
Mitochondrial PCGs Provide Novel Insights into Subspecies Classification, Codon Usage and Selection of Cervus canadensis Distributed in Qinghai and Gansu, China
 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
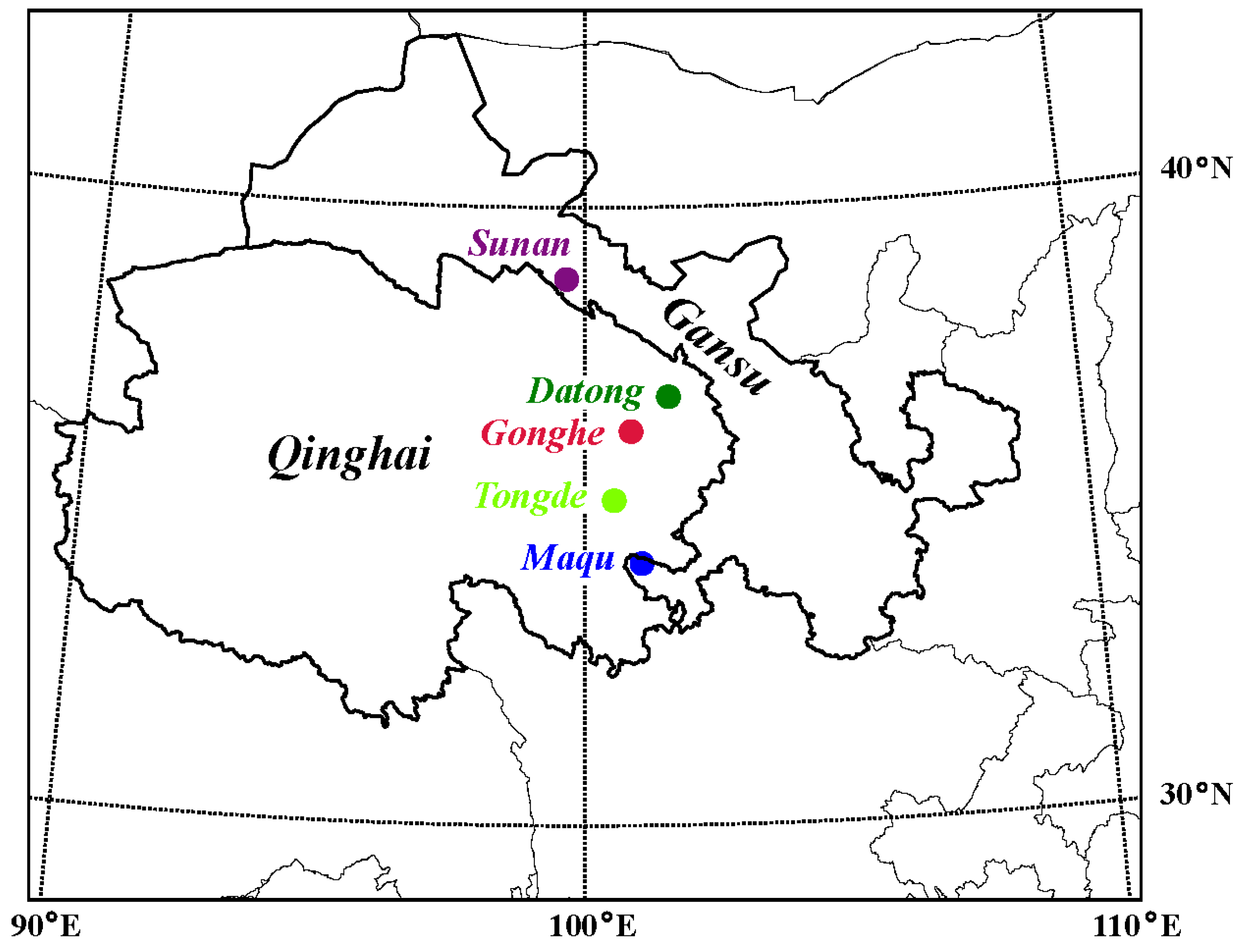
2.1. Sample Collection, DNA Extraction and Sequencing
2.2. Mitochondrial Genome Assembly and Annotation
2.3. Phylogenetic Analysis
2.4. Analysis of Mitochondrial Characteristics
3. Results and Discussion
3.1. Genome Composition and Organization
3.2. Phylogeny of Cervus canadensis
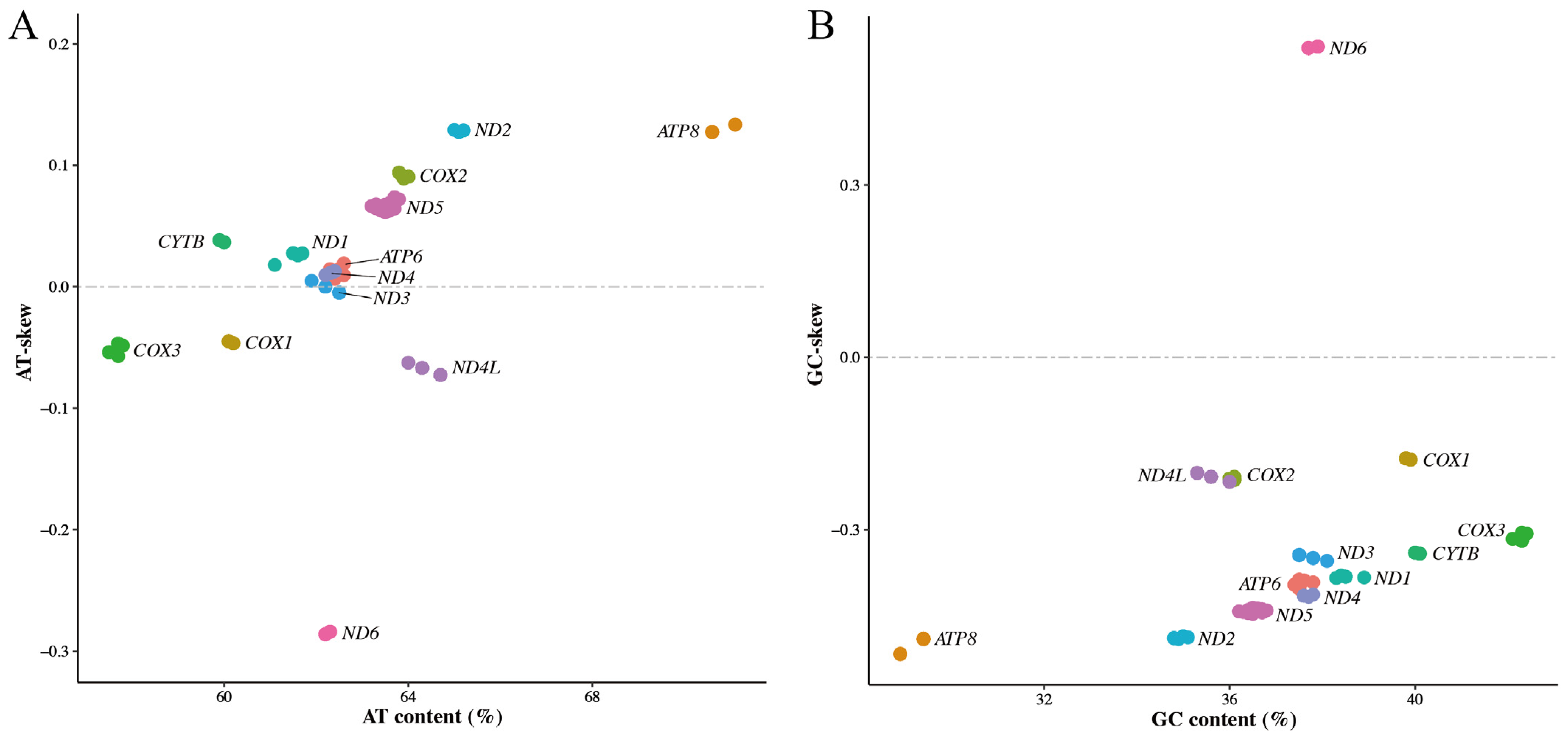
3.3. Nucleotide Bias of Mitochondrial Protein-Coding Genes
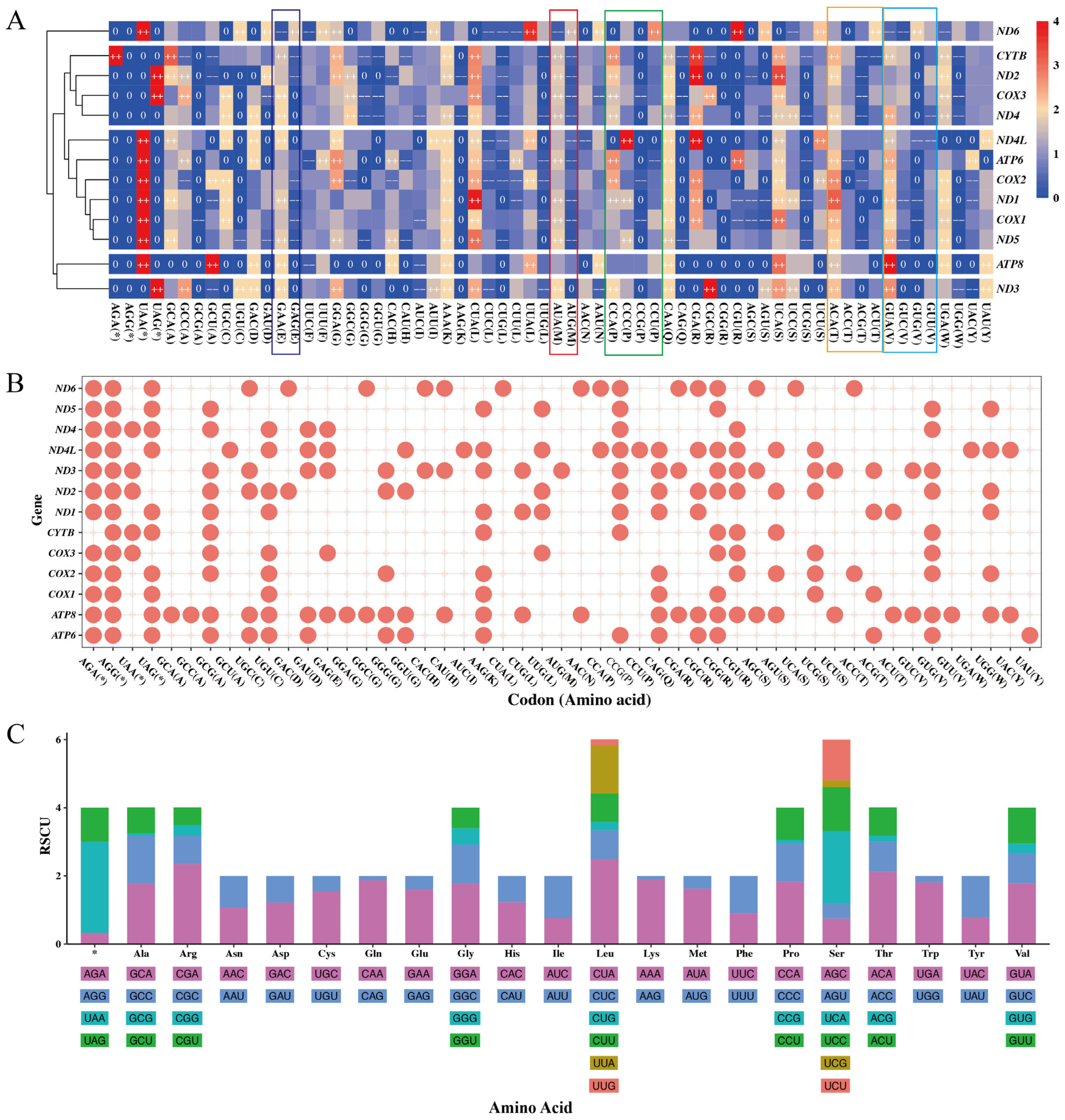
3.4. Codon Usage of Mitochondrial Protein-Coding Genes
3.5. Effective Number of Codons
3.6. PR2 Bias Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NCBI | National Center for Biotechnology Information |
| PCG | Protein-coding gene |
| IUCN | International Union for Conservation of Nature |
| PR2 | Parity Rule 2 |
| ENC | Effective Number of Codons |
| RSCU | Relative Synonymous Codon Usage |
| CUP | Codon Usage Pattern |
| CAM | Codon Aversion Motif |
References
- Polziehn, R.; Strobeck, C. Phylogeny of wapiti, red deer, sika deer, and other North American cervids as determined from mitochondrial DNA. Mol. Phylogenet. Evol. 1998, 10, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Ludt, C.J.; Schroeder, W.; Rottmann, O.; Kuehn, R. Mitochondrial DNA phylogeography of red deer (Cervus elaphus). Mol. Phylogenet. Evol. 2004, 31, 1064–1083. [Google Scholar] [CrossRef]
- Randi, E.; Mucci, N.; Claro-Hergueta, F.; Bonnet, A.; Douzery, E.J.P. A mitochondrial DNA control region phylogeny of the Cervinae: Speciation in Cervus and implications for conservation. Anim. Conserv. 2001, 4, 1–11. [Google Scholar] [CrossRef]
- Mahmut, H.; Masuda, R.; Onuma, M.; Takahashi, M.; Nagata, J.; Suzuki, M.; Ohtaishi, N. Molecular Phylogeography of the Red Deer (Cervus elaphus) Populations in Xinjiang of China: Comparison with other Asian, European, and North American Populations. Zool. Sci. 2002, 19, 485–495. [Google Scholar] [CrossRef]
- Kuwayama, R.; Ozawa, T. Phylogenetic Relationships among European Red Deer, Wapiti, and Sika Deer Inferred from Mitochondrial DNA Sequences. Mol. Phylogenet. Evol. 2000, 15, 115–123. [Google Scholar] [CrossRef]
