
Comparative Genomics Unveils Functional Diversity, Pangenome Openness, and Underlying Biological Drivers among Bacillus subtilis Group

Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Genome Data Quality Control and Taxonomic Confirmation
2.3. Pangenome Assembly
2.4. Pangenome Functional Enrichment Analysis
2.5. Calculation of Pangenome Openness Parameters
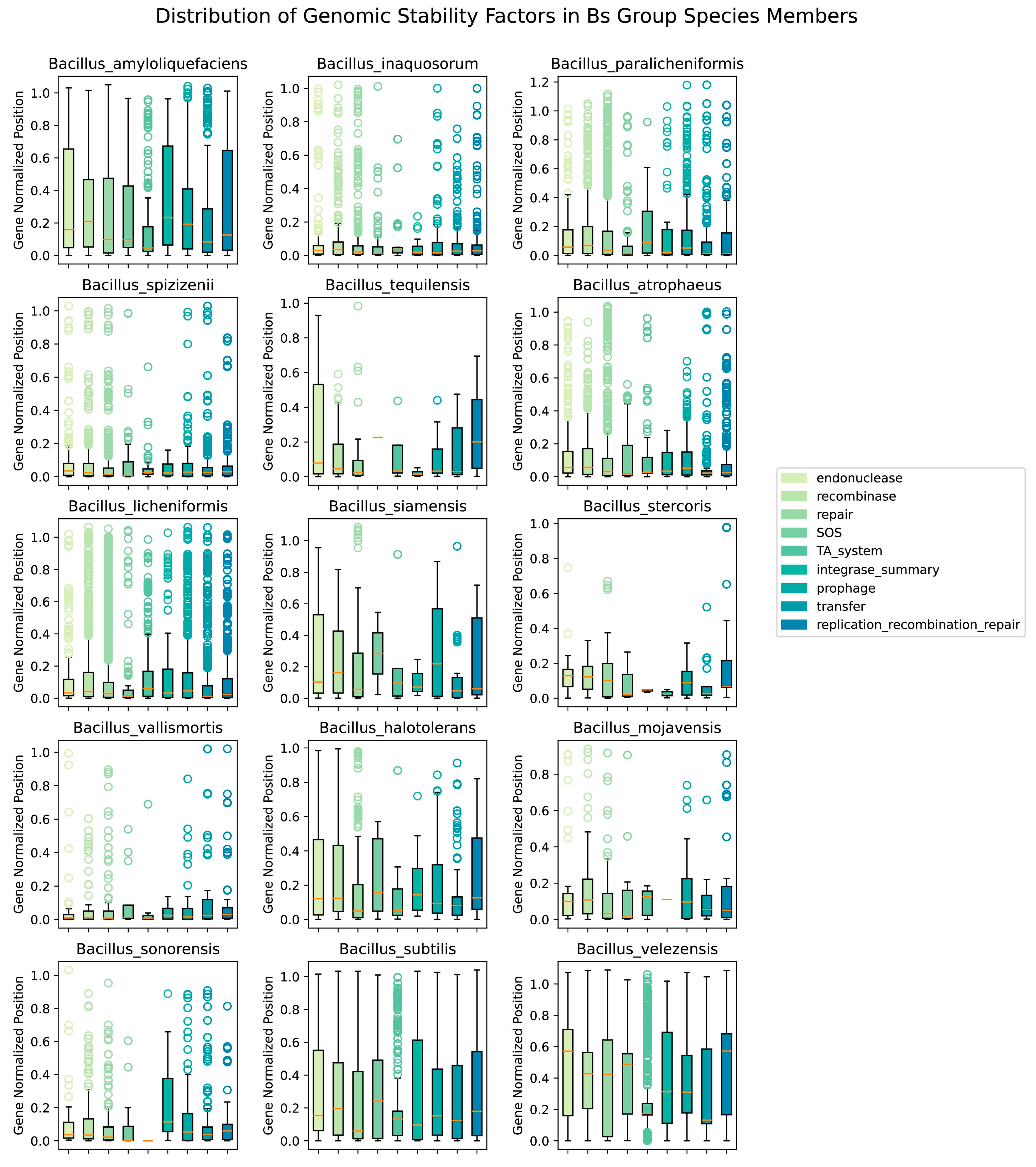
2.6. Acquisition and Analysis of Genome Stability Factor Data
3. Results
3.1. Significant Differences in Pangenomes Was Observed among Members of the Bs Group
3.2. COG Enrichment Based on Pangenome Revealed High Conservation of Core Genes in Bs Group
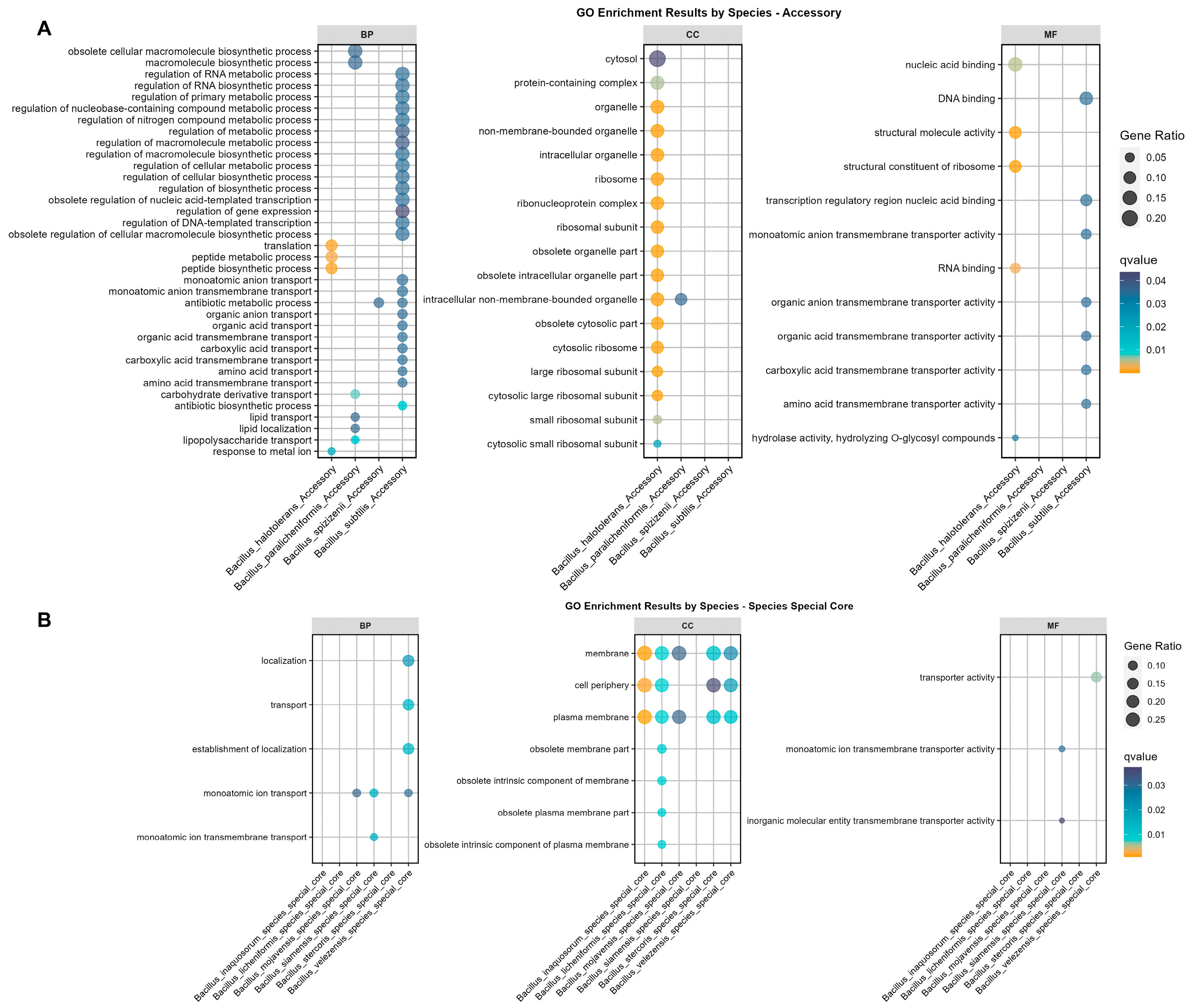
3.3. GO Enrichment Based on Pangenome Revealed Significant Differences of Accessory Genes and Species-Specific Core Genes in the Bs Group
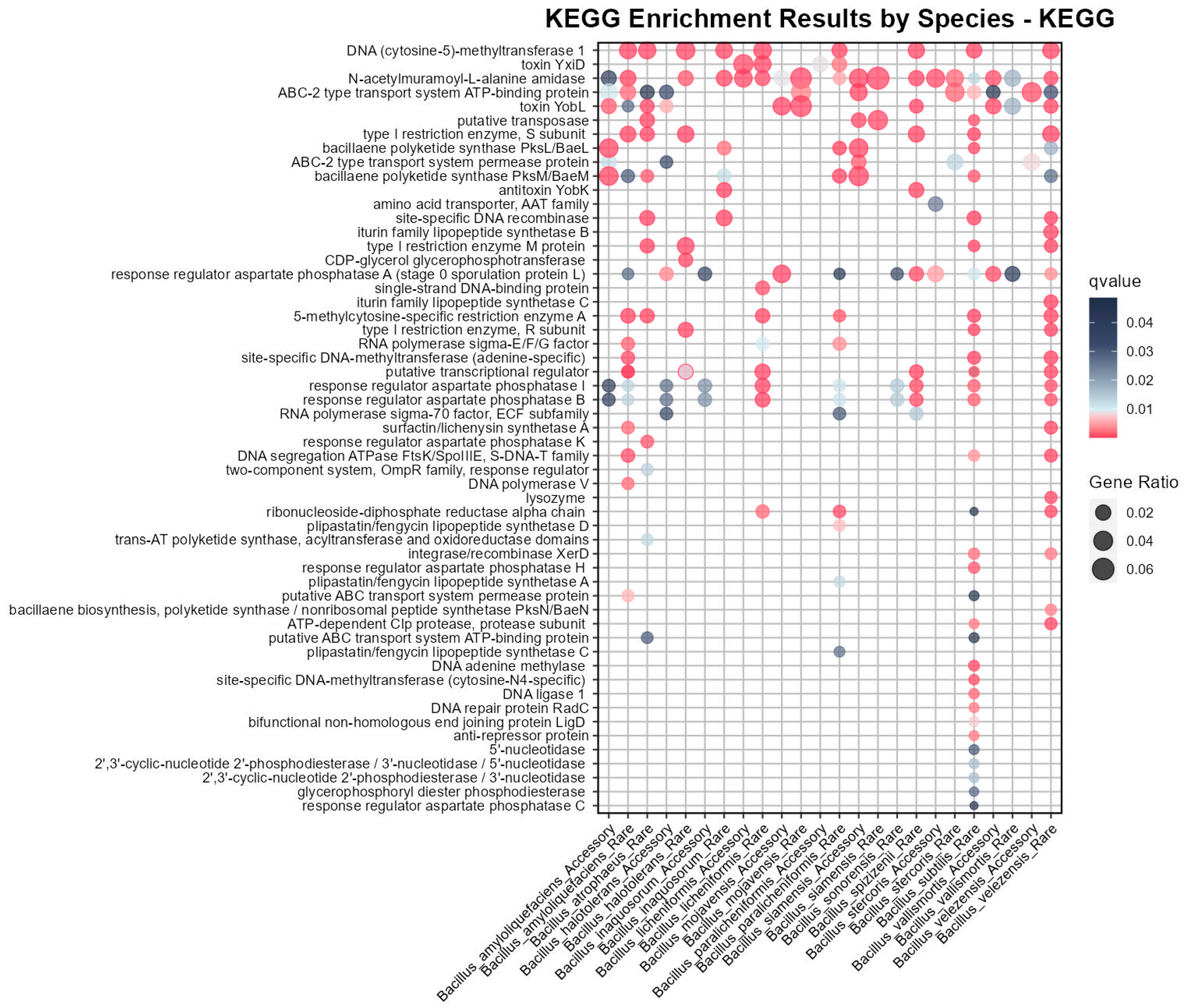
3.4. KEGG Enrichment Based on Pangenome Revealed Medicinal Potential in the Bs Group
3.5. Core Genes and Rare Genes Jointly Contributed to the Pangenome Openness among Bs Group
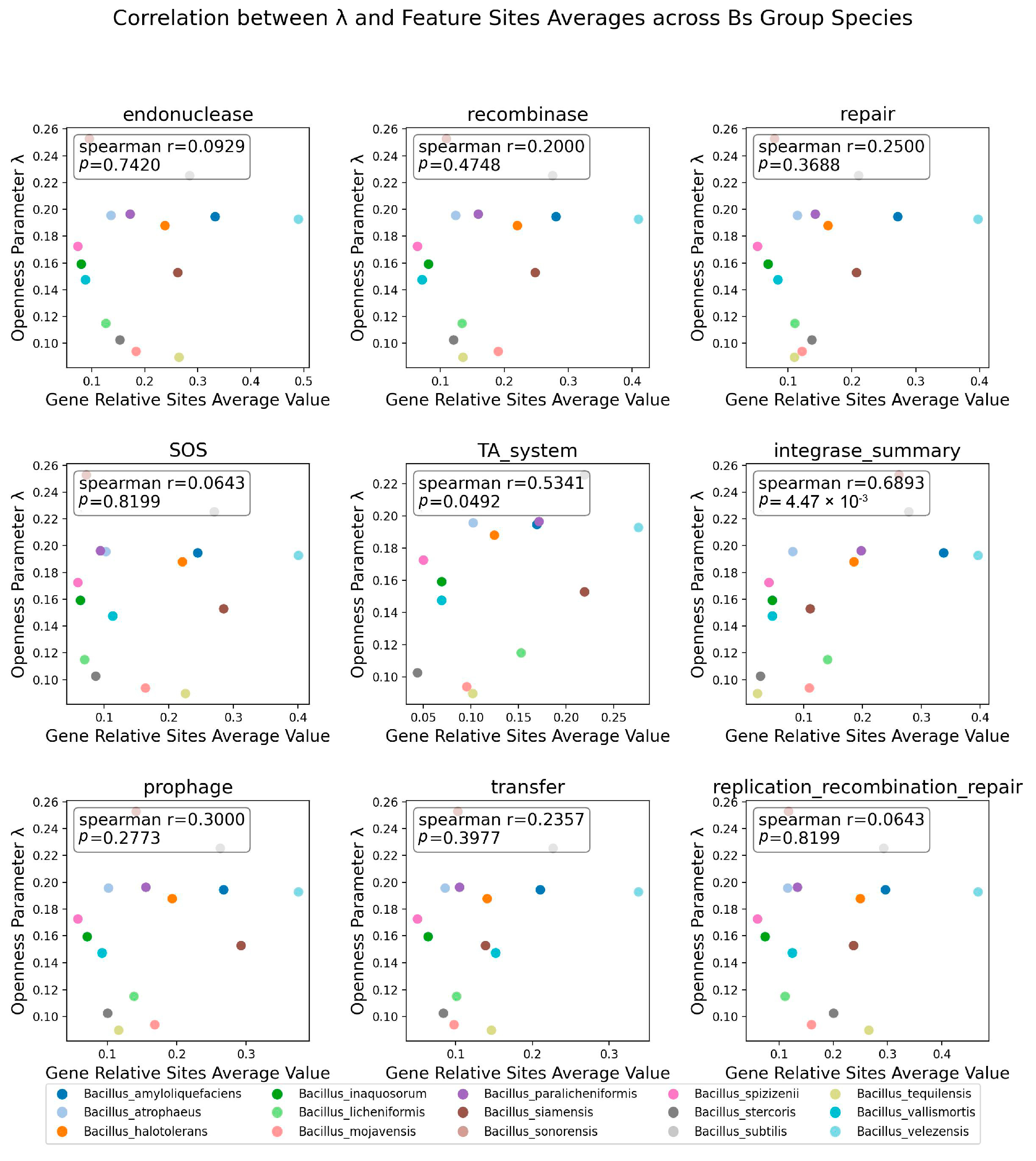
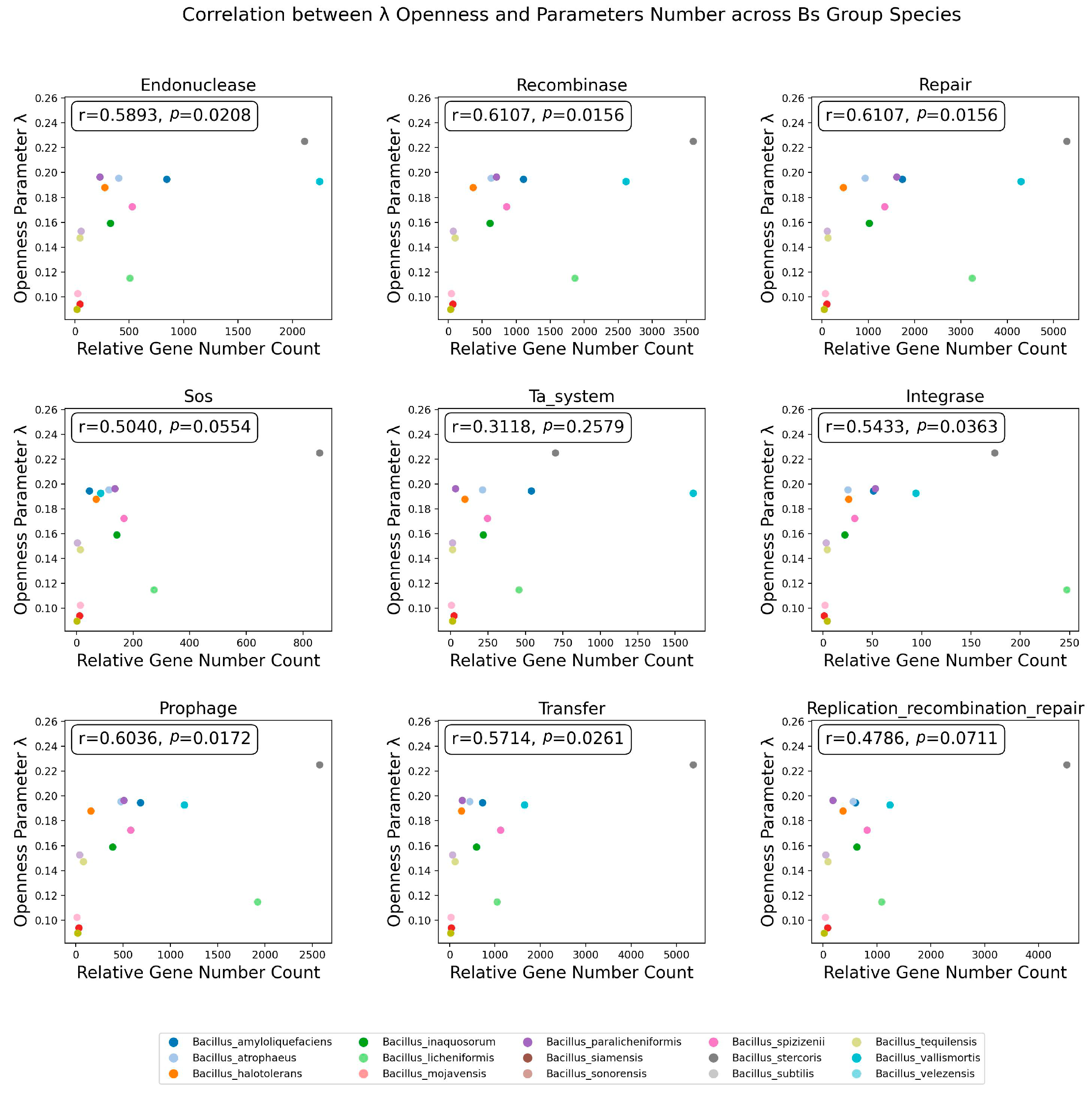
3.6. The Distribution of Integrase Genes Was Significantly Correlated with the Openness of the Bs Group
3.7. The Quantity of Prophages and Other Factors Was Significantly Correlated with Openness
4. Discussion
4.1. Comparative Pangenomes and Enrichment Analysis Facilitated Resource Mining in the Bs Group
4.2. Genome Stability Factors and Mobile Elements Influence the Openness of the Species Pangenomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patel, S.; Gupta, R.S. A Phylogenomic and Comparative Genomic Framework for Resolving the Polyphyly of the Genus Bacillus: Proposal for Six New Genera of Bacillus Species, Peribacillus Gen. Nov., Cytobacillus Gen. Nov., Mesobacillus Gen. Nov., Neobacillus Gen. Nov., Metabacillus Gen. Nov. and Alkalihalobacillus Gen. Nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. [Google Scholar] [PubMed]
