Cuminal Inhibits Trichothecium roseum Growth by Triggering Cell Starvation: Transcriptome and Proteome Analysis




Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. In Vitro Antifungal Activity Measurement
2.1.1. Antigermination Test and Minimum Inhibitory Concentration (MIC)
2.1.2. Antifungal Activity of Cuminal Vapor against Mycelial Growth
2.2. In Vivo Antifungal Activity
2.3. Electron Microscope Observations
2.4. Fungal Sample Preparation
2.5. RNA Sequencing and Data Processing
2.5.1. RNA Extraction and Sequencing Strategies
2.5.2. Transcriptome Assembly and Annotations
2.5.3. Transcriptome Analysis
2.6. iTRAQ-Labeled Proteome Analysis
2.6.1. Protein Extraction, Digestion and Labeling
2.6.2. Peptide Fractionation and Mass-Spectrometry Analysis
2.6.3. Protein Identification and Quantification
2.6.4. Bioinformatics Analysis of DEPs
2.7. Protein–Protein Interaction Analysis
2.8. Statistical Analysis
3. Results
3.1. Antifungal Activity in Vitro
3.2. In Vivo Apple Fruit Assay
3.3. Effects on Conidial Morphology and Ultrastructure
3.4. Fungal Transcriptome Profiles upon Cuminal Exposure
3.5. GO Analysis of DEGs
3.6. KEGG Metabolic Pathways of DEGs
3.7. Global Proteome Changes in Response to Cuminal Exposure
3.8. GO Annotation Analysis of the DEPs
3.9. KEGG Pathway Analysis of DEPs
3.10. Integration Analysis of Transcriptome and Proteome
3.10.1. Correlations of Proteome with Transcriptome
3.10.2. Pathway Integration of DEGs and DEPs
3.10.3. PPI upon Cuminal Exposure
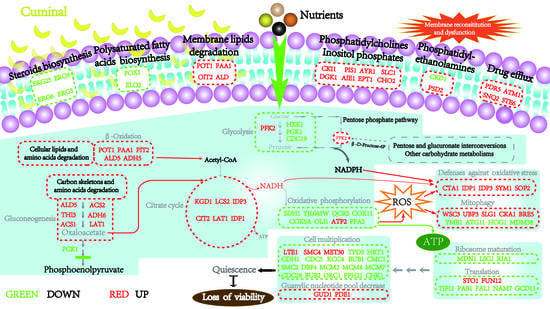
3.11. T. roseum Responses to Cuminal Stress
3.11.1. Central Carbon Metabolism and Energy Acquisition
3.11.2. Membrane Constitution and Drug Effluxing
3.11.3. Stress Mediation and Antioxidative Defenses
3.11.4. Ribosome Formation and Protein Biosynthesis
3.11.5. Cell Cycle and Multiplication
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Niu, L.-L.; Bi, Y.; Bai, X.-D.; Zhang, S.-G.; Xue, H.-L.; Li, Y.-C.; Wang, Y.; Calderón-Urrea, A. Damage to trichothecium roseum caused by sodium silicate is independent from ph. Can. J. Microbiol. 2015, 62, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Shiping, T.; Jie, Z.; Yonghong, G. Harpin induces local and systemic resistance against trichothecium roseum in harvested hami melons. Postharvest Biol. Technol. 2005, 38, 183–187. [Google Scholar] [CrossRef]
- Li, W.; Bi, Y.; Ge, Y.; Li, Y.; Wang, J.; Wang, Y. Effects of postharvest sodium silicate treatment on pink rot disease and oxidative stress-antioxidative system in muskmelon fruit. Eur. Food Res. Technol. 2012, 234, 137–145. [Google Scholar] [CrossRef]
- Oh, S.-Y.; Nam, K.-W.; Yoon, D.-H. Identification of Acremonium acutatum and Trichothecium roseum isolated from grape with white stain symptom in korea. Mycobiology 2014, 42, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.-H.; Shen, S.-S.; Kim, J.-W. Occurrence of pink mold rot of strawberry caused by trichothecium roseum in korea. Plant Pathol. J. 2010, 26, 296. [Google Scholar] [CrossRef]
- Hamid, M.I.; Hussain, M.; Ghazanfar, M.; Raza, M.; Liu, X. Trichothecium roseum causes fruit rot of tomato, orange, and apple in pakistan. Plant Dis. 2014, 98, 1271. [Google Scholar] [CrossRef]
- Lai, T.; Wang, Y.; Fan, Y.; Zhou, Y.; Bao, Y.; Zhou, T. The response of growth and patulin production of postharvest pathogen penicillium expansum to exogenous potassium phosphite treatment. Int. J. Food Microbiol. 2017, 244, 1–10. [Google Scholar] [CrossRef]
- Tian, J.; Wang, Y.; Zeng, H.; Li, Z.; Zhang, P.; Tessema, A.; Peng, X. Efficacy and possible mechanisms of perillaldehyde in control of aspergillus niger causing grape decay. Int. J. Food Microbiol. 2015, 202, 27–34. [Google Scholar] [CrossRef]
- Administration, F.F.A.D. (Food and Drug Administration). Code of Federal Regulations (cfr); Title 21—Food and Drugs. Chapter i—Food and Drug Administration, Department of Health and Human Services, Subchapter b-Food for Human Consumption (Continued), part 182—Substances Generally Recognized as Safe (gras), Subpart a—General Provisions, Subpart 182.20—Essential oils, oleoresins, and natural extractives; Office of the Federal Register: Washington, DC, USA, 2016. Available online: Https://www.Accessdata.Fda.Gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.Cfm?Fr¼182.20) (accessed on 8 May 2019).