- Lorenzini, R.; Garofalo, L. Insights into the evolutionary history of Cervus (Cervidae, tribe Cervini) based on Bayesian analysis of mitochondrial marker sequences, with first indications for a new species. J. Zool. Syst. Evol. Res. 2015, 53, 340–349. [Google Scholar] [CrossRef]
- Mackiewicz, P.; Matosiuk, M.; Świsłocka, M.; Zachos, F.E.; Hajji, G.M.; Saveljev, A.P.; Seryodkin, I.V.; Farahvash, T.; Rezaei, H.R.; Torshizi, R.V. Phylogeny and evolution of the genus Cervus (Cervidae, Mammalia) as revealed by complete mitochondrial genomes. Sci. Rep. 2022, 12, 16381. [Google Scholar] [CrossRef]
- Xiao, B.; Wang, T.; Lister, A.M.; Yuan, J.; Hu, J.; Song, S.; Lin, H.; Wang, S.; Wang, C.; Wei, D.; et al. Ancient and modern mitogenomes of red deer reveal its evolutionary history in northern China. Quat. Sci. Rev. 2023, 301, 107924. [Google Scholar] [CrossRef]
- Doan, K.; Niedziałkowska, M.; Stefaniak, K.; Sykut, M.; Jędrzejewska, B.; Ratajczak-Skrzatek, U.; Piotrowska, N.; Ridush, B.; Zachos, F.E.; Popović, D. Phylogenetics and phylogeography of red deer mtDNA lineages during the last 50,000 years in Eurasia. Zool. J. Linn. Soc. 2022, 194, 431–456. [Google Scholar] [CrossRef]
- Ohitaishi, N.; Gao, Y. A review of the distribution of all species of deer (Tragulidae, Moschidae and Cervidae) in China. Mammal Rev. 1990, 20, 125–144. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, J.; Ding, M.; Hayat Khattak, R.; Teng, L.; Liu, Z. Growth Stages and Inter-Species Gut Microbiota Composition and Function in Captive Red Deer (Cervus elaphus alxaicus) and Blue Sheep (Pseudois nayaur). Animals 2023, 13, 553. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, J.; Sun, Y.; Hou, Z.; Teng, L. Complete mitochondrial genome of a wild Alashan Red Deer (Cervus elaphus alxaicus). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 3313–3314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xu, H.; Li, D.; Xie, M.; Ni, Q.; Zhang, M.; Yao, Y. Complete mitochondrial genome and phylogenetic analysis of Sichuan deer (Cervus elaphus macneilli). Conserv. Genet. Resour. 2018, 10, 431–435. [Google Scholar] [CrossRef]
- Catanese, G.; Catanese, G.; Infante, C.; Manchado, M.; Catanese, G.; Infante, C.; Manchado, M. Complete mitochondrial DNA sequences of the frigate tuna Auxis thazard and the bullet tuna Auxis rochei. DNA Seq. 2008, 19, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wu, L.; Chen, J.; Zhao, Y.; Han, C.; Huang, T.; Yang, G. Insight into the characteristics of an important evolutionary model bird (Geospiza magnirostris) mitochondrial genome through comparison. Biocell 2022, 46, 1733. [Google Scholar] [CrossRef]