- Herrmann, L.W.; Letti, L.A.J.; Penha, R.d.O.; Soccol, V.T.; Rodrigues, C.; Soccol, C.R. Bacillus Genus Industrial Applications and Innovation: First Steps towards a Circular Bioeconomy. Biotechnol. Adv. 2024, 70, 108300. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Xu, X.; Wu, Y.; Niu, T.; Liu, Y.; Li, J.; Du, G.; Liu, L. Advances and Prospects of Bacillus subtilis Cellular Factories: From Rational Design to Industrial Applications. Metab. Eng. 2018, 50, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Bhatia, S.K.; Park, H.A.; Kim, S.Y.; Sudheer, P.D.V.N.; Yang, Y.-H.; Choi, K.-Y. Bacillus subtilis as a Robust Host for Biochemical Production Utilizing Biomass. Crit. Rev. Biotechnol. 2021, 41, 827–848. [Google Scholar] [CrossRef] [PubMed]
- Earl, A.M.; Losick, R.; Kolter, R. Ecology and Genomics of Bacillus subtilis. Trends Microbiol. 2008, 16, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Um, S.; Fraimout, A.; Sapountzis, P.; Oh, D.-C.; Poulsen, M. The Fungus-Growing Termite Macrotermes Natalensis Harbors Bacillaene-Producing Bacillus sp. That Inhibit Potentially Antagonistic Fungi. Sci. Rep. 2013, 3, 3250. [Google Scholar] [CrossRef] [PubMed]
- Brito, P.H.; Chevreux, B.; Serra, C.R.; Schyns, G.; Henriques, A.O.; Pereira-Leal, J.B. Genetic Competence Drives Genome D iversity in Bacillus subtilis. Genome Biol. Evol. 2018, 10, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.-L.; Narayan, K.; Castaing, J.-P.; Tian, F.; Subramaniam, S.; Ramamurthi, K.S. A Versatile Nano Display Platform from Bacterial Spore Coat Proteins. Nat. Commun. 2015, 6, 6777. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Yokoyama, S. Trends in the Application of Bacillus in Fermented Foods. Curr. Opin. Biotechnol. 2019, 56, 36–42. [Google Scholar] [CrossRef]