- Aziz, M.; Karboune, S. Natural antimicrobial/antioxidant agents in meat and poultry products as well as fruits and vegetables: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 486–511. [Google Scholar] [CrossRef]
- Prakash, A.; Baskaran, R.; Paramasivam, N.; Vadivel, V. Essential oil based nanoemulsions to improve the microbial quality of minimally processed fruits and vegetables: A review. Food Res. Int. 2018, 111, 509–523. [Google Scholar] [CrossRef]
- Boubaker, H.; Karim, H.; El Hamdaoui, A.; Msanda, F.; Leach, D.; Bombarda, I.; Vanloot, P.; Abbad, A.; Boudyach, E.; Aoumar, A.A.B. Chemical characterization and antifungal activities of four thymus species essential oils against postharvest fungal pathogens of citrus. Ind. Crop. Prod. 2016, 86, 95–101. [Google Scholar] [CrossRef]
- Ding, P.; Lee, Y. Use of essential oils for prolonging postharvest life of fresh fruits and vegetables. Int. Food Res. J. 2019, 26. [Google Scholar]
- Pandey, A.K.; Kumar, P.; Singh, P.; Tripathi, N.N.; Bajpai, V.K. Essential oils: Sources of antimicrobials and food preservatives. Front. Microbiol. 2017, 7, 2161. [Google Scholar] [CrossRef] [PubMed]
- Baldim, J.L.; Silveira, J.G.F.; Almeida, A.P.; Carvalho, P.L.N.; Rosa, W.; Schripsema, J.; Chagas-Paula, D.A.; Soares, M.G.; Luiz, J.H.H. The synergistic effects of volatile constituents of ocimum basilicum against foodborne pathogens. Ind. Crop. Prod. 2018, 112, 821–829. [Google Scholar] [CrossRef]
- Marchese, A.; Barbieri, R.; Coppo, E.; Orhan, I.E.; Daglia, M.; Nabavi, S.F.; Izadi, M.; Abdollahi, M.; Nabavi, S.M.; Ajami, M. Antimicrobial activity of eugenol and essential oils containing eugenol: A mechanistic viewpoint. Crit. Rev. Microbiol. 2017, 43, 668–689. [Google Scholar] [CrossRef]
- OuYang, O.; Duan, X.; Li, L.; Tao, N. Cinnamaldehyde exerts its antifungal activity by disrupting the cell wall integrity of geotrichum citri-aurantii. Front. Microbiol. 2019, 10, 55. [Google Scholar] [CrossRef]
- Li, W.-R.; Li, H.-L.; Shi, Q.-S.; Sun, T.-L.; Xie, X.-B.; Song, B.; Huang, X.-M. The dynamics and mechanism of the antimicrobial activity of tea tree oil against bacteria and fungi. Appl. Microbiol. Biotechnol. 2016, 100, 8865–8875. [Google Scholar] [CrossRef]
- Li, Y.; Shao, X.; Xu, J.; Wei, Y.; Xu, F.; Wang, H. Tea tree oil exhibits antifungal activity against botrytis cinerea by affecting mitochondria. Food Chem. 2017, 234, 62–67. [Google Scholar] [CrossRef]
- Xu, J.; Shao, X.; Wei, Y.; Xu, F.; Wang, H. Itraq proteomic analysis reveals that metabolic pathways involving energy metabolism are affected by tea tree oil in botrytis cinerea. Front. Microbiol. 2017, 8, 1989. [Google Scholar] [CrossRef]
- Marchant, J. When antibiotics turn toxic. Nature 2018, 555, 431–433. [Google Scholar] [CrossRef]
- Yang, W.; Ding, D.; Zhang, C.; Zhou, J.; Su, X. Itraq-based proteomic profiling of vibrio parahaemolyticus under various culture conditions. Proteome Sci. 2015, 13, 19. [Google Scholar] [CrossRef]
- Zhang, F.; Zhong, H.; Han, X.; Guo, Z.; Yang, W.; Liu, Y.; Yang, K.; Zhuang, Z.; Wang, S. Proteomic profile of aspergillus flavus in response to water activity. Fungal Biol. 2015, 119, 114–124. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Men, J.-L.; Chang, M.-C.; Feng, C.-P.; Yuan, L.-G. Itraq-based quantitative proteome revealed metabolic changes of flammulina velutipes mycelia in response to cold stress. J. Proteom. 2017, 156, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wang, Z.; Zhang, X.; Liu, Z.; Li, G.; Wang, S.; Ding, Y. Complementary proteome and transcriptome profiling in developing grains of a notched-belly rice mutant reveals key pathways involved in chalkiness formation. Plant Cell Physiol. 2017, 58, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, Q.; Ding, R.; Xia, Y.; Xiong, L.; Bi, Y.; Prusky, D. Acidolysis-dominated pretreatment elevates distillation yield and impacts composition, antioxidant and antifungal activities of essential oil from cuminum cyminum seeds. RSC Adv. 2018, 8, 32283–32295. [Google Scholar] [CrossRef]