- Li, Z.; Han, Y.; Li, Y.; Wu, W.; Lei, J.; Wang, D.; Lin, Y.; Wang, X. Whole Mitochondrial Genome Sequencing and Phylogenetic Tree Construction for Procypris mera (Lin 1933). Animals 2024, 14, 2672. [Google Scholar] [CrossRef]
- Fragkoulis, G.; Hangas, A.; Fekete, Z.; Michell, C.; Moraes Carlos, T.; Willcox, S.; Griffith, J.D.; Goffart, S.; Pohjoismäki, J.L.O. Linear DNA-driven recombination in mammalian mitochondria. Nucleic Acids Res. 2024, 52, 3088–3105. [Google Scholar] [CrossRef]
- Rokas, A.; Ladoukakis, E.; Zouros, E. Animal mitochondrial DNA recombination revisited. Trends Ecol. Evol. 2003, 18, 411–417. [Google Scholar] [CrossRef]
- Golosova, O.S.; Kholodova, M.V.; Volodin, I.A.; Seryodkin, I.V.; Okhlopkov, I.M.; Argunov, A.V.; Sipko, T.P. Genetic Diversity of the Eastern Subspecies of Red Deer (Cervus elaphus) in Russia Revealed by mtDNA and Microsatellite Polymorphism. Biol. Bull. Rev. 2023, 13, 482–494. [Google Scholar] [CrossRef]
- Han, S.; Ding, H.; Peng, H.; Dai, C.; Zhang, S.; Yang, J.; Gao, J.; Kan, X. Sturnidae sensu lato Mitogenomics: Novel Insights into Codon Aversion, Selection, and Phylogeny. Animals 2024, 14, 2777. [Google Scholar] [CrossRef]
- Hao, J.; Liang, Y.; Wang, T.; Su, Y. Correlations of gene expression, codon usage bias, and evolutionary rates of the mitochondrial genome show tissue differentiation in Ophioglossum vulgatum. BMC Plant Biol. 2025, 25, 134. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Luo, Y.; Sha, A.; Xiao, W.; Xiong, Z.; Chen, X.; He, J.; Peng, L.; Zou, L. Analysis of synonymous codon usage patterns in mitochondrial genomes of nine Amanita species. Front. Microbiol. 2023, 14, 1134228. [Google Scholar] [CrossRef]
- Wada, K.; Okumura, K.; Nishibori, M.; Kikkawa, Y.; Yokohama, M. The complete mitochondrial genome of the domestic red deer (Cervus elaphus) of New Zealand and its phylogenic position within the family Cervidae. Anim. Sci. J. 2010, 81, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ba, H.; Yang, F. Complete mitochondrial genome of Cervus elaphus songaricus (Cetartiodactyla: Cervinae) and a phylogenetic analysis with related species. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 620–621. [Google Scholar] [CrossRef]