- Parati, M.; Khalil, I.; Tchuenbou-Magaia, F.; Adamus, G.; Mendrek, B.; Hill, R.; Radecka, I. Building a Circular Economy around Poly(D/L-γ-Glutamic Acid)—A Smart Microbial Biopolymer. Biotechnol. Adv. 2022, 61, 108049. [Google Scholar] [CrossRef]
- Gao, G.-R.; Wei, S.-Y.; Ding, M.-Z.; Hou, Z.-J.; Wang, D.-J.; Xu, Q.-M.; Cheng, J.-S.; Yuan, Y.-J. Enhancing Fengycin Production in the Co-Culture of Bacillus subtilis and Corynebacterium glutamicum by Engineering Proline Transporter. Bioresour. Technol. 2023, 383, 129229. [Google Scholar] [CrossRef]
- Zhang, G.; An, Y.; Zabed, H.M.; Yun, J.; Parvez, A.; Zhao, M.; Zhang, C.; Ravikumar, Y.; Li, J.; Qi, X. Rewiring Bacillus subtilis and Bioprocess Optimization for Oxidoreductive Reaction-Mediated Biosynthesis of D-Tagatose. Bioresour. Technol. 2023, 389, 129843. [Google Scholar] [CrossRef] [PubMed]
- Rooney, A.P.; Price, N.P.J.; Ehrhardt, C.; Swezey, J.L.; Bannan, J.D. Phylogeny and Molecular Taxonomy of the Bacillus subtilis Species Complex and Description of Bacillus subtilis Subsp. Inaquosorum Subsp. Nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 2429–2436. [Google Scholar] [CrossRef]
- Fan, B.; Blom, J.; Klenk, H.-P.; Borriss, R. Bacillus amyloliquefaciens, Bacillus velezensis, and Bacillus siamensis Form an “Operational Group B. amyloliquefaciens” within the B. subtilis Species Complex. Front. Microbiol. 2017, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, C.A.; Bowman, M.J.; Zeigler, D.R. Promotion of Bacillus subtilis Subsp. inaquosorum, Bacillus subtilis Subsp. spizizenii and Bacillus subtilis Subsp. stercoris to Species Status. Antonie Van Leeuwenhoek 2020, 113, 1–12. [Google Scholar] [CrossRef]
- Harwood, C.R.; Mouillon, J.-M.; Pohl, S.; Arnau, J. Secondary Metabolite Production and the Safety of Industrially Important Members of the Bacillus subtilis Group. FEMS Microbiol. Rev. 2018, 42, 721–738. [Google Scholar] [CrossRef]