- Chitarra, G.; Breeuwer, P.; Nout, M.; Van Aelst, A.; Rombouts, F.; Abee, T. An antifungal compound produced by bacillus subtilis ym 10–20 inhibits germination of penicillium roqueforti conidiospores. J. Appl. Microbiol. 2003, 94, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Stojković, D.; Soković, M.; Glamočlija, J.; Džamić, A.; Ćirić, A.; Ristić, M.; Grubišić, D. Chemical composition and antimicrobial activity of Vitex agnus-castus L. Fruits and leaves essential oils. Food Chem. 2011, 128, 1017–1022. [Google Scholar] [CrossRef]
- Aguilar-González, A.E.; Palou, E.; López-Malo, A. Antifungal activity of essential oils of clove (Syzygium aromaticum) and/or mustard (Brassica nigra) in vapor phase against gray mold (Botrytis cinerea) in strawberries. Innov. Food Sci. Emerg. Technol. 2015, 32, 181–185. [Google Scholar] [CrossRef]
- Dwivedy, A.K.; Singh, V.K.; Prakash, B.; Dubey, N.K. Nanoencapsulated illicium verum hook. F. Essential oil as an effective novel plant-based preservative against aflatoxin b1 production and free radical generation. Food Chem. Toxicol. 2018, 111, 102–113. [Google Scholar] [CrossRef]
- Hansen, K.D.; Brenner, S.E.; Dudoit, S. Biases in illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010, 38, e131. [Google Scholar] [CrossRef]
- Yan, M.; Dai, W.; Cai, E.; Deng, Y.Z.; Chang, C.; Jiang, Z.; Zhang, L.-H. Transcriptome analysis of sporisorium scitamineum reveals critical environmental signals for fungal sexual mating and filamentous growth. Bmc Genom. 2016, 17, 354. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359. [Google Scholar] [CrossRef]
- Zhang, P.; Li, C.; Zhang, P.; Jin, C.; Pan, D.; Bao, Y. Itraq-based proteomics reveals novel members involved in pathogen challenge in sea cucumber apostichopus japonicus. PLoS ONE 2014, 9, e100492. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Cui, W.; Pan, J.; Xie, Y.; Wang, J.; Shen, W. Proteomic analysis provides insights into the molecular bases of hydrogen gas-induced cadmium resistance in medicago sativa. J. Proteom. 2017, 152, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P. String v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2014, 43, D447–D452. [Google Scholar] [CrossRef]
- Westfall, C.S.; Levin, P.A. Comprehensive analysis of central carbon metabolism illuminates connections between nutrient availability, growth rate, and cell morphology in escherichia coli. PLoS Genet. 2018, 14, e1007205. [Google Scholar] [CrossRef]
- Nidelet, T.; Brial, P.; Camarasa, C.; Dequin, S. Diversity of flux distribution in central carbon metabolism of s. Cerevisiae strains from diverse environments. Microb. Cell Fact. 2016, 15, 58. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, X.-F.; Wang, X.-F.; Zhang, D.-P. Arabidopsis 3-ketoacyl-coa thiolase-2 (kat2), an enzyme of fatty acid β-oxidation, is involved in aba signal transduction. Plant Cell Physiol. 2011, 52, 528–538. [Google Scholar] [CrossRef]
- Hishikawa, D.; Hashidate, T.; Shimizu, T.; Shindou, H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 2014, 55, 799–807. [Google Scholar] [CrossRef]
- Siroli, L.; Patrignani, F.; Gardini, F.; Lanciotti, R. Effects of sub-lethal concentrations of thyme and oregano essential oils, carvacrol, thymol, citral and trans-2-hexenal on membrane fatty acid composition and volatile molecule profile of listeria monocytogenes, escherichia coli and salmonella enteritidis. Food Chem. 2015, 182, 185–192. [Google Scholar] [CrossRef]
- Souid, A.-K.; Gao, C.; Wang, L.; Milgrom, E.; Shen, W.-C.W. Elm1 is required for multidrug resistance in saccharomyces cerevisiae. Genetics 2006, 173, 1919–1937. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iwaki, T.; Fujita, Y.; Tanaka, N.; Giga-Hama, Y.; Takegawa, K. Mitochondrial abc transporter atm1p is required for protection against oxidative stress and vacuolar functions in schizosaccharomyces pombe. Biosci. Biotechnol. Biochem. 2005, 69, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- O’Doherty, P.J.; Lyons, V.; Higgins, V.; Rogers, P.J.; Bailey, T.D.; Wu, M.J. Transcriptomic insights into the molecular response of saccharomyces cerevisiae to linoleic acid hydroperoxide. Free Radic. Res. 2013, 47, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Baral, B. Evolutionary trajectories of entomopathogenic fungi abc transporters. In Advances in Genetics; Elsevier: San Diego, CA, USA, 2017; Volume 98, pp. 117–154. [Google Scholar]