- Shao, Y.; Xing, X.; Zha, D.; Yang, F. Complete mitochondrial genome sequence of tarim red deer (Cervus elaphus yarkandensis). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 130–131. [Google Scholar] [CrossRef]
- Shao, Y.; Su, W.; Liu, H.; Zha, D.; Zhang, R.; Xing, X. Complete mitochondrial genome sequence of northeastern red deer (Cervus elaphus xanthopygus). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 547–548. [Google Scholar] [CrossRef]
- Frank, K.; Barta, E.; Bana, N.A.; Nagy, J.; Horn, P.; Orosz, L.; Steger, V. Complete mitochondrial genome sequence of a Hungarian red deer (Cervus elaphus hippelaphus) from high-throughput sequencing data and its phylogenetic position within the family Cervidae. Acta Biol. Hung. 2016, 67, 133–147. [Google Scholar] [CrossRef]
- Liu, H.; Wang, T.; He, J.; Tu, J.; Yang, X.; Yang, F.; Xing, X. The complete mitochondrial genome of Cervus elaphus kansuensis (Artiodactyla: Cervidae) and its phylogenetic analysis. Mitochondrial DNA Part B 2019, 4, 1720–1722. [Google Scholar] [CrossRef]
- Kim, H.J.; Hwang, J.Y.; Park, K.J.; Park, H.C.; Kang, H.E.; Park, J.; Sohn, H.J. The first complete mitogenome of Cervus canadensis nannodes (Merriam, 1905). Mitochondrial DNA B Resour. 2020, 5, 2294–2296. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Heng, L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Li, M.; Wang, J.; Dai, R.; Smagghe, G.; Wang, X.; You, S. Comparative analysis of codon usage patterns and phylogenetic implications of five mitochondrial genomes of the genus Japanagallia Ishihara, 1955 (Hemiptera, Cicadellidae, Megophthalminae). PeerJ 2023, 11, e16058. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Xi, Y.-Q.; Yin, X.-M. Phylogenetic relationships of Brachycera (Insecta: Diptera) inferred from mitochondrial genome sequences. Zool. J. Linn. Soc. 2022, 196, 720–739. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Hassanin, A.; Delsuc, F.; Ropiquet, A.; Hammer, C.; Vuuren, B.J.V.; Matthee, C.; Ruiz-Garcia, M.; Catzeflis, F.; Areskoug, V.; Nguyen, T.T.; et al. Pattern and timing of diversification of Cetartiodactyla (Mammalia, Laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes. Comptes Rendus Biol. 2012, 335, 32–50. [Google Scholar] [CrossRef]
- Kumar, C.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Xiong, M.; Luo, A.; Wang, X.; Wu, S.A. Characterization of the first mitochondrial genome of Aclerdidae (Hemiptera: Coccoidea) with a novel gene arrangement. Zool. Syst. 2022, 47, 293–304. [Google Scholar] [CrossRef]
