B.; Davidson, W.S. Genome-wide association study (GWAS) for growth rate and age at sexual maturation in Atlantic salmon (Salmo salar). PLoS ONE 2015, 10, e0119730. [Google Scholar] [CrossRef]
- Leeds, T.D.; Vallejo, R.L.; Weber, G.M.; Gonzalez-Pena, D.; Silverstein, J.T. Response to five generations of selection for growth performance traits in rainbow trout (Oncorhynchus mykiss). Aquaculture 2016, 465, 341–351. [Google Scholar] [CrossRef]
- Omeka, W.K.M.; Liyanage, D.S.; Lee, S.; Lim, C.; Yang, H.; Sandamalika, W.M.G.; Udayantha, H.M.V.; Kim, G.; Ganeshalingam, S.; Jeong, T. Genome-wide association study (GWAS) of growth traits in olive flounder (Paralichthys olivaceus). Aquaculture 2022, 555, 738257. [Google Scholar] [CrossRef]
- Oikonomou, S.; Samaras, A.; Tekeoglou, M.; Loukovitis, D.; Dimitroglou, A.; Kottaras, L.; Papanna, K.; Papaharisis, L.; Tsigenopoulos, C.S.; Pavlidis, M. Genomic selection and genome-wide association analysis for stress response, disease resistance and body weight in European seabass. Animals 2022, 12, 277. [Google Scholar] [CrossRef]
- Zhou, Z.; Shao, G.; Shen, Y.; He, F.; Tu, X.; Ji, J.; Ao, J.; Chen, X. Extreme-phenotype genome-wide association analysis for growth traits in spotted sea bass (Lateolabrax maculatus) using whole-genome resequencing. Animals 2024, 14, 2995. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, L.; Wu, X.; Li, B.; Huang, W.; Weng, Z.; Lin, Z.; Song, L.; Guo, Y.; Meng, Z. Identification of candidate growth-related SNPs and genes using GWAS in brown-marbled grouper (Epinephelus fuscoguttatus). Mar. Biotechnol. 2020, 22, 153–166. [Google Scholar] [CrossRef]
- Houston, R.D.; Bean, T.P.; Macqueen, D.J.; Gundappa, M.K.; Jin, Y.H.; Jenkins, T.L.; Selly, S.L.C.; Martin, S.A.M.; Stevens, J.R.; Santos, E.M. Harnessing genomics to fast-track genetic improvement in aquaculture. Nat. Rev. Genet. 2020, 21, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Lyu, D.; Yu, Y.; Wang, Q.; Luo, Z.; Zhang, Q.; Zhang, X.; Xiang, J.; Li, F. Identification of growth-associated genes by genome-wide association study and their potential application in the breeding of Pacific white shrimp (Litopenaeus vannamei). Front. Genet. 2021, 12, 611570. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.F.; Mastrochirico-Filho, V.A.; Gallardo-Hidalgo, J.; Campos-Montes, G.R.; Medrano-Mendoza, T.; Rivero-Martínez, P.V.; Caballero-Zamora, A.; Hashimoto, D.T.; Yáñez, J.M. A high-density linkage map and sex-determination loci in Pacific white shrimp (Litopenaeus vannamei). BMC Genom. 2024, 25, 565. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, W.; Jiang, S.; Qiao, H.; Xiong, Y.; Jin, S.; Fu, H. Genome-wide association and transcriptomic analysis and the identification of growth-related genes in Macrobrachium nipponense. BMC Genom. 2024, 25, 1182. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.-Y.; Xu, Y.-F.; Feng, W.-R.; He, Q.-H.; Hua, G.-A.; Li, W.-J.; Xu, P.; Zhou, J.; Tang, Y.-K. Genomic analysis of hypoxia-tolerant population of the Chinese mitten crab (Eriocheir sinensis). Fish Shellfish Immunol. 2024, 154, 109931. [Google Scholar] [CrossRef]
