Simple Summary
Kongshan Cattle, a breed native to Sichuan Province, China, recognized for their resilience to adverse conditions, are the focus of a critical conservation effort due to their declining numbers. This study used whole-genome resequencing to analyze their genetic structure comprehensively. The resequencing data revealed an average of 17.5 billion clean bases per sample, demonstrating high data quality with significant SNP discoveries—approximately 14 million SNPs per sample. We also identified key gene variants that could contribute to the breed’s unique traits, such as their noted stress resistance. These findings are crucial for future conservation strategies and highlight the importance of preserving the genetic diversity of local cattle breeds like Kongshan Cattle.
Abstract
Kongshan Cattle, indigenous to Sichuan Province and recognized as China’s 56th local cattle breed in 2024, exhibit unique adaptations including superior resistance to harsh conditions. Despite a declining population due to the influx of foreign breeds, there is a significant focus on preserving their genetic diversity through advanced genomic techniques. This study utilized whole-genome resequencing, a cost-effective and information-rich method, to perform a comprehensive genetic assessment of the Kongshan Cattle. High-quality resequencing data yielded an average of 17.5 billion clean bases per sample, with high proportions of Q20 and Q30 bases and a balanced GC content. SNP analysis revealed an average of 14 million SNPs per sample, with a notable transition-to-transversion ratio and a significant portion of heterozygosity. Further analysis of genomic and coding regions identified substantial insertions and deletions, particularly in coding sequences affecting gene functionality. A detailed examination of these genetic variations highlighted genes, including NEIL2 and PNKP, which are integral to stress resistance pathways, indicating potential adaptive advantages. This study not only underscores the genetic diversity of Kongshan Cattle but also contributes to broader efforts in germplasm conservation.
1. Introduction
Kongshan Cattle, native to Tongjiang County in Sichuan Province, were officially recognized in 2024 as China’s 56th local cattle breed by the National Livestock Genetic Resources Committee. They exhibit superior resistance to moisture and adverse conditions, outperforming cattle from plains and hilly areas, and have fewer diseases [1]. Although Kongshan Cattle are slender and produce less meat, their meat quality aligns well with the preferences of Chinese consumers, making them an integral part of China’s cattle biodiversity [1]. The population of Kongshan Cattle sharply declined in the late 20th century following the introduction of large quantities of foreign cattle breeds for domestic improvement. Given the urgency to protect such valuable local breeds, in-depth research and analysis of their genetic diversity and structure are necessary.
The advent of advanced sequencing technologies, particularly second-generation techniques, like high-throughput sequencing, resequencing, de novo sequencing, and exome sequencing, now represent the most widely used and effective methods in genomics [2]. Resequencing, being both cost-effective and rich in genetic information, plays a critical role in analyzing the genetic structure of species [3]. This technique has been extensively utilized in cattle genetic studies, as demonstrated by Naveed Iqbal et al. [4], Elisa Peripolli et al. [5], and Shunjin Zhang et al. [6], who have identified genomic variants and signatures of selection in various cattle breeds through whole-genome resequencing.
Given the significant research potential of Kongshan Yellow Cattle, and the lack of comprehensive genomic data available for this breed, analyzing their whole genome to assess genetic diversity and structure could stabilize their genetic foundation. Thus, this research not only provides a scientific basis for genome studies and functional gene screening for germplasm conservation but also aids in establishing complete pedigree data and stabilizing the genetic characteristics of Kongshan Yellow Cattle.
2. Materials and Methods
2.1. Sample Collection, DNA Extraction and Sequencing
Blood samples were collected from 40 Kongshan cattle by licensed veterinarians in Tongjiang County, Bazhong City, Sichuan Province, China. The samples were drawn from the jugular vein under sterile conditions using 10 mL EDTA tubes to ensure the integrity and quality of the samples for genomic analysis. Total genomic DNA was extracted using the standard procedure provided by an Animal Genomic DNA Kit (Tiangen, Beijing, China). DNA quality and quantity were assessed using NanoVue Plus (GE, USA). DNA libraries (350 bp) suitable for Illumina/BGI sequencing were prepared following the manufacturer’s specifications. Sequencing was conducted on an Illumina HiSeq XTen/NovaSeq/BGI platform by Biomarker Technologies (Beijing, China), generating 150 bp reads. Raw reads were filtered to exclude pair-end reads with >10% “N” bases and reads where over 50% of bases had a quality score below 20 (Phred-like score). After adapter removal, high-quality sequences were retained for analysis.