- De, A.K.; Muthiyan, R.; Sunder, J.; Sawhney, S.; Sujatha, T.; Bhattacharya, D. The whole mitochondrial genome signature of Teressa goat, an indigenous goat germplasm of Andaman and Nicobar Islands, India. Small Rumin. Res. 2022, 217, 106848. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, J.; Wu, Q.; Xu, X.; Wang, P.; Wang, Z. Insights into the evolution of Brachyura (Crustacea: Decapoda) from mitochondrial sequences and gene order rearrangements. Int. J. Biol. Macromol. 2021, 170, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ren, Z.; Wang, L.; Li, Q.; Qin, W. Molecular phylogeny of wapiti (red deer) populations in Xinjiang. Acta Agric. Boreali-Occident. Sin. 2008, 17, 25–29. [Google Scholar] [CrossRef]
- An, J.; Yin, X.; Chen, R.; Boyko, C.B.; Liu, X. Integrative taxonomy of the subfamily Orbioninae (Crustacea: Isopoda) based on mitochondrial and nuclear data with evidence that supports Epicaridea as a suborder. Mol. Phylogenet. Evol. 2023, 180, 107681. [Google Scholar] [CrossRef]
- Das, P.J.; Kumar, S.; Choudhury, M.; Banik, S.; Pegu, S.R.; Kumar, S.; Deb, R.; Gupta, V.K. Characterization of the complete mitochondrial genome and identification of signature sequence of Indian wild pig. Gene 2024, 897, 148070. [Google Scholar] [CrossRef]
- Ma, Y.; Zheng, B.; Li, J.; Meng, W.; Xu, K.; Ye, Y. Characterization of the complete mitochondrial genome of Desmaulus extinctorium (Littorinimorpha, Calyptraeoidea, Calyptraeidae) and molecular phylogeny of Littorinimorpha. PLoS ONE 2024, 19, e0301389. [Google Scholar] [CrossRef]
- Ding, H.; Bi, D.; Han, S.; Yi, R.; Zhang, S.; Ye, Y.; Gao, J.; Yang, J.; Kan, X. Mitogenomic Codon Usage Patterns of Superfamily Certhioidea (Aves, Passeriformes): Insights into Asymmetrical Bias and Phylogenetic Implications. Animals 2022, 13, 96. [Google Scholar] [CrossRef]
- Chakraborty, S.; Mazumder, T.H.; Uddin, A. Compositional dynamics and codon usage pattern of BRCA1 gene across nine mammalian species. Genomics 2018, 111, 167–176. [Google Scholar] [CrossRef]
- Ran, R.; Zhang, X.; Wan, T.; Yi, W. Method analysis of codon usage bias and its related research. Grassl. Prataculture 2022, 34, 5–10+43. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.F.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Khandia, R.; Singhal, S.; Kumar, U.; Ansari, A.I.; Singh, R.K. Analysis of Nipah Virus Codon Usage and Adaptation to Hosts. Front. Microbiology 2019, 10, 886. [Google Scholar] [CrossRef]