- Limón-Pacheco, J.; Gonsebatt, M.E. The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutat. Res./Genet. Toxicol. Environ. Mutagenesis 2009, 674, 137–147. [Google Scholar]
- Walton, P.A.; Brees, C.; Lismont, C.; Apanasets, O.; Fransen, M. The peroxisomal import receptor pex5 functions as a stress sensor, retaining catalase in the cytosol in times of oxidative stress. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, M.J.; Yoon, W.; Kim, E.Y.; Kim, H.; Lee, Y.; Min, B.; Kang, K.S.; Son, J.H.; Park, H.T. Isocitrate protects dj-1 null dopaminergic cells from oxidative stress through nadp+-dependent isocitrate dehydrogenase (idh). PLoS Genet. 2017, 13, e1006975. [Google Scholar] [CrossRef]
- Trott, A.; Morano, K.A. Sym1 is the stress-induced saccharomyces cerevisiae ortholog of the mammalian kidney disease gene mpv17 and is required for ethanol metabolism and tolerance during heat shock. Eukaryot. Cell 2004, 3, 620–631. [Google Scholar] [CrossRef]
- Krick, S.; Shi, S.; Ju, W.; Faul, C.; Tsai, S.-Y.; Mundel, P.; Böttinger, E.P. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of omi/htra2 protease. Proc. Natl. Acad. Sci. USA 2008, 105, 14106–14111. [Google Scholar] [CrossRef]
- Boroumand, F.; Mahmoudinasab, H.; Saadat, M. Association of the sod2 (rs2758339 and rs5746136) polymorphisms with the risk of heroin dependency and the sod2 expression levels. Gene 2018, 649, 27–31. [Google Scholar] [CrossRef]
- Angeles de la Torre-Ruiz, M.; Pujol, N.; Sundaran, V. Coping with oxidative stress. The yeast model. Curr. Drug Targets 2015, 16, 2–12. [Google Scholar] [CrossRef]
- Kanki, T.; Furukawa, K.; Yamashita, S.-I. Mitophagy in yeast: Molecular mechanisms and physiological role. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Jing, D.; Kuroda, K.; Ueda, M. Activation of signaling pathways related to cell wall integrity and multidrug resistance by organic solvent in saccharomyces cerevisiae. Curr. Genet. 2014, 60, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Nostramo, R.; Varia, S.N.; Zhang, B.; Emerson, M.M.; Herman, P.K. The catalytic activity of the ubp3 deubiquitinating protease is required for efficient stress granule assembly in saccharomyces cerevisiae. Mol. Cell. Biol. 2016, 36, 173–183. [Google Scholar] [PubMed]
- Mollet, S.; Cougot, N.; Wilczynska, A.; Dautry, F.; Kress, M.; Bertrand, E.; Weil, D. Translationally repressed mrna transiently cycles through stress granules during stress. Mol. Biol. Cell 2008, 19, 4469–4479. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Liu, D. Functional disruption of the pentatricopeptide protein slg1 affects mitochondrial rna editing, plant development, and responses to abiotic stresses in arabidopsis. Plant J. 2012, 70, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, H.; Hu, S.; Lu, X.; Yuan, C.; Zhang, C.; Wang, P.; Xiao, W.; Xiao, L.; Xue, G.-P. Plastid casein kinase 2 knockout reduces abscisic acid (aba) sensitivity, thermotolerance, and expression of aba-and heat-stress-responsive nuclear genes. J. Exp. Bot. 2014, 65, 4159–4175. [Google Scholar] [CrossRef]
- Gutierrez-Beltran, E.; Moschou, P.N.; Smertenko, A.P.; Bozhkov, P.V. Tudor staphylococcal nuclease links formation of stress granules and processing bodies with mrna catabolism in arabidopsis. Plant Cell 2015, 27, 926–943. [Google Scholar] [CrossRef]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal degradation of yme1l and oma1 adapts mitochondrial proteolytic activity during stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef]
- Mao, K.; Chew, L.H.; Inoue-Aono, Y.; Cheong, H.; Nair, U.; Popelka, H.; Yip, C.K.; Klionsky, D.J. Atg29 phosphorylation regulates coordination of the atg17-atg31-atg29 complex with the atg11 scaffold during autophagy initiation. Proc. Natl. Acad. Sci. USA 2013, 110, E2875–E2884. [Google Scholar] [CrossRef]