2.2. SNP and InDel Calling
All clean reads were aligned to the reference genome using the MEM algorithm of Burrows–Wheeler Aligner (bwa-mem2 v2.2). The aligned reads were sorted and duplicates removed using samtools (v1.7) [7]. Subsequently, samtools (v1.7) was utilized to sort the aligned reads and remove duplicates, ensuring data integrity for downstream analysis [8] and filtered by parameters including QD < 2.0, MQ < 40.0, FS > 60.0, QUAL < 30.0, MQrankSum < −12.5, and ReadPosRankSum < −8.0. Following further filtration, SNP annotation was conducted using snpEff (v3.6c) [9], categorizing SNPs into regions such as intergenic, upstream/downstream, and coding (synonymous or nonsynonymous). InDels in coding exons were classified by their potential to cause frameshift mutations.
2.3. Functional Analysis of Variant Genes
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses on variant genes were performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID 2021) [10,11].
2.4. Statistical Analyses
Data were analyzed via one-way ANOVA in SAS 9.0, with Duncan’s method for post hoc comparisons. Results were presented as mean ± SD, with significance indicated by * p < 0.05.
3. Results
3.1. Comprehensive Quality Assessment of Resequencing Data for Kongshan Cattle
After performing quality checks and filtering on the raw data, each sample of Kongshan Cattle yielded an average of 17.5 billion high-quality bases (clean bases). Among these, the proportions of Q20 and Q30 bases were notably high at 97.88% and 94.43%, respectively, while the GC content was balanced at 42.71%. These results indicate a high quality of sequencing data, with the base quality and GC content showing no significant deviations (Table 1).

Table 1.
Basic information on the resequencing data quality for Kongshan Cattle.
3.2. Genomic SNP Variant Analysis: Transition, Transversion, and Zygosity Rates
Quality control filtering of mutation sites revealed an average of 14,058,387.00 SNPs per sample. Of these, 9,957,241.00 were transition-type SNPs and 4,101,146.00 were transversion-type SNPs, resulting in a transition-to-transversion ratio of 2.42. The count of heterozygous SNPs was 3,266,503.78, while the number of homozygous SNPs was 10,791,883.23, constituting 23.19% of the total SNPs as heterozygous (Table 2).
Table 2.
Statistical information on SNP-related indicators for the Kongshan Cattle population.
3.3. Analysis of Fragment Insertions and Deletions (InDels) in Genomic and Coding Regions
Quality control filtering identified a total of 1,818,975 sites with fragment insertions and 2,987,128 sites with fragment deletions. Among these, there were 4026 coding region sites with insertions and 6225 with deletions (Table 3). Additionally, the highest number of deletions in coding regions involved fragments more significant than 10 bp, followed by insertions and deletions of 1 bp fragments. In the genomic regions, sites with 1 bp insertions and deletions were most frequent, followed by sites with deletions of fragments larger than 10 bp (Figure 1). Additionally, we performed an analysis of all differential variants between samples, which is summarized in Table S1, and an analysis of all differential small InDels between samples, as detailed in Table S2. These analyses highlight the diverse genomic landscape of the population studied
Table 3.
Statistical information on small InDel-related indicators for the Kongshan Cattle population.