- Ronzon, T.; Iost, S.; Philippidis, G. An Output-Based Measurement of EU Bioeconomy Services: Marrying Statistics with Policy Insight. Struct. Chang. Econ. Dyn. 2022, 60, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Muras, A.; Romero, M.; Mayer, C.; Otero, A. Biotechnological Applications of Bacillus licheniformis. Crit. Rev. Biotechnol. 2021, 41, 609–627. [Google Scholar] [CrossRef]
- Luo, Z.; Yan, Y.; Du, S.; Zhu, Y.; Pan, F.; Wang, R.; Xu, Z.; Xu, X.; Li, S.; Xu, H. Recent Advances and Prospects of Bacillus amyloliquefaciens as Microbial Cell Factories: From Rational Design to Industrial Applications. Crit. Rev. Biotechnol. 2023, 43, 1073–1091. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Shin, H.; Chen, R.R.; Wang, N.S.; Li, J.; Du, G.; Chen, J. Developing Bacillus spp. as a Cell Factory for Production of Microbial Enzymes and Industrially Important Biochemicals in the Context of Systems and Synthetic Biology. Appl. Microbiol. Biotechnol. 2013, 97, 6113–6127. [Google Scholar] [CrossRef]
- Elshaghabee, F.M.F.; Rokana, N.; Gulhane, R.D.; Sharma, C.; Panwar, H. Bacillus As Potential Probiotics: Status, Concerns, and Future Perspectives. Front. Microbiol. 2017, 8, 1490. [Google Scholar] [CrossRef] [PubMed]
- Mingmongkolchai, S.; Panbangred, W. Bacillus Probiotics: An Alternative to Antibiotics for Livestock Production. J. Appl. Microbiol. 2018, 124, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Steinke, K.; Mohite, O.S.; Weber, T.; Kovács, Á.T. Phylogenetic Distribution of Secondary Metabolites in the Bacillus subtilis Species Complex. mSystems 2021, 6, e00057-21. [Google Scholar] [CrossRef] [PubMed]