- Santos, J.L.; Nick, F.; Adhitama, N.; Fields, P.D.; Stillman, J.H.; Kato, Y.; Watanabe, H.; Ebert, D. Trehalose mediates salinity-stress tolerance in natural populations of a freshwater crustacean. Curr. Biol. 2024, 34, 4160–4169. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, Y.; Zhang, M.; Yu, X.; Qiu, B.; Wu, F.; Tocher, D.R.; Zhang, J.; Ye, S.; Cui, W. High-resolution chromosome-level genome of Scylla paramamosain provides molecular insights into adaptive evolution in crabs. BMC Biol. 2024, 22, 255. [Google Scholar] [CrossRef]
- Ye, S.; Zhou, X.; Ouyang, M.; Cui, W.; Xiang, Z.; Zhang, Y.; Yuan, Y.; Ikhwanuddin, M.; Li, S.; Zheng, H. Development and validation of a 40 K liquid SNP array for the mud crab (Scylla paramamosain). Aquaculture 2025, 594, 741394. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2023, 2, e107. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Yin, D.; Fu, Y.; Yuan, X.; Li, X.; Liu, X.; Zhao, S. HIBLUP: An integration of statistical models on the BLUP framework for efficient genetic evaluation using big genomic data. Nucleic Acids Res. 2023, 51, 3501–3512. [Google Scholar] [CrossRef]
- Peterson, B.G.; Carl, P.; Boudt, K.; Bennett, R.; Ulrich, J.; Zivot, E.; Lestel, M.; Balkissoon, K.; Wuertz, D.; Christidis, A.A.; et al. PerformanceAnalytics: Econometric Tools for Performance and Risk Analysis. R Package Version 2.0.4. Available online: https://CRAN.R-project.org/package=PerformanceAnalytics (accessed on 16 June 2025).
- Wickham, H.; Sievert, C. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps. R Package Version 1.0.12. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 16 June 2025).
- Zheng, J.-S.; Lai, C.-Q.; Parnell, L.D.; Lee, Y.-C.; Shen, J.; Smith, C.E.; Casas-Agustench, P.; Richardson, K.; Li, D.; Noel, S.E. Genome-wide interaction of genotype by erythrocyte n-3 fatty acids contributes to phenotypic variance of diabetes-related traits. BMC Genom. 2014, 15, 781. [Google Scholar] [CrossRef]
- Fang, L.; Wang, Q.; Hu, Y.; Jia, Y.; Chen, J.; Liu, B.; Zhang, Z.; Guan, X.; Chen, S.; Zhou, B. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 2017, 49, 1089–1098. [Google Scholar] [CrossRef]
- Naderi, E.; Crijns, A.P.G.; Steenbakkers, R.J.H.M.; van den Hoek, J.G.M.; Boezen, H.M.; Alizadeh, B.Z.; Langendijk, J.A. A two-stage genome-wide association study of radiation-induced acute toxicity in head and neck cancer. J. Transl. Med. 2021, 19, 481. [Google Scholar] [CrossRef]
- Shim, H.; Chasman, D.I.; Smith, J.D.; Mora, S.; Ridker, P.M.; Nickerson, D.A.; Krauss, R.M.; Stephens, M. A multivariate genome-wide association analysis of 10 LDL subfractions, and their response to statin treatment, in 1868 Caucasians. PLoS ONE 2015, 10, e0120758. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Lang, A.-G.; Buchner, A. G* Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Gatti, D.M.; Svenson, K.L.; Shabalin, A.; Wu, L.-Y.; Valdar, W.; Simecek, P.; Goodwin, N.; Cheng, R.; Pomp, D.; Palmer, A. Quantitative trait locus mapping methods for diversity outbred mice. G3 Genes Genomes Genet. 2014, 4, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, Y.; Liu, J.; Chen, F.; Guo, Y.; Ke, H.; Wang, X.; Luo, M.; Fu, S. Genome-wide association study reveals genomic loci of sex differentiation and gonadal development in Plectropomus leopardus. Front. Genet. 2023, 14, 1229242. [Google Scholar] [CrossRef]