- Alonso-Monge, R.; Navarro-García, F.; Román, E.; Negredo, A.I.; Eisman, B.; Nombela, C.; Pla, J. The hog1 mitogen-activated protein kinase is essential in the oxidative stress response and chlamydospore formation in candida albicans. Eukaryot. Cell 2003, 2, 351–361. [Google Scholar] [CrossRef]
- Yu, P.-L.; Chen, L.-H.; Chung, K.-R. How the pathogenic fungus alternaria alternata copes with stress via the response regulators ssk1 and sho1. PLoS ONE 2016, 11, e0149153. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, T.; Kanoh, J.; Ishikawa, F. Fission yeast vps1 and atg8 contribute to oxidative stress resistance. Genes Cells 2010, 15, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Nowikovsky, K.; Reipert, S.; Devenish, R.; Schweyen, R. Mdm38 protein depletion causes loss of mitochondrial k+/h+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 2007, 14, 1647. [Google Scholar] [CrossRef]
- Lempiäinen, H.; Shore, D. Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 2009, 21, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Henras, A.; Soudet, J.; Gerus, M.; Lebaron, S.; Caizergues-Ferrer, M.; Mougin, A.; Henry, Y. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 2008, 65, 2334–2359. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.; Ferreira-Cerca, S.; Hurt, E. Eukaryotic Ribosome Biogenesis at a Glance. J. Cell Sci. 2013, 126, 4815–4821. [Google Scholar] [CrossRef]
- De Klerk, E.; AC’t Hoen, P. Alternative mrna transcription, processing, and translation: Insights from rna sequencing. Trends Genet. 2015, 31, 128–139. [Google Scholar] [CrossRef]
- Jun, K.O.; Song, C.-H.; Kim, Y.-B.; An, J.; Oh, J.H.; Choi, S.K. Activation of translation via reduction by thioredoxin–thioredoxin reductase in saccharomyces cerevisiae. FEBS Lett. 2009, 583, 2804–2810. [Google Scholar] [CrossRef]
- Algire, M.A.; Maag, D.; Savio, P.; Acker, M.G.; Tarun, S.Z.; Sachs, A.B.; Asano, K.; Nielsen, K.H.; Olsen, D.S.; Phan, L. Development and characterization of a reconstituted yeast translation initiation system. Rna 2002, 8, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Brune, C.; Munchel, S.E.; Fischer, N.; Podtelejnikov, A.V.; Weis, K. Yeast poly (a)-binding protein pab1 shuttles between the nucleus and the cytoplasm and functions in mrna export. Rna 2005, 11, 517–531. [Google Scholar] [CrossRef]
- Ghaemmaghami, S.; Huh, W.-K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737. [Google Scholar] [CrossRef] [PubMed]
- Borck, G.; Shin, B.-S.; Stiller, B.; Mimouni-Bloch, A.; Thiele, H.; Kim, J.-R.; Thakur, M.; Skinner, C.; Aschenbach, L.; Smirin-Yosef, P. Eif2γ mutation that disrupts eif2 complex integrity links intellectual disability to impaired translation initiation. Mol. Cell 2012, 48, 641–646. [Google Scholar] [CrossRef]
- Batt, C.A.; Tortorello, M. Encyclopedia of Food Microbiology, 2nd ed.; Elsevier Ltd.: London, UK, 2014; p. 1014. [Google Scholar]
- Bastia, D.; Srivastava, P.; Zaman, S.; Choudhury, M.; Mohanty, B.K.; Bacal, J.; Langston, L.D.; Pasero, P.; O’Donnell, M.E. Phosphorylation of cmg helicase and tof1 is required for programmed fork arrest. Proc. Natl. Acad. Sci. USA 2016, 113, E3639–E3648. [Google Scholar] [CrossRef] [PubMed]
- Falk, J.E.; Campbell, I.W.; Joyce, K.; Whalen, J.; Seshan, A.; Amon, A. Lte1 promotes exit from mitosis by multiple mechanisms. Mol. Biol. Cell 2016, 27, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.F.-I.; Singh, M.; Thompson, J.; Yates, J.R., III; Hagstrom, K.A. An smc-like protein binds and regulates caenorhabditis elegans condensins. PLoS Genet. 2017, 13, e1006614. [Google Scholar] [CrossRef] [PubMed]
- Su, N.-Y.; Ouni, I.; Papagiannis, C.V.; Kaiser, P. A dominant suppressor mutation of the met30 cell cycle defect suggests regulation of the s. Cerevisiae met4 cbf1 transcription complex by met32. J. Biol. Chem. 2008. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Broach, J.R. Tor proteins and protein phosphatase 2a reciprocally regulate tap42 in controlling cell growth in yeast. EMBO J. 1999, 18, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Ribar, B.; Prakash, L.; Prakash, S. Ela1 and cul3 are required along with elc1 for rna polymerase ii polyubiquitylation and degradation in DNA-damaged yeast cells. Mol. Cell. Biol. 2007, 27, 3211–3216. [Google Scholar] [CrossRef]
