
Marine Bacterial Community Structures of Selected Coastal Seawater and Sediment Sites in Qatar

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Descriptions and Sample Collection Protocol
2.2. Sample Preprocessing and Physicochemical Analysis
2.3. Bacteriological Analysis
2.4. DNA Extraction, PCR Amplification, and 16S rRNA Amplicon Sequencing
3. Results and Discussion
3.1. Environmental Factors and Their Relationships with Marine Bacterial Communities
3.2. Indicator Bacteria Concentrations in Seawater
3.3. Indicator Bacteria Concentrations in Sediment
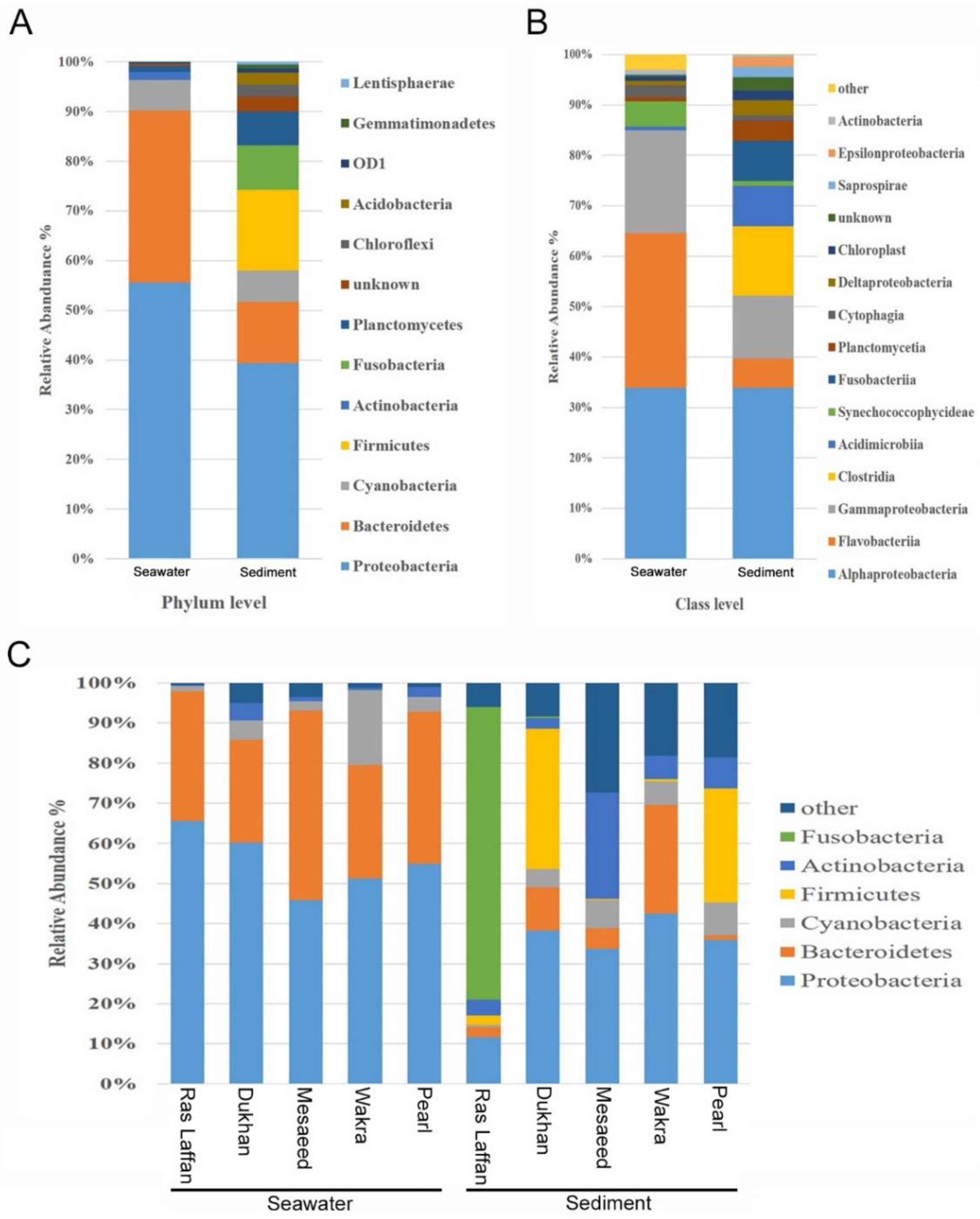
3.4. Diversity and Taxonomic Composition of Marine Bacteria at Qatar Coastal Locations
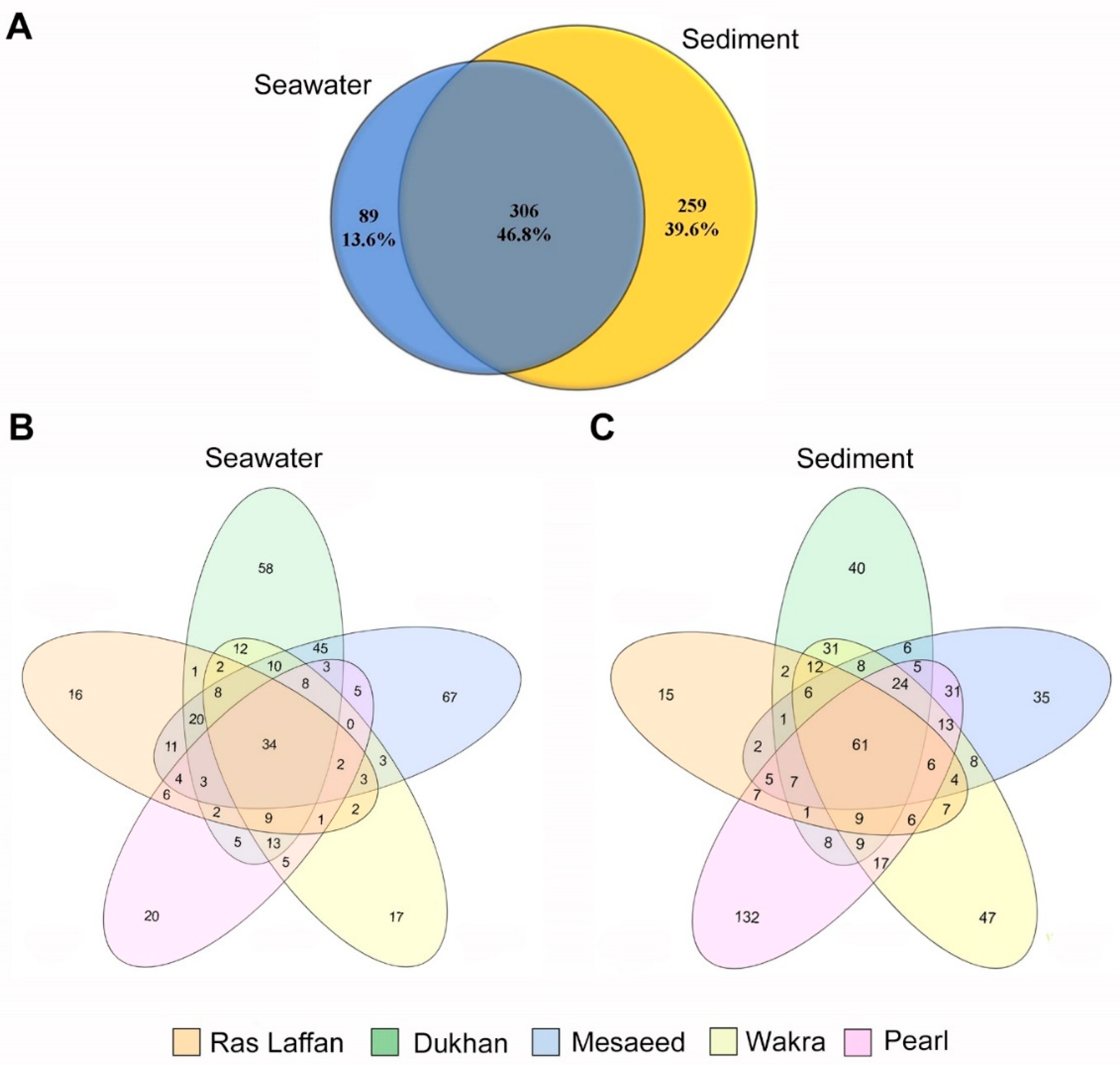