- Silva, E.F.P.; Gaia, R.C.; Mulim, H.A.; Pinto, L.F.B.; Iung, L.H.S.; Brito, L.F.; Pedrosa, V.B. Genome-wide association study of conformation traits in Brazilian holstein cattle. Animals 2024, 14, 2472. [Google Scholar] [CrossRef]
- Fonseca, P.A.; Suárez-Vega, A.; Marras, G.; Cánovas, A. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. Gigascience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Carlson, M.; Pagès, H. AnnotationForge: Tools For Building SQLite-Based Annotation Data Packages. R Package Version 1.44.0. 2019. Available online: https://bioconductor.org/packages/AnnotationForge (accessed on 16 June 2025).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.I. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Browning, B.L.; Tian, X.; Zhou, Y.; Browning, S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef]
- Smith, J.L.; Wilson, M.L.; Nilson, S.M.; Rowan, T.N.; Schnabel, R.D.; Decker, J.E.; Seabury, C.M. Genome-wide association and genotype by environment interactions for growth traits in US Red Angus cattle. BMC Genom. 2022, 23, 517. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Tang, Y.; Liu, F.; Lu, L.; Liu, A.; Ye, H. Evaluation of the Effect of Adipokinetic Hormone/Corazonin-Related Peptide (ACP) on Ovarian Development in the Mud Crab, Scylla paramamosain. Animals 2024, 14, 3706. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Q.; Fang, S.; Li, S.; Zheng, H.; Zhang, Y.; Ikhwanuddin, M.; Ma, H. mRNA profile provides novel insights into stress adaptation in mud crab megalopa, Scylla paramamosain after salinity stress. BMC Genom. 2020, 21, 559. [Google Scholar] [CrossRef] [PubMed]
- Waiho, K.; Shi, X.; Fazhan, H.; Li, S.; Zhang, Y.; Zheng, H.; Liu, W.; Fang, S.; Ikhwanuddin, M.; Ma, H. High-density genetic linkage maps provide novel insights into ZW/ZZ sex determination system and growth performance in mud crab (Scylla paramamosain). Front. Genet. 2019, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, W.; Chen, W.; Ma, C.; Zhang, F.; Jiang, K.; Liu, J.; Diao, L.; Qian, H.; Zhao, J. Genome survey, high-resolution genetic linkage map construction, growth-related quantitative trait locus (QTL) identification and gene location in Scylla paramamosain. Sci. Rep. 2019, 9, 2910. [Google Scholar] [CrossRef]
- Yengo, L.; Vedantam, S.; Marouli, E.; Sidorenko, J.; Bartell, E.; Sakaue, S.; Graff, M.; Eliasen, A.U.; Jiang, Y.; Raghavan, S. A saturated map of common genetic variants associated with human height. Nature 2022, 610, 704–712. [Google Scholar] [CrossRef]
- Jonsdottir, A.B.; Sveinbjornsson, G.; Thorolfsdottir, R.B.; Tamlander, M.; Tragante, V.; Olafsdottir, T.; Rognvaldsson, S.; Sigurdsson, A.; Eggertsson, H.P.; Aegisdottir, H.M. Missense variants in FRS3 affect body mass index in populations of diverse ancestries. Nat. Commun. 2025, 16, 2694. [Google Scholar] [CrossRef]
- Zhang, X.; Brody, J.A.; Graff, M.; Highland, H.M.; Chami, N.; Xu, H.; Wang, Z.; Ferrier, K.R.; Chittoor, G.; Josyula, N.S. Whole genome sequencing analysis of body mass index identifies novel African ancestry-specific risk allele. Nat. Commun. 2025, 16, 3470. [Google Scholar] [CrossRef]