- Yaderets, V.; Karpova, N.; Glagoleva, E.; Shibaeva, A.; Dzhavakhiya, V. Bacillus subtilis RBT-7/32 and Bacillus licheniformis RBT-11/17 as New Promising Strains for Use in Probiotic Feed Additives. Microorganisms 2023, 11, 2729. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Ma, J.; Yin, Z.; Liu, K.; Yao, G.; Xu, W.; Fan, L.; Du, B.; Ding, Y.; Wang, C. Comparative Genomic Analysis of Bacillus paralicheniformis MDJK30 with Its Closely Related Species Reveals an Evolutionary Relationship between B. paralicheniformis and B. licheniformis. BMC Genom. 2019, 20, 283. [Google Scholar] [CrossRef] [PubMed]
- Kiesewalter, H.T.; Lozano-Andrade, C.N.; Wibowo, M.; Strube, M.L.; Maróti, G.; Snyder, D.; Jørgensen, T.S.; Larsen, T.O.; Cooper, V.S.; Weber, T.; et al. Genomic and Chemical Diversity of Bacillus subtilis Secondary Metabolites against Plant Pathogenic Fungi. mSystems 2021, 6, e00770-20. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Li-Qun, Z.; Zeng, Q.; Atiq, M.; Ahmad, K.; Tariq, A.; Al-Ansari, N.; Blom, J.; Fenske, L.; Alodaini, H.A.; et al. Comprehensive Genomic Analysis of Bacillus paralicheniformis Strain BP9, Pan-Genomic and Genetic Basis of Biocontrol Mechanism. Comput. Struct. Biotechnol. J. 2023, 21, 4647–4662. [Google Scholar] [CrossRef] [PubMed]
- Giani, A.M.; Gallo, G.R.; Gianfranceschi, L.; Formenti, G. Long Walk to Genomics: History and Current Approaches to Genome Sequencing and Assembly. Comput. Struct. Biotechnol. J. 2020, 18, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.S. A Review of Pangenome Tools and Recent Studies. In The Pangenome: Diversity, Dynamics and Evolution of Genomes; Tettelin, H., Medini, D., Eds.; Springer: Cham, Switzerland, 2020; ISBN 978-3-030-38280-3. [Google Scholar]
- Hyun, J.C.; Monk, J.M.; Palsson, B.O. Comparative Pangenomics: Analysis of 12 Microbial Pathogen Pangenomes Reveals Conserved Global Structures of Genetic and Functional Diversity. BMC Genom. 2022, 23, 7. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A.; et al. Producing Polished Prokaryotic Pangenomes with the Panaroo Pipeline. Genome Biol. 2020, 21, 180. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative Genomics: The Bacterial Pan-Genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.P.; Roux, S.; Schulz, F.; Babinski, M.; Xu, Y.; Hu, B.; Chain, P.S.G.; Nayfach, S.; Kyrpides, N.C. Identification of Mobile Genetic Elements with geNomad. Nat. Biotechnol. 2023, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.L.; Mullet, J.; Hindi, F.; Stoll, J.E.; Gupta, S.; Choi, M.; Keenum, I.; Vikesland, P.; Pruden, A.; Zhang, L. mobileOG-Db: A Manually Curated Database of Protein Families Mediating the Life Cycle of Bacterial Mobile Genetic Elements. Appl. Environ. Microbiol. 2022, 88, e00991-22. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Fan, X.; Lou, Z.; Wang, H.; Olatunde, A.; Rengasamy, K.R.R. Iturin: Cyclic Lipopeptide with Multifunction Biological Potential. Crit. Rev. Food Sci. Nutr. 2022, 62, 7976–7988. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhi, Y.; Xu, Y. Systematically Engineering the Biosynthesis of a Green Biosurfactant Surfactin by Bacillus subtilis 168. Metab. Eng. 2019, 52, 87–97. [Google Scholar] [CrossRef]
- Piewngam, P.; Zheng, Y.; Nguyen, T.H.; Dickey, S.W.; Joo, H.-S.; Villaruz, A.E.; Glose, K.A.; Fisher, E.L.; Hunt, R.L.; Li, B.; et al. Pathogen Elimination by Probiotic Bacillus via Signalling Interference. Nature 2018, 562, 532–553. [Google Scholar] [CrossRef]
- Gangwal, A.; Kumar, N.; Sangwan, N.; Dhasmana, N.; Dhawan, U.; Sajid, A.; Arora, G.; Singh, Y. Giving a Signal: How Protein Phosphorylation Helps Bacillus Navigate through Different Life Stages. FEMS Microbiol. Rev. 2023, 47, fuad044. [Google Scholar] [CrossRef] [PubMed]