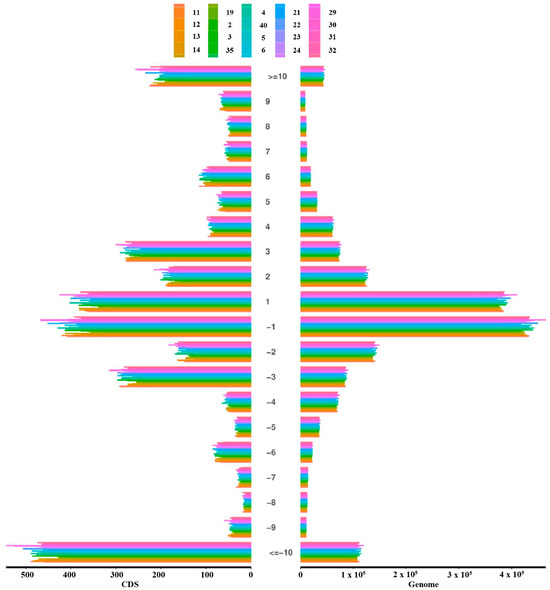
Figure 1.
Genome-wide and coding region InDel length distribution.
3.4. Mining Genetic Variations at the DNA Level
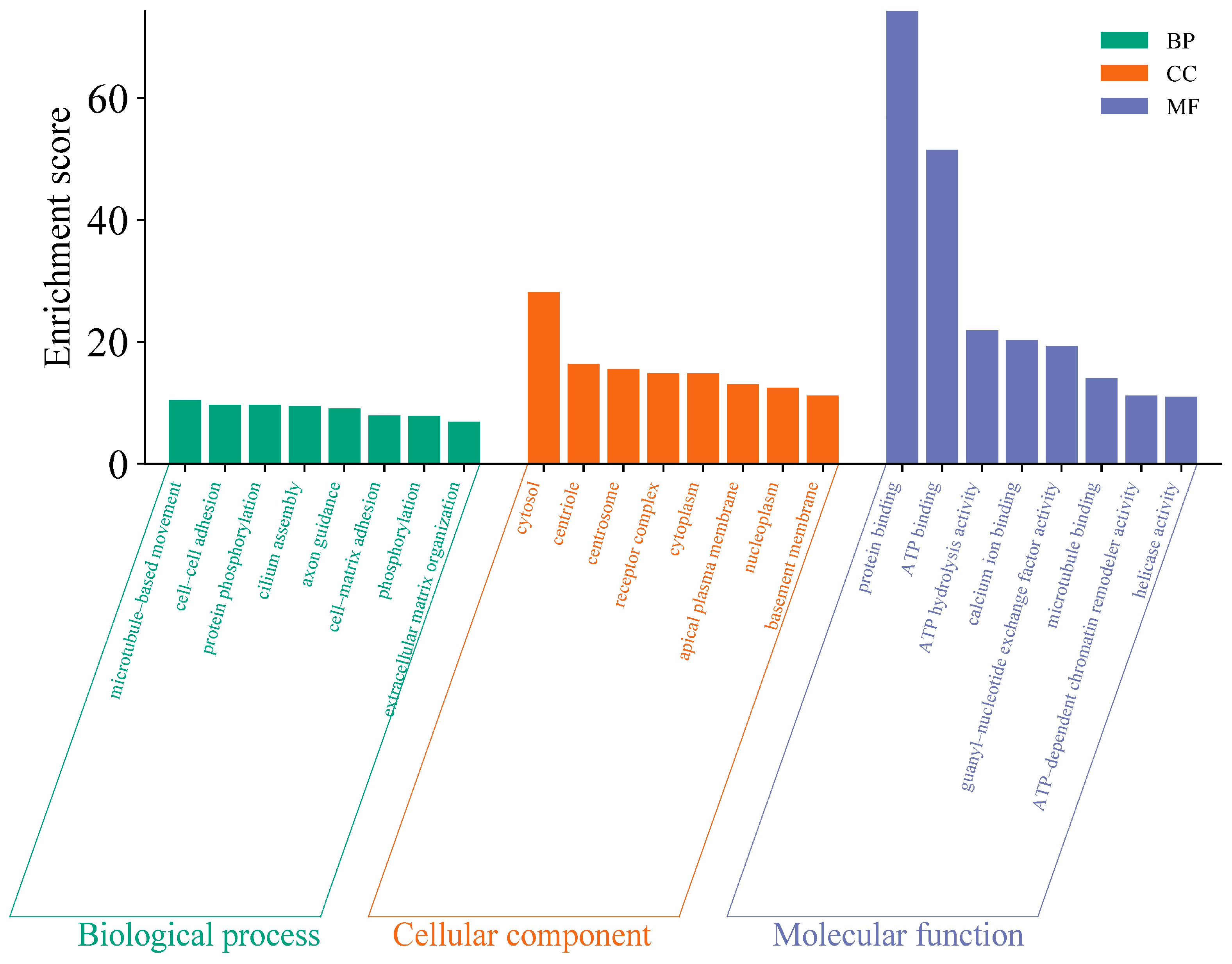
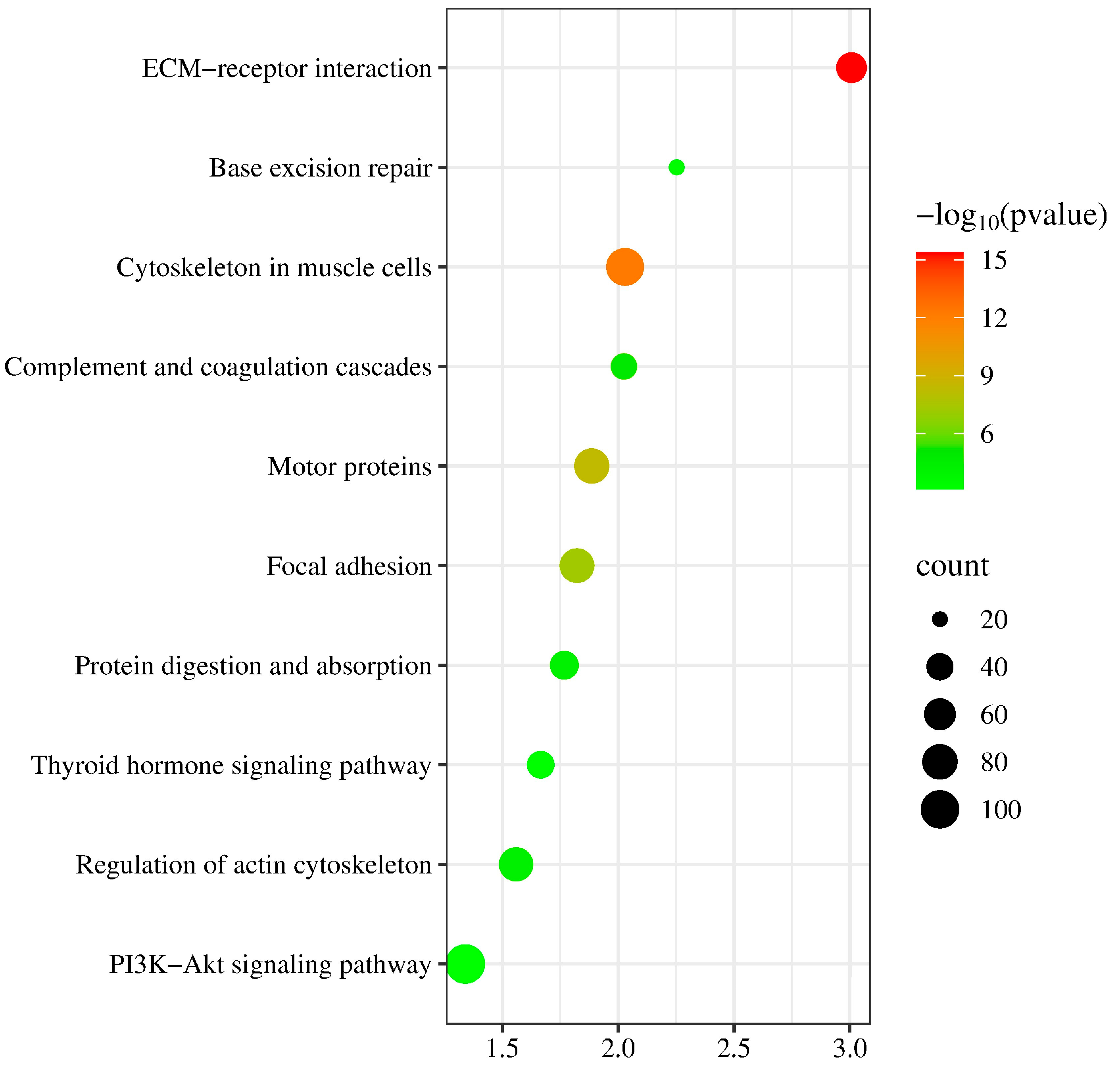
Due to mutations in the coding sequence (CDS) regions affecting gene functionality, this study compiled statistics on the differential genes caused by mutations, as shown in Table 4. The average number of genes with non-synonymous SNPs was 14,302.38, and those with insertions or deletions averaged 3278 (Table S3). To further explore the functions of these variant genes, we first overlapped all variant genes across 40 individuals, revealing that 4873 known genes exhibited mutations in all individuals. To investigate the functions of these variant genes, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. The GO results indicated that the mutated genes were predominantly involved in processes such as microtubule-based movement and cell–cell adhesion (Figure 2) (Table S4), while KEGG analysis