- Wang, J.; Fan, T.; Du, Z.; Xu, L.; Chen, Y.; Zhang, L.; Gao, H.; Li, J.; Ma, Y.; Gao, X. Genome-wide association analysis identifies the PMEL gene affecting coat color and birth weight in Simmental × Holstein. Animals 2023, 13, 3821. [Google Scholar] [CrossRef]
- Han, M.; Wang, X.; Du, H.; Cao, Y.; Zhao, Z.; Niu, S.; Bao, X.; Rong, Y.; Ao, X.; Guo, F. Genome-wide association study identifies candidate genes affecting body conformation traits of Zhongwei goat. BMC Genom. 2025, 26, 37. [Google Scholar] [CrossRef]
- Tu, T.-C.; Lin, C.-J.; Liu, M.-C.; Hsu, Z.-T.; Chen, C.-F. Genomic prediction and genome-wide association study for growth-related traits in taiwan country chicken. Animals 2025, 15, 376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, H.; Brandt, D.Y.C.; Hu, B.; Sheng, J.; Wang, M.; Luo, H.; Li, Y.; Guo, S.; Sheng, B. The genetic architecture of phenotypic diversity in the Betta fish (Betta splendens). Sci. Adv. 2022, 8, eabm4955. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Jiao, Z.; Yu, F.-X. The Hippo signaling pathway in development and regeneration. Cell Rep. 2024, 43, 113926. [Google Scholar] [CrossRef]
- Pan, D. Hippo signaling in organ size control. Genes Dev. 2007, 21, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef]
- Liu, A.; O’Connell, J.; Wall, F.; Carthew, R.W. Scaling between cell cycle duration and wing growth is regulated by Fat-Dachsous signaling in Drosophila. eLife 2024, 12, RP91572. [Google Scholar] [CrossRef]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, X.; Kou, Q.; Sun, Y.; Liu, C.; Li, S.; Yu, Y.; Zhang, C.; Jin, S.; Xiang, J. Genome of a giant isopod, Bathynomus jamesi, provides insights into body size evolution and adaptation to deep-sea environment. BMC Biol. 2022, 20, 113. [Google Scholar] [CrossRef]
- Lv, J.; Li, R.; Su, Z.; Gao, B.; Ti, X.; Yan, D.; Liu, G.; Liu, P.; Wang, C.; Li, J. A chromosome-level genome of Portunus trituberculatus provides insights into its evolution, salinity adaptation and sex determination. Mol. Ecol. Resour. 2022, 22, 1606–1625. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, G.; Liu, J.; Zhang, L.; Huang, S.; Wang, Y.; Yang, Z.; Ge, H. Analysis of transcriptome difference between rapid-growing and slow-growing in Penaeus vannamei. Gene 2021, 787, 145642. [Google Scholar] [CrossRef]
- Skalny, A.V.; Aschner, M.; Tsatsakis, A.; Rocha, J.B.T.; Santamaria, A.; Spandidos, D.A.; Martins, A.C.; Lu, R.; Korobeinikova, T.V.; Chen, W. Role of vitamins beyond vitamin D 3 in bone health and osteoporosis. Int. J. Mol. Med. 2024, 53, 9. [Google Scholar] [CrossRef] [PubMed]
- Fratoni, V.; Brandi, M.L. B vitamins, homocysteine and bone health. Nutrients 2015, 7, 2176–2192. [Google Scholar] [CrossRef]
- Tan, J.; Neupert, S.; Paluzzi, J.-P. Functional characterization of CCHamides and deorphanization of their receptors in the yellow fever mosquito, Aedes aegypti. Gen. Comp. Endocrinol. 2024, 359, 114618. [Google Scholar] [CrossRef] [PubMed]