- Darmon, E.; Leach, D.R.F. Bacterial Genome Instability. Microbiol. Mol. Biol. Rev. 2014, 78, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Padan, E.; Venturi, M.; Gerchman, Y.; Dover, N. Na+/H+ Antiporters. Biochim. Biophys. Acta BBA-Bioenerg. 2001, 1505, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, A.; Gelsinger, D.R.; Ma, B.; Wierzchos, J.; Ravel, J.; Davila, A.; Casero, M.C.; DiRuggiero, J. Functional Interactions of Archaea, Bacteria and Viruses in a Hypersaline Endolithic Community. Environ. Microbiol. 2016, 18, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, W.; Teng, D.; Yang, X.; Zhang, Y.; Li, Y. Metagenomic Insights into Microbial Community Structure, Function, and Salt Adaptation in Saline Soils of Arid Land, China. Microorganisms 2022, 10, 2183. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Luo, J.; Chen, Y.; Wu, R.; Liu, H.; Zhou, Z.; Akhtar, M.; Xiao, Y.; Shi, D. A Buffalo Rumen-Derived Probiotic (SN-6) Could Effectively Increase Simmental Growth Performance by Regulating Fecal Microbiota and Metabolism. Front. Microbiol. 2022, 13, 935884. [Google Scholar] [CrossRef]
- Helfrich, M.; Entian, K.-D.; Stein, T. Antibiotic Profiling of Wild-Type Bacilli Led to the Discovery of New Lanthipeptide Subtilin-Producing Bacillus spizizenii Strains Whose 16S rDNA Sequences Differ from the B. spizizenii Typing Strain. Int. Microbiol. 2022, 25, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Chauhan, S.M.; Mohite, O.S.; Hyun, J.C.; Ardalani, O.; Jahn, L.J.; Sommer, M.O.; Palsson, B.O. Pangenome Analysis Reveals the Genetic Basis for Taxonomic Classification of the Lactobacillaceae Family. Food Microbiol. 2023, 115, 104334. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Jabbar, K.S.; Ruohtula, T.; Honkanen, J.; Avila-Pacheco, J.; Siljander, H.; Stražar, M.; Oikarinen, S.; Hyöty, H.; Ilonen, J.; et al. Mobile Genetic Elements from the Maternal Microbiome Shape Infant Gut Microbial Assembly and Metabolism. Cell 2022, 185, 4921–4936.e15. [Google Scholar] [CrossRef]
- Arnold, B.J.; Huang, I.-T.; Hanage, W.P. Horizontal Gene Transfer and Adaptive Evolution in Bacteria. Nat. Rev. Microbiol. 2022, 20, 206–218. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Formal Genome Number | CheckM Number | GTDB Number | LPSN Message |