showed a concentration in pathways like complement and coagulation cascades and base excision repair (Figure 3) (Table S5). These pathways are closely associated with the animal’s stress resistance, suggesting that mutations in these genes may enhance the stress resistance of Kongshan Cattle.
Table 4.
Statistics of gene types affected by mutations.
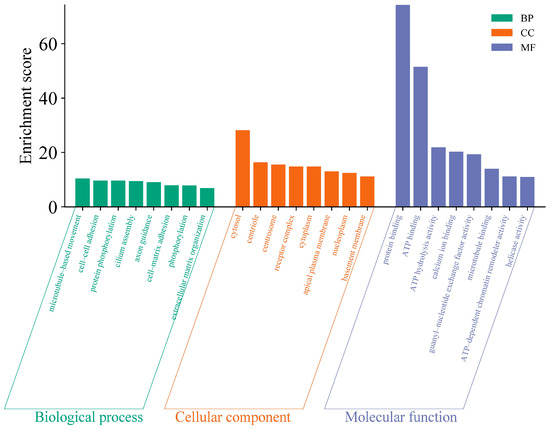
Figure 2.
Gene ontology (GO) analysis of part of variant genes.
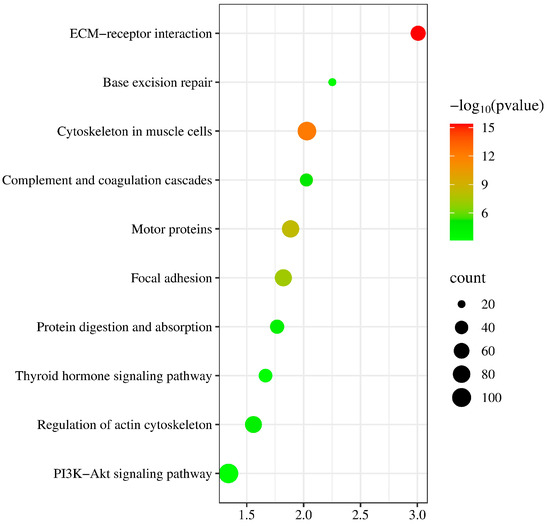
Figure 3.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of part of variant genes.
4. Discussion
Kongshan Cattle, historically known as “Bashan cattle”, originate from the northern alpine regions of Tongjiang County, Bazhong City, Sichuan Province, at the southern extremity of the Daba Mountains [1]. These cattle exhibit strong resilience and utility, traits developed through centuries of domestication by local farmers from wild ancestors in the Daba Mountains. Despite their historical significance, the Kongshan Cattle population has experienced a significant decline recently, underscoring an immediate need for breed protection. A survey identified four primary factors contributing to this decline [1]. Firstly, the local cattle industry generally sees lower breeding efficiency compared to beef cattle, leading farmers to sell calves and cows at reduced prices in pursuit of short-term market benefits. Secondly, existing conservation areas and farms have not been fully effective, struggling with inadequate personnel, funding, and imperfect conservation mechanisms. Thirdly, the absence of a dedicated breeding base for Kongshan Cattle means that sustaining the breed through local resources is not feasible. Lastly, the breed’s unique advantages and characteristics have been ineffectively developed or marketed, resulting in inadequate market presence [1]. Given these challenges, conducting in-depth research and analyses of their genetic diversity and structure is imperative to ensuring the preservation and revitalization of this valuable local breed.