- Shahid, S.; Amir, M.B.; Ding, T.B.; Liu, T.X.; Smagghe, G.; Shi, Y. RNAi of Neuropeptide CCHamide-1 and Its Receptor Indicates Role in Feeding Behavior in the Pea Aphid, Acyrthosiphon pisum. Insects 2024, 15, 939. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Cui, L.; Wu, M.; Wang, S.; Song, Y.; Liu, Z.; Xue, Z.; Chen, W.; Zhang, Y.; Li, H. DCX-EMAP is a core organizer for the ultrastructure of Drosophila mechanosensory organelles. J. Cell Biol. 2023, 222, e202209116. [Google Scholar] [CrossRef]
- Iwama, K.; Takaori, T.; Fukushima, A.; Tohyama, J.; Ishiyama, A.; Ohba, C.; Mitsuhashi, S.; Miyatake, S.; Takata, A.; Miyake, N. Novel recessive mutations in MSTO1 cause cerebellar atrophy with pigmentary retinopathy. J. Hum. Genet. 2018, 63, 263–270. [Google Scholar] [CrossRef]
- Vargas-Albores, F.; Yepiz-Plascencia, G. Beta glucan binding protein and its role in shrimp immune response. Aquaculture 2000, 191, 13–21. [Google Scholar] [CrossRef]
- Yoshiyama, T.; Namiki, T.; Mita, K.; Kataoka, H.; Niwa, R. Neverland is an evolutionally conserved Rieske-domain protein that is essential for ecdysone synthesis and insect growth. Development 2006, 133, 2565–2574. [Google Scholar] [CrossRef]
- Liu, S.; Wang, X.; Bu, X.; Zhang, C.; Qiao, F.; Qin, C.; Li, E.; Qin, J.G.; Chen, L. Influences of dietary vitamin D3 on growth, antioxidant capacity, immunity and molting of Chinese mitten crab (Eriocheir sinensis) larvae. J. Steroid Biochem. Mol. Biol. 2021, 210, 105862. [Google Scholar] [CrossRef]
- Han, J.; Kim, D.-H.; Kim, H.-S.; Nelson, D.R.; Lee, J.-S. Genome-wide identification of 52 cytochrome P450 (CYP) genes in the copepod Tigriopus japonicus and their B [α] P-induced expression patterns. Comp. Biochem. Physiol. Part D Genom. Proteom. 2017, 23, 49–57. [Google Scholar] [CrossRef]
- Baldwin, W.S.; Marko, P.B.; Nelson, D.R. The cytochrome P450 (CYP) gene superfamily in Daphnia pulex. BMC Genom. 2009, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ma, L.; Zhang, F.; Zhang, L.; Yin, J.; Wang, W.; Zhao, M. Identification and Evolution Analysis of the Genes Involved in the 20-Hydroxyecdysone Metabolism in the Mud Crab, Scylla paramamosain: A Preliminary Study. Genes 2024, 15, 1586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, Y.; Xiang, M.; Shang, Q.; Gao, X. The retardant effect of 2-tridecanone, mediated by cytochrome P450, on the development of cotton bollworm, Helicoverpa armigera. BMC Genom. 2016, 17, 954. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Mo, H.; Gao, J.; Yao, M.; Du, Y.; Liu, K.; Zhang, Q.; Yu, J.; Li, Y.; Wang, L. Fibroblast growth factor 1 (FGF1) improves glucose homeostasis, modulates gut microbial composition, and reduces inflammatory responses in rainbow trout (Oncorhynchus mykiss) fed a high-fat diet. Int. J. Biol. Macromol. 2024, 281, 136226. [Google Scholar] [CrossRef]
- Hsu, C.C.; Wu, K.L.H.; Peng, J.M.; Wu, Y.N.; Chen, H.T.; Lee, M.S.; Cheng, J.H. Low-energy extracorporeal shockwave therapy improves locomotor functions, tissue regeneration, and modulating the inflammation induced FGF1 and FGF2 signaling to protect damaged tissue in spinal cord injury of rat model: An experimental animal study. Int. J. Surg. 2024, 110, 7563–7572. [Google Scholar] [CrossRef]