- Han, S.; Bi, D.; Yi, R.; Ding, H.; Wu, L.; Kan, X. Plastome evolution of Aeonium and Monanthes (Crassulaceae): Insights into the variation of plastomic tRNAs, and the patterns of codon usage and aversion. Planta 2022, 256, 35. [Google Scholar] [CrossRef]
- Miller, J.B.; Hippen, A.A.; Belyeu, J.R.; Whiting, M.F.; Ridge, P.G. Missing something? Codon aversion as a new character system in phylogenetics. Cladistics 2017, 33, 545–556. [Google Scholar] [CrossRef]
- Miller, J.B.; McKinnon, L.M.; Whiting, M.F.; Ridge, P.G. CAM: An alignment-free method to recover phylogenies using codon aversion motifs. PeerJ 2019, 7, e6984. [Google Scholar] [CrossRef]
- Guan, D.; Ma, L.; Khan, M.S.; Zhang, X.; Xu, S.; Xie, J. Analysis of codon usage patterns in Hirudinaria manillensis reveals a preference for GC-ending codons caused by dominant selection constraints. BMC Genom. 2018, 19, 542. [Google Scholar] [CrossRef]
- He, B.; Dong, H.; Jiang, C.; Cao, F.; Tao, S.; Xu, L.A. Analysis of codon usage patterns in Ginkgo biloba reveals codon usage tendency from A/U-ending to G/C-ending. Sci. Rep. 2016, 6, 35927. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.K.; Lei, T.; Jing-Jing, G.U.; Song, C.; Xin, Q.I. Codon bias analysis of the mitochondrial genome reveals natural selection in the nonbiting midge Microtendipes umbrosus Freeman, 1955 (Diptera: Chironomidae). Pan-Pac. Entomol. 2023, 99, 217–225. [Google Scholar] [CrossRef]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef]
- Sueoka, N. Near homogeneity of PR2-bias fingerprints in the human genome and their implications in phylogenetic analyses. J. Mol. Evol. 2001, 53, 469–476. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Longitude | Latitude | Number | Sample Type |
|---|---|---|---|---|
| Sunan | 99.6 | 38.8 | 20 | ossified antler |
| Datong | 101.7 | 36.9 | 10 | intravenous blood |
| Gonghe | 100.9 | 36.4 | 23 | frozen velvet antler |
| Tongde | 100.6 | 35.3 | 6 | ossified antler |
| Maqu | 101.1 | 34.2 | 30 | intravenous blood |
| Accession Number | Length (bp) | Species | Subspecies | Location |
|---|---|---|---|---|
| JN632599 | 16,349 | Axis axis | -- | -- |
| OR143296 | 16,351 | Axis axis | -- | -- |
| CM102251 | 16,474 | C. albirostris | -- | -- |
| HM049636 | 16,478 | C. albirostris | -- | -- |