- Rawal, C.C.; Riccardo, S.; Pesenti, C.; Ferrari, M.; Marini, F.; Pellicioli, A. Reduced kinase activity of polo kinase cdc5 affects chromosome stability and DNA damage response in s. Cerevisiae. Cell Cycle 2016, 15, 2906–2919. [Google Scholar] [CrossRef]
- Okuzaki, D.; Watanabe, T.; Tanaka, S.; Nojima, H. The saccharomyces cerevisiae bud-neck proteins kcc4 and gin4 have distinct but partially-overlapping cellular functions. Genes Genet. Syst. 2003, 78, 113–126. [Google Scholar] [CrossRef]
- Freeman, L.; Aragon-Alcaide, L.; Strunnikov, A. The condensin complex governs chromosome condensation and mitotic transmission of rdna. J. Cell Biol. 2000, 149, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F. Concerted loading of mcm2–7 double hexamers around DNA during DNA replication origin licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.C.; Dautel, M.; Di Genova, B.M.; Amberg, D.C.; Castilho, B.A.; Sattlegger, E. The gcn2 regulator yih1 interacts with the cyclin dependent kinase cdc28 and promotes cell cycle progression through g2/m in budding yeast. PLoS ONE 2015, 10, e0131070. [Google Scholar] [CrossRef]
- D’Aquino, K.E.; Monje-Casas, F.; Paulson, J.; Reiser, V.; Charles, G.M.; Lai, L.; Shokat, K.M.; Amon, A. The protein kinase kin4 inhibits exit from mitosis in response to spindle position defects. Mol. Cell 2005, 19, 223–234. [Google Scholar] [CrossRef]
- Amable, L.; Grankvist, N.; Largen, J.W.; Ortsäter, H.; Sjöholm, Å.; Honkanen, R.E. Disruption of serine/threonine protein phosphatase 5 (pp5: Ppp5c) in mice reveals a novel role for pp5 in the regulation of ultraviolet light-induced phosphorylation of serine/threonine protein kinase chk1 (chek1). J. Biol. Chem. 2011, 286, 40413–40422. [Google Scholar] [CrossRef] [PubMed]
- Saint-Marc, C.; Daignan-Fornier, B. Gud1 (ydl238c) encodes saccharomyces cerevisiae guanine deaminase, an enzyme expressed during post-diauxic growth. Yeast 2004, 21, 1359–1363. [Google Scholar] [CrossRef]
- Nikawa, J.-L.; Sass, P.; Wigler, M. Cloning and characterization of the low-affinity cyclic amp phosphodiesterase gene of saccharomyces cerevisiae. Mol. Cell. Biol. 1987, 7, 3629–3636. [Google Scholar] [CrossRef]
- Massimi, M.; Cardarelli, S.; Galli, F.; Giardi, M.F.; Ragusa, F.; Panera, N.; Cinque, B.; Cifone, M.G.; Biagioni, S.; Giorgi, M. Increase of intracellular cyclic amp by pde4 inhibitors affects hepg2 cell cycle progression and survival. J. Cell. Biochem. 2017, 118, 1401–1411. [Google Scholar] [CrossRef]
- Moghaddam, M.; Miran, S.N.K.; Pirbalouti, A.G.; Mehdizadeh, L.; Ghaderi, Y. Variation in essential oil composition and antioxidant activity of cumin (Cuminum cyminum L.) fruits during stages of maturity. Ind. Crop. Prod. 2015, 70, 163–169. [Google Scholar] [CrossRef]
- Petretto, G.; Fancello, F.; Bakhy, K.; Faiz, C.A.; Sibawayh, Z.; Chessa, M.; Zara, S.; Sanna, M.; Maldini, M.; Rourke, J.P. Chemical composition and antimicrobial activity of essential oils from Cuminum cyminum L. Collected in different areas of morocco. Food Biosci. 2018, 22, 50–58. [Google Scholar] [CrossRef]
- Tak, J.-H.; Isman, M.B. Enhanced cuticular penetration as the mechanism of synergy for the major constituents of thyme essential oil in the cabbage looper, trichoplusia ni. Ind. Crop. Prod. 2017, 101, 29–35. [Google Scholar] [CrossRef]
- Chansang, A.; Champakaew, D.; Junkum, A.; Jitpakdi, A.; Amornlerdpison, D.; Aldred, A.K.; Riyong, D.; Wannasan, A.; Intirach, J.; Muangmoon, R. Synergy in the adulticidal efficacy of essential oils for the improvement of permethrin toxicity against Aedes aegypti L.(diptera: Culicidae). Parasites Vectors 2018, 11, 417. [Google Scholar] [CrossRef] [PubMed]