Rapid advancements in high-throughput sequencing technologies have fundamentally transformed the study of population genetics across both model and non-model species [12]. With the decreasing costs of sequencing and the completion of livestock genome sequencing projects, whole-genome resequencing has emerged as a crucial tool for investigating genetic variations in livestock. This technique provides a wealth of genetic variation information, including single nucleotide polymorphisms (SNPs) and insertions/deletions (InDels), creating a genomic information repository for exploring livestock phenotypic traits and genetic improvements, thus facilitating in-depth research and utilization of livestock genetic resources [13,14,15]. For instance, Chugang Mei et al. conducted whole-genome resequencing on six phenotypically and geographically diverse domestic Chinese cattle breeds (Qinchuan, Nanyang, Luxi, Yanbian, Yunnan, and Leiqiong cattle), as well as two non-Chinese breeds (Japanese Black and Red Angus cattle). They discovered that the level of genetic variation in Chinese cattle depends on the degree of indicine content and identified many potential selective sweep regions related to breed-specific characteristics, including genes associated with coat color and meat production/quality [16]. Similarly, Xiwen Guan et al. analyzed Dabieshan Cattle from China and detected candidate genes related to fertility, feed efficiency, immune response, heat resistance, and coat color through selective sweeps [17]. Assessments of genomic diversity and signatures of selection using whole-genome sequencing data have also been performed by Xiaoting Xia on Jiaxian Red cattle and Xiaohui Ma on Bohai Black cattle [18,19].
Whole-genome resequencing allows researchers to discover a vast array of genetic variants. For instance, resequencing of the globally renowned Limousin cattle breed identified a total of 13,943,766 variants, including 311,852 bi-allelic SNPs and 92,229 indels [20]. Similarly, resequencing of the famous Holstein dairy cattle breed revealed 365,169 indels [21]. In our study of Kongshan Cattle, the high-quality data acquired after initial quality control showed ratios of Q20 and Q30 bases above 90% and a GC content around 50%, indicating a low error rate in base recognition during sequencing and a high feasibility of the experimental results. This thorough resequencing approach yielded an average of 14,058,387 SNPs per sample. We identified 1,818,975 sites with fragment insertions and 2,987,128 sites with deletions, indicating high genetic diversity within the Kongshan breed. Of these, the SNP analysis revealed 9,957,241 transition-type SNPs and 4,101,146 transversion-type SNPs, resulting in a transition-to-transversion ratio of 2.42. Quality control filtering further highlighted the significant numbers of these variants, especially in coding regions. Like Holstein cattle, where most indels (97.96%) were less than 10 bp with a decreasing trend in length [21], many indels in Kongshan Cattle were also less than 10 bp, although a notable number exceeded 10 bp. The higher proportion of homozygous SNPs suggests a significant genomic divergence between the sampled Kongshan Cattle and the reference genome, underscoring potential unique evolutionary adaptations.