|---|---|---|---|---|
| B. amyloliquefaciens | 176 | 176 | 176 | TRUE |
| B. halotolerans | 56 | 56 | 56 | TRUE |
| B. mojavensis | 11 | 11 | 11 | TRUE |
| B. siamensis | 11 | 11 | 11 | TRUE |
| B. tequilensis | 5 | 5 | 5 | TRUE |
| B. velezensis | 446 | 446 | 446 | TRUE |
| B. atrophaeus | 93 | 93 | 93 | TRUE |
| B. licheniformis | 274 | 274 | 274 | TRUE |
| B. paralicheniformis | 136 | 136 | 136 | TRUE |
| B. sonorensis | 26 | 26 | 26 | TRUE |
| B. vallismortis | 14 | 14 | 13 | TRUE |
| B. spizizenii | 142 | 140 | 140 | TRUE |
| B. stercoris | 10 | 8 | 8 | TRUE |
| B. inaquosorum | 106 | 106 | 106 | TRUE |
| B. subtilius | 585 | 585 | 571 | TRUE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Shi, Y.; Zheng, M.; Zheng, J. Comparative Genomics Unveils Functional Diversity, Pangenome Openness, and Underlying Biological Drivers among Bacillus subtilis Group. Microorganisms 2024, 12, 986. https://doi.org/10.3390/microorganisms12050986
Wang T, Shi Y, Zheng M, Zheng J. Comparative Genomics Unveils Functional Diversity, Pangenome Openness, and Underlying Biological Drivers among Bacillus subtilis Group. Microorganisms. 2024; 12(5):986. https://doi.org/10.3390/microorganisms12050986
Chicago/Turabian StyleWang, Taiquan, Yiling Shi, Mengzhuo Zheng, and Jinshui Zheng. 2024. "Comparative Genomics Unveils Functional Diversity, Pangenome Openness, and Underlying Biological Drivers among Bacillus subtilis Group" Microorganisms 12, no. 5: 986. https://doi.org/10.3390/microorganisms12050986
APA StyleWang, T., Shi, Y., Zheng, M., & Zheng, J. (2024). Comparative Genomics Unveils Functional Diversity, Pangenome Openness, and Underlying Biological Drivers among Bacillus subtilis Group. Microorganisms, 12(5), 986. https://doi.org/10.3390/microorganisms12050986