| JN632690 | 16,512 | C. albirostris | -- | HT * |
| MF966595 | 16,473 | C. albirostris | -- | China |
| CM033226 | 16,429 | C. canadensis | -- | America |
| GU457434 | 16,416 | C. canadensis | xanthopygus | -- |
| HQ191429 | 16,419 | C. canadensis | songaricus | -- |
| KJ025072 | 16,419 | C. canadensis | songaricus | -- |
| KP172593 | 16,428 | C. canadensis | alxaicus | China |
| KU942399 | 16,503 | C. canadensis | alxaicus | China |
| KX449334 | 16,350 | C. canadensis | macneilli | China |
| MH513320 | 16,430 | C. canadensis | kansuensis | China |
| MT430939 | 16,428 | C. canadensis | nannodes | Republic of Korea |
| MT534583 | 16,428 | C. canadensis | -- | Republic of Korea |
| OL679923 | 16,428 | C. canadensis | sibiricus | Russia |
| OL679924 | 16,353 | C. canadensis | xanthopygus | Russia |
| OP764651 | 16,430 | C. canadensis | alashanicus | China |
| OP764652 | 16,436 | C. canadensis | alashanicus | China |
| OP764653 | 16,430 | C. canadensis | xanthopygus | China |
| AB245427 | 16,357 | C. elaphus | -- | New Zealand |
| KT290948 | 16,354 | C. elaphus | hippelaphus | Hungary |
| MF872247 | 16,357 | C. elaphus | -- | Denmark |
| MF872248 | 16,357 | C. elaphus | -- | Denmark |
| OL679912 | 16,351 | C. elaphus | -- | Poland |
| OL679913 | 16,351 | C. elaphus | -- | Poland |
| OL679914 | 16,352 | C. elaphus | -- | Poland |
| OL679915 | 16,352 | C. elaphus | -- | Poland |
| OL679916 | 16,355 | C. elaphus | -- | Poland |
| OL679917 | 16,355 | C. elaphus | -- | Poland |
| OL679918 | 16,354 | C. elaphus | -- | Poland |
| OL679919 | 16,350 | C. elaphus | maral | Iran |
| OL679920 | 16,351 | C. elaphus | -- | Poland |
| OL679921 | 16,351 | C. elaphus | italicus | Italy |
| OL679922 | 16,353 | C. elaphus | barbarus | Tunisia |
| GU457435 | 16,351 | C. hanglu | yarkandensis | -- |
| MW430050 | 16,354 | C. hanglu | hanglu | India |
| MW430051 | 16,354 | C. hanglu | hanglu | India |
| ON416884 | 16,351 | C. hanglu | hanglu | India |
| ON416885 | 16,351 | C. hanglu | hanglu | India |
| Gene (Anticodon) | Length (bp) | Strand | Start Codon | Stop Codon | Gene (Anticodon) | Length (bp) | Strand | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|---|---|
| trnF (gaa) | 69 | + | trnK (uuu) | 69 | + | ||||
| rrnS | 956 | + | ATP8 | 201 | + | ATG | TAA | ||
| trnV (uac) | 67 | + | ATP6 | 681 | + | ATG | TAA | ||
| rrnL | 1574 a | + | COX3 | 804 | + | ATG | TAG | ||
| trnL (uaa) | 75 | + | trnG (ucc) | 69 | + | ||||