Mutations in coding regions can significantly alter gene functions. Typically, GO and KEGG analyses are employed to explore the functionality of these variant genes. For instance, studies involving whole-genome resequencing of Holstein cattle identified key pathways and genes related to lipid synthesis, such as ACSBG2, which catalyzes the conversion of fatty acids, including long-chain and very-long-chain fatty acids, into their active form, acyl-CoAs, for cellular lipid synthesis [21]. Another example is Glucose 6-phosphate dehydrogenase (G-6PDH), the primary enzyme in the pentose phosphate pathway. This pathway serves as an alternative route for glucose metabolism, producing NADPH necessary for fatty acid synthesis and ribose residues for nucleotide and nucleic acid biosynthesis [21]. In our study, KEGG analysis highlighted significant activity in pathways like complement and coagulation cascades and base excision repair (BER). Research has shown that the complement and coagulation systems are two interconnected protein cascades in plasma, playing crucial roles in host defense and hemostasis, respectively. The activation of the complement system on bacteria supports cellular immune responses and leads directly to bacterial destruction via the formation of the Membrane Attack Complex (MAC) [22]. Increasing evidence suggests that cross-talk between these pathways can rapidly amplify their responses, potentially leading to extensive and prolonged systemic inflammation [23]. Base excision repair is critical for correcting DNA damage caused by oxidation, deamination, and alkylation. The essential role of BER has been underscored by studies showing the inactivation of critical proteins involved in its steps [24]. Notably, the major AP endonuclease in mammalian cells, APE1 (also known as HAP1 and Apex), is vital for survival [25]. This enzyme carries out both AP endonuclease activity and a redox function that are essential for activating several transcription factors and protecting against oxidative stress [26]. These findings are consistent with the observed stress resilience in Kongshan Cattle. The cattle’s resistance may be enhanced by these genetic variations, suggesting that Kongshan Cattle possess robust mechanisms to counter environmental and physiological stresses.
5. Conclusions
In this study, we uncovered significant genomic diversity within Kongshan Cattle, demonstrated by an average of 14 million SNPs per sample, substantial heterozygosity, and numerous insertions and deletions impacting gene functionality. Our analysis pinpointed genes linked to stress resistance, underscoring potential adaptive traits that bolster the breed’s resilience. These genetic discoveries not only emphasize the critical need to protect the genetic diversity of Kongshan Cattle as their populations decline but also bolster efforts to stabilize their genetic lineage.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ani14213056/s1. Genomic Insights and Conservation Priorities for Kongshan Cattle: A Whole-Genome Resequencing Approach, Table S1: Analysis of All Differential Variants between Samples; Table S2: Analysis of All Differential Small InDels between Samples; Table S3: Overlap of Variant Genes Across 40 Kongshan Cattle Individuals; Table S4: GO Enrichment Analysis of Variant Genes; Table S5: KEGG Enrichment Analysis of Variant Genes.
Author Contributions
Conceptualization, W.S., H.R. and S.L.; methodology, H.R., L.M., B.Z. and M.L.; software, H.R., L.M., B.Z. and M.L.; validation, H.R., L.M., B.Z. and M.L.; data curation, H.R., L.M., B.Z. and M.L.; writing—original draft preparation, W.S. and H.R.; writing—review and editing, X.J., J.W., S.C. and S.L.; supervision, W.S. and S.L.; project administration, W.S. and S.L.; funding acquisition, W.S. and S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China Youth Science Foundation Project (32202670); Research and Public Service on Major Livestock and Poultry Genetic Resources (Breeding Key Project), Key R&D Project of Sichuan Province (2021YFYZ0001); Innovation of High-Quality Cattle Breeding Materials and Methods and New Variety Cultivation, Key R&D Project of Sichuan Province (2021YFYZ0001); the 74th Batch of General Grants for Postdoctoral Researchers in China (2023M742516); the National Key R&D Program of China (2021YFD1200403); and the Sichuan innovation team of national modern agricultural industry technology system (SCCXTD-2024-13).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board of the Institutional Animal Care and Use Committee from the Sichuan Agricultural University, China (Certification No 20220425).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data underlying this article will be shared upon reasonable request to the corresponding author.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