- Yun, D.G.; Lee, D.G. Silymarin exerts antifungal effects via membrane-targeted mode of action by increasing permeability and inducing oxidative stress. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1859, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, D.A.; Harris, F.; Mura, M.; Dennison, S.R. The increasing role of phosphatidylethanolamine as a lipid receptor in the action of host defence peptides. Prog. Lipid Res. 2015, 59, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.A.; Bleackley, M.R.; Lee, T.-H.; Shafee, T.M.; Poon, I.K.; Hulett, M.D.; Aguilar, M.-I.; van der Weerden, N.L.; Anderson, M.A. The plant defensin nad1 introduces membrane disorder through a specific interaction with the lipid, phosphatidylinositol 4, 5 bisphosphate. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 1099–1109. [Google Scholar] [CrossRef]
- Sant, D.; Tupe, S.; Ramana, C.; Deshpande, M. Fungal cell membrane—Promising drug target for antifungal therapy. J. Appl. Microbiol. 2016, 121, 1498–1510. [Google Scholar] [CrossRef]
- Lass-Flörl, C. Triazole antifungal agents in invasive fungal infections. Drugs 2011, 71, 2405–2419. [Google Scholar] [CrossRef]
- Dupont, S.; Lemetais, G.; Ferreira, T.; Cayot, P.; Gervais, P.; Beney, L. Ergosterol biosynthesis: A fungal pathway for life on land? Evol. Int. J. Org. Evol. 2012, 66, 2961–2968. [Google Scholar] [CrossRef]
- Geißel, B.; Loiko, V.; Klugherz, I.; Zhu, Z.; Wagener, N.; Kurzai, O.; van den Hondel, C.A.; Wagener, J. Azole-induced cell wall carbohydrate patches kill aspergillus fumigatus. Nat. Commun. 2018, 9, 3098. [Google Scholar] [CrossRef]
- Martin, C.E.; Oh, C.-S.; Jiang, Y. Regulation of long chain unsaturated fatty acid synthesis in yeast. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2007, 1771, 271–285. [Google Scholar] [CrossRef]
- Uemura, H. Synthesis and production of unsaturated and polyunsaturated fatty acids in yeast: Current state and perspectives. Appl. Microbiol. Biotechnol. 2012, 95, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guerzoni, M.E.; Lanciotti, R.; Cocconcelli, P.S. Alteration in cellular fatty acid composition as a response to salt, acid, oxidative and thermal stresses in lactobacillus helveticus. Microbiology 2001, 147, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Wolfger, H.; Mamnun, Y.M.; Kuchler, K. Fungal abc proteins: Pleiotropic drug resistance, stress response and cellular detoxification. Res. Microbiol. 2001, 152, 375–389. [Google Scholar] [CrossRef]
- Davies, K.J. Oxidative Stress: The Paradox of Aerobic Life; Biochemical Society Symposia, Portland Press Limited: London, UK, 1995; pp. 1–31. [Google Scholar]
- Rodríguez-Serrano, M.; Romero-Puertas, M.C.; Sanz-Fernández, M.; Hu, J.; Sandalio, L.M. Peroxisomes extend peroxules in a fast response to stress via a reactive oxygen species-mediated induction of the peroxin pex11a. Plant Physiol. 2016, 171, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, M.; Fransen, M. Peroxisomal metabolism and oxidative stress. Biochimie 2014, 98, 56–62. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377. [Google Scholar] [CrossRef]
- Di Rita, A.; D’Acunzo, P.; Simula, L.; Campello, S.; Strappazzon, F.; Cecconi, F. Ambra1-mediated mitophagy counteracts oxidative stress and apoptosis induced by neurotoxicity in human neuroblastoma sh-sy5y cells. Front. Cell. Neurosci. 2018, 12, 92. [Google Scholar] [CrossRef]
- Lee, S.H.; Du, J.; Stitham, J.; Atteya, G.; Lee, S.; Xiang, Y.; Wang, D.; Jin, Y.; Leslie, K.L.; Spollett, G. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. Embo Mol. Med. 2016, 8, 779–795. [Google Scholar] [CrossRef]
- Hawk, M.A.; Gorsuch, C.L.; Fagan, P.; Lee, C.; Kim, S.E.; Hamann, J.C.; Mason, J.A.; Weigel, K.J.; Tsegaye, M.A.; Shen, L. Ripk1-mediated induction of mitophagy compromises the viability of extracellular-matrix-detached cells. Nat. Cell Biol. 2018, 20, 272. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Büttner, S.; Thevissen, K. Regulated cell death as a therapeutic target for novel antifungal peptides and biologics. Oxid. Med. Cell. Longev. 2018, 5473817. [Google Scholar] [CrossRef]