| ND1 | 957 | + | ATG | TAA | ND3 | 357 | + | ATA | TAG |
| trnI (gau) | 69 b | + | trnR (ucg) | 69 | + | ||||
| trnQ (uug) | 72 | − | ND4L | 297 | + | ATG | TAA | ||
| trnM (cau) | 69 | + | ND4 | 1378 | + | ATG | T-- | ||
| ND2 | 1044 | + | ATA | TAG | trnH (gug) | 69 c | + | ||
| trnW (uca) | 68 | + | trnS (gcu) | 60 | + | ||||
| trnA (ugc) | 69 | − | trnL (uag) | 70 | + | ||||
| trnN (guu) | 73 | − | ND5 | 1821 d | + | ATA | TAA | ||
| trnC (gca) | 68 | − | ND6 | 528 | − | ATG | TAA | ||
| trnY (gua) | 69 | − | trnE (uuc) | 69 | − | ||||
| COX1 | 1546 | + | ATG | TAA | CYTB | 1140 | + | ATG | AGA |
| trnS (uga) | 70 | − | trnT (ugu) | 70 | + | ||||
| trnD (guc) | 68 | + | trnP (ugg) | 66 | − | ||||
| COX2 | 684 | + | ATG | TAA | Control Region | 522 e | + |
| RSCU Value | Number of Codons | Percentage (%) | ||||
|---|---|---|---|---|---|---|
| A-Ending 1 | G-Ending 2 | U-Ending 3 | C-Ending 4 | Total | ||
| 0 | 27 | 113 | 42 | 30 | 212 | 25.48 |
| 0~0.6 | 4 | 63 | 27 | 31 | 125 | 15.02 |
| 0.6~1.6 | 45 | 26 | 113 | 117 | 301 | 36.18 |
| >1.6 | 132 | 6 | 26 | 30 | 194 | 23.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, S.; Tang, L.; Yang, S.; Chen, X.; Feng, Y.; Wang, X.; Su, W.; Xing, X. Mitochondrial PCGs Provide Novel Insights into Subspecies Classification, Codon Usage and Selection of Cervus canadensis Distributed in Qinghai and Gansu, China. Animals 2025, 15, 1486. https://doi.org/10.3390/ani15101486
Dong S, Tang L, Yang S, Chen X, Feng Y, Wang X, Su W, Xing X. Mitochondrial PCGs Provide Novel Insights into Subspecies Classification, Codon Usage and Selection of Cervus canadensis Distributed in Qinghai and Gansu, China. Animals. 2025; 15(10):1486. https://doi.org/10.3390/ani15101486
Chicago/Turabian StyleDong, Shiwu, Lixin Tang, Sukun Yang, Xu Chen, Yang Feng, Xinhao Wang, Weilin Su, and Xiumei Xing. 2025. "Mitochondrial PCGs Provide Novel Insights into Subspecies Classification, Codon Usage and Selection of Cervus canadensis Distributed in Qinghai and Gansu, China" Animals 15, no. 10: 1486. https://doi.org/10.3390/ani15101486
APA StyleDong, S., Tang, L., Yang, S., Chen, X., Feng, Y., Wang, X., Su, W., & Xing, X. (2025). Mitochondrial PCGs Provide Novel Insights into Subspecies Classification, Codon Usage and Selection of Cervus canadensis Distributed in Qinghai and Gansu, China. Animals, 15(10), 1486. https://doi.org/10.3390/ani15101486