- He, F.; Wang, W.; Shi, Y.; Fang, D.; Aguo, Y.; Gan, J.; Deng, X.; Fu, M.; Yi, J. Growth and Development Analysis of Kongshan Cattle. Agric. Biotechnol. 2022, 11, 62–65. [Google Scholar]
- Bayes, M.; Heath, S.; Gut, I.G. Applications of second generation sequencing technologies in complex disorders. Curr. Top. Behav. Neurosci. 2012, 12, 321–343. [Google Scholar] [PubMed]
- Martin, J.; Schackwitz, W.; Lipzen, A. Genomic Sequence Variation Analysis by Resequencing. Methods Mol. Biol. 2018, 1775, 229–239. [Google Scholar]
- Iqbal, N.; Liu, X.; Yang, T.; Huang, Z.; Hanif, Q.; Asif, M.; Khan, Q.M.; Mansoor, S. Genomic variants identified from whole-genome resequencing of indicine cattle breeds from Pakistan. PLoS ONE 2019, 14, e0215065. [Google Scholar] [CrossRef]
- Peripolli, E.; Reimer, C.; Ha, N.T.; Geibel, J.; Machado, M.A.; Panetto, J.; do Egito, A.A.; Baldi, F.; Simianer, H.; da Silva, M. Genome-wide detection of signatures of selection in indicine and Brazilian locally adapted taurine cattle breeds using whole-genome re-sequencing data. BMC Genom. 2020, 21, 624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yao, Z.; Li, X.; Zhang, Z.; Liu, X.; Yang, P.; Chen, N.; Xia, X.; Lyu, S.; Shi, Q.; et al. Assessing genomic diversity and signatures of selection in Pinan cattle using whole-genome sequencing data. BMC Genom. 2022, 23, 460. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, J.; Shen, M.; Xie, X.L.; Liu, G.J.; Xu, Y.X.; Lv, F.H.; Yang, H.; Yang, Y.L.; Liu, C.B.; et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Li, X.; Ye, J.; Han, X.; Qiao, R.; Li, X.; Lv, G.; Wang, K. Whole-genome sequencing identifies potential candidate genes for reproductive traits in pigs. Genomics 2020, 112, 199–206. [Google Scholar] [CrossRef]
- Rubin, C.J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef]
- Mei, C.; Wang, H.; Liao, Q.; Wang, L.; Cheng, G.; Wang, H.; Zhao, C.; Zhao, S.; Song, J.; Guang, X.; et al. Genetic Architecture and Selection of Chinese Cattle Revealed by Whole Genome Resequencing. Mol. Biol. Evol. 2018, 35, 688–699. [Google Scholar] [CrossRef]
- Guan, X.; Zhao, S.; Xiang, W.; Jin, H.; Chen, N.; Lei, C.; Jia, Y.; Xu, L. Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing. Biology 2022, 11, 1327. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, S.; Zhang, H.; Zhang, Z.; Chen, N.; Li, Z.; Sun, H.; Liu, X.; Lyu, S.; Wang, X.; et al. Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data. BMC Genom. 2021, 22, 43. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cheng, H.; Liu, Y.; Sun, L.; Chen, N.; Jiang, F.; You, W.; Yang, Z.; Zhang, B.; Song, E.; et al. Assessing Genomic Diversity and Selective Pressures in Bohai Black Cattle Using Whole-Genome Sequencing Data. Animals 2022, 12, 665. [Google Scholar] [CrossRef]
- Mariadassou, M.; Ramayo-Caldas, Y.; Charles, M.; Féménia, M.; Renand, G.; Rocha, D. Detection of selection signatures in Limousin cattle using whole-genome resequencing. Anim. Genet. 2020, 51, 815–819. [Google Scholar] [CrossRef]
- Jiang, J.; Gao, Y.; Hou, Y.; Li, W.; Zhang, S.; Zhang, Q.; Sun, D. Whole-genome resequencing of Holstein bulls for indel discovery and identification of genes associated with milk composition traits in dairy cattle. PLoS ONE 2016, 11, e0168946. [Google Scholar] [CrossRef]
- Berends, E.T.; Kuipers, A.; Ravesloot, M.M.; Urbanus, R.T.; Rooijakkers, S.H. Bacteria under stress by complement and coagulation. FEMS Microbiol. Rev. 2014, 38, 1146–1171. [Google Scholar] [CrossRef]
- Satyam, A.; Graef, E.R.; Lapchak, P.H.; Tsokos, M.G.; Dalle Lucca, J.J.; Tsokos, G.C. Complement and coagulation cascades in trauma. Acute Med. Surg. 2019, 6, 329–335. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Meira, L.B. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair 2006, 5, 189–209. [Google Scholar] [CrossRef]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).