- Hu, G.; McQuiston, T.; Bernard, A.; Park, Y.-D.; Qiu, J.; Vural, A.; Zhang, N.; Waterman, S.R.; Blewett, N.H.; Myers, T.G. The role of transcriptional ‘futile cycles’ in autophagy and microbial pathogenesis. Microb. Cell 2015, 2, 302. [Google Scholar] [CrossRef] [PubMed]
- Plesofsky, N.; Higgins, L.; Markowski, T.; Brambl, R. Glucose starvation alters heat shock response, leading to death of wild type cells and survival of map kinase signaling mutant. PLoS ONE 2016, 11, e0165980. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.T. Nadph producing enzymes as promising drug targets for chagas disease. Curr. Med. Chem. 2019, 26, 6564–6571. [Google Scholar] [CrossRef] [PubMed]
- Herms, A.; Bosch, M.; Reddy, B.J.; Schieber, N.L.; Fajardo, A.; Rupérez, C.; Fernández-Vidal, A.; Ferguson, C.; Rentero, C.; Tebar, F. Ampk activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 2015, 6, 7176. [Google Scholar] [CrossRef]
- Handtke, S.; Albrecht, D.; Otto, A.; Becher, D.; Hecker, M.; Voigt, B. The proteomic response of bacillus pumilus cells to glucose starvation. Proteomics 2018, 18, 1700109. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ros signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Calabrese, V.; Lodi, R.; Tonon, C.; D’Agata, V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.G.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in friedreich’s ataxia. J. Neurol. Sci. 2005, 233, 145–162. [Google Scholar] [CrossRef]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2784–2790. [Google Scholar] [CrossRef]
- Solé, C.; Nadal-Ribelles, M.; de Nadal, E.; Posas, F. A novel role for lncrnas in cell cycle control during stress adaptation. Curr. Genet. 2015, 61, 299–308. [Google Scholar] [CrossRef]
- Meyer, C.; Garzia, A.; Mazzola, M.; Gerstberger, S.; Molina, H.; Tuschl, T. The tia1 rna-binding protein family regulates eif2ak2-mediated stress response and cell cycle progression. Mol. Cell 2018, 69, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Fugger, K.; Chu, W.K.; Haahr, P.; Kousholt, A.N.; Beck, H.; Payne, M.J.; Hanada, K.; Hickson, I.D.; Sørensen, C.S. Fbh1 co-operates with mus81 in inducing DNA double-strand breaks and cell death following replication stress. Nat. Commun. 2013, 4, 1423. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.P.; Heller, J.; Daskalov, A.; Videira, A.; Glass, N.L. Regulated forms of cell death in fungi. Front. Microbiol. 2017, 8, 1837. [Google Scholar] [CrossRef]
- Aird, K.M.; Worth, A.J.; Snyder, N.W.; Lee, J.V.; Sivanand, S.; Liu, Q.; Blair, I.A.; Wellen, K.E.; Zhang, R. Atm couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 2015, 11, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Alberti, S. Cell adaptation upon stress: The emerging role of membrane-less compartments. Curr. Opin. Cell Biol. 2017, 47, 34–42. [Google Scholar] [CrossRef]
- Liao, R.S.; Rennie, R.P.; Talbot, J.A. Sublethal injury and resuscitation of candida albicans after amphotericin b treatment. Antimicrob. Agents Chemother. 2003, 47, 1200–1206. [Google Scholar] [CrossRef]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Zhang, W.; Bi, Y.; Han, Y.; Zong, Y.; Prusky, D. Cuminal Inhibits Trichothecium roseum Growth by Triggering Cell Starvation: Transcriptome and Proteome Analysis. Microorganisms 2020, 8, 256. https://doi.org/10.3390/microorganisms8020256
Zhang Z, Zhang W, Bi Y, Han Y, Zong Y, Prusky D. Cuminal Inhibits Trichothecium roseum Growth by Triggering Cell Starvation: Transcriptome and Proteome Analysis. Microorganisms. 2020; 8(2):256. https://doi.org/10.3390/microorganisms8020256
Chicago/Turabian StyleZhang, Zhong, Wenting Zhang, Yang Bi, Ye Han, Yuanyuan Zong, and Dov Prusky. 2020. "Cuminal Inhibits Trichothecium roseum Growth by Triggering Cell Starvation: Transcriptome and Proteome Analysis" Microorganisms 8, no. 2: 256. https://doi.org/10.3390/microorganisms8020256
APA StyleZhang, Z., Zhang, W., Bi, Y., Han, Y., Zong, Y., & Prusky, D. (2020). Cuminal Inhibits Trichothecium roseum Growth by Triggering Cell Starvation: Transcriptome and Proteome Analysis. Microorganisms, 8(2), 256. https://doi.org/10.3390/microorganisms8020256