- Sherbet, G.V. Hippo Signalling in Cell Proliferation, Migration and Angiogenesis. In Molecular Approach to Cancer Management; Academic Press: Cambridge, MA, USA, 2017; pp. 68–80. [Google Scholar]
- Rubenstein, J.L.; Nord, A.S.; Ekker, M. DLX genes and proteins in mammalian forebrain development. Development 2024, 151, dev202684. [Google Scholar] [CrossRef]
- Duan, S.G.; Liu, A.; Wang, C.; Zhang, R.L.; Lu, J.; Wang, M.Q. Homeotic Protein Distal-Less Regulates NlObp8 and NlCsp10 to Impact the Recognition of Linalool in the Brown Planthopper Nilaparvata lugens. J. Agric. Food Chem. 2023, 71, 10291–10303. [Google Scholar] [CrossRef]
- Tai, H.H.; Cho, H.; Tong, M.; Ding, Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: Structure and biological functions. Curr. Pharm. Des. 2006, 12, 955–962. [Google Scholar] [CrossRef]
- Yang, Y.; Shao, Y.; Gao, X.; Hu, Z.; Wang, Y.; Ma, C.; Jin, G.; Zhu, F.; Dong, G.; Zhou, G. RGS10 deficiency alleviated intestinal mucosal inflammation through suppression of Th1/Th17 cell immune responses in ulcerative colitis. Immunology 2025, 174, 139–152. [Google Scholar] [CrossRef]
- Liu, Y.; He, Y.; Cao, J.; Lu, H.; Zou, R.; Zuo, Z.; Li, R.; Zhang, Y.; Sun, J. Correlative analysis of transcriptome and proteome in Penaeus vannamei reveals key signaling pathways are involved in IFN-like antiviral regulation mediated by interferon regulatory factor (PvIRF). Int. J. Biol. Macromol. 2023, 253, 127138. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.-G.; Feng, G.-P.; Yao, Z.-L.; Li, S.-H.; Zhou, J.-F.; Fang, W.-H.; Chen, Y.-H.; Li, X.-C. PvML1 suppresses bacterial infection by recognizing LPS and regulating AMP expression in shrimp. Front. Immunol. 2022, 13, 1088862. [Google Scholar] [CrossRef] [PubMed]
- Betancourt, J.L.; Rodríguez-Ramos, T.; Dixon, B. Pattern recognition receptors in Crustacea: Immunological roles under environmental stress. Front. Immunol. 2024, 15, 1474512. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, A.; Huang, Y.; Lu, L.; Guo, S.; Ye, H. Role of crustacean female sex hormone in regulating immune response in the mud crab, Scylla paramamosain. Fish Shellfish Immunol. 2023, 142, 109094. [Google Scholar] [CrossRef]
- Limkul, S.; Phiwthong, T.; Massu, A.; Jaree, P.; Thawonsuwan, J.; Teaumroong, N.; Boonanuntanasarn, S.; Somboonwiwat, K.; Boonchuen, P. The interferon-like proteins, Vagos, in Fenneropenaeus merguiensis elicit antimicrobial responses against WSSV and VPAHPND infection. Fish Shellfish Immunol. 2022, 131, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Kichaev, G.; Bhatia, G.; Loh, P.-R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019, 104, 65–75. [Google Scholar] [CrossRef]
- Zhu, Z.; Guo, Y.; Shi, H.; Liu, C.-L.; Panganiban, R.A.; Chung, W.; O’Connor, L.J.; Himes, B.E.; Gazal, S.; Hasegawa, K. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK Biobank. J. Allergy Clin. Immunol. 2020, 145, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Ni, G.; Van Der Werf, J.; Zhou, X.; Hyppönen, E.; Wray, N.R.; Lee, S.H. Genotype–covariate correlation and interaction disentangled by a whole-genome multivariate reaction norm model. Nat. Commun. 2019, 10, 2239. [Google Scholar] [CrossRef]
- de Kinderen, M.A.J.; Sölkner, J.; Mészáros, G.; Alemu, S.W.; Esatu, W.; Bastiaansen, J.W.M.; Komen, H.; Dessie, T. Genotype by environment interactions (G* E) of chickens tested in Ethiopia using body weight as a performance trait. Animals 2023, 13, 3121. [Google Scholar] [CrossRef]
- Gamazon, E.R.; Park, D.S. SNP-based heritability estimation: Measurement noise, population stratification, and stability. In The Genetic Architecture of Neuropsychiatric Traits: Mechanism, Polygenicity, and Genome Gunction; Gamazon, E.R., Ed.; Universiteit van Amsterdam: Amsterdam, The Netherlands, 2016; pp. 277–294. [Google Scholar]
- Zhu, H.; Zhou, X. Statistical methods for SNP heritability estimation and partition: A review. Comput. Struct. Biotechnol. J. 2020, 18, 1557–1568. [Google Scholar] [CrossRef]
- Wang, M.; Xu, S. Statistical power in genome-wide association studies and quantitative trait locus mapping. Heredity 2019, 123, 287–306. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait a | No b | Mean (±SD) c/g | Female: Mean (±SD) c/g | Male: Mean (±SD) c/g | CV (%) d | Female: CV (%) d | Male: CV (%) d | h2 e |
|---|---|---|---|---|---|---|---|---|
| BW | 320 | 218.74 ± 74.37 | 213.52 ± 74.31 | 219.49 ± 66.93 | 34.00 | 34.80 | 30.49 | 0.32 |
| TruW | 317 | 132.07 ± 45.35 | 141.93 ± 48.44 | 119.66 ± 30.44 | 34.34 | 34.12 | 25.43 | 0.20 |
| WEC | 319 | 157.66 ± 53.28 | 168.15 ± 58.42 | 145.79 ± 39.14 | 33.79 | 34.74 | 26.84 | 0.25 |
| AppW | 317 | 86.67 ± 38.59 | 69.24 ± 25.97 | 97.23 ± 35.08 | 44.53 | 37.51 | 36.08 | 0.38 |
| CheW | 313 | 61.08 ± 30.90 | 44.56 ± 15.75 | 69.54 ± 25.52 | 50.59 | 35.35 | 36.70 | 0.40 |
| Chr | Nsnp | QTL Region | Ngene |
|---|---|---|---|
| 2 | 1 | 37,665,876–37,865,876 | 4 |
| 3 | 3 | 40,271,752–40,471,757 | 9 |
| 15 | 1 | 22,304,281–22,504,281 | 15 |
| 21 | 0 | 14,509,234–14,885,809 | 1 |
| 25 | 1 | 17,254,201–17,454,201 | 1 |
| 26 | 1 | 8,689,084–8,889,084 | 6 |
| 30 | 1 | 14,347,661–14,547,661 | 9 |
| 34 | 0 | 15,078,189–15,522,120 | 1 |
| 36 | 1 | 2,632,612–2,832,612 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Zhang, Y.; Jia, P.; Zhou, S.; Qin, Q.; Zhang, W.; Huang, K.; Wang, X.; Ye, H. Genome-Wide Association Study for Weight-Related Traits in Scylla paramamosain Using Whole-Genome Resequencing. Animals 2025, 15, 1829. https://doi.org/10.3390/ani15131829
Chen L, Zhang Y, Jia P, Zhou S, Qin Q, Zhang W, Huang K, Wang X, Ye H. Genome-Wide Association Study for Weight-Related Traits in Scylla paramamosain Using Whole-Genome Resequencing. Animals. 2025; 15(13):1829. https://doi.org/10.3390/ani15131829
Chicago/Turabian StyleChen, Lin, Yaodong Zhang, Peitan Jia, Siyi Zhou, Qionghui Qin, Weiren Zhang, Kewei Huang, Xiaopeng Wang, and Haihui Ye. 2025. "Genome-Wide Association Study for Weight-Related Traits in Scylla paramamosain Using Whole-Genome Resequencing" Animals 15, no. 13: 1829. https://doi.org/10.3390/ani15131829
APA StyleChen, L., Zhang, Y., Jia, P., Zhou, S., Qin, Q., Zhang, W., Huang, K., Wang, X., & Ye, H. (2025). Genome-Wide Association Study for Weight-Related Traits in Scylla paramamosain Using Whole-Genome Resequencing. Animals, 15(13), 1829. https://doi.org/10.3390/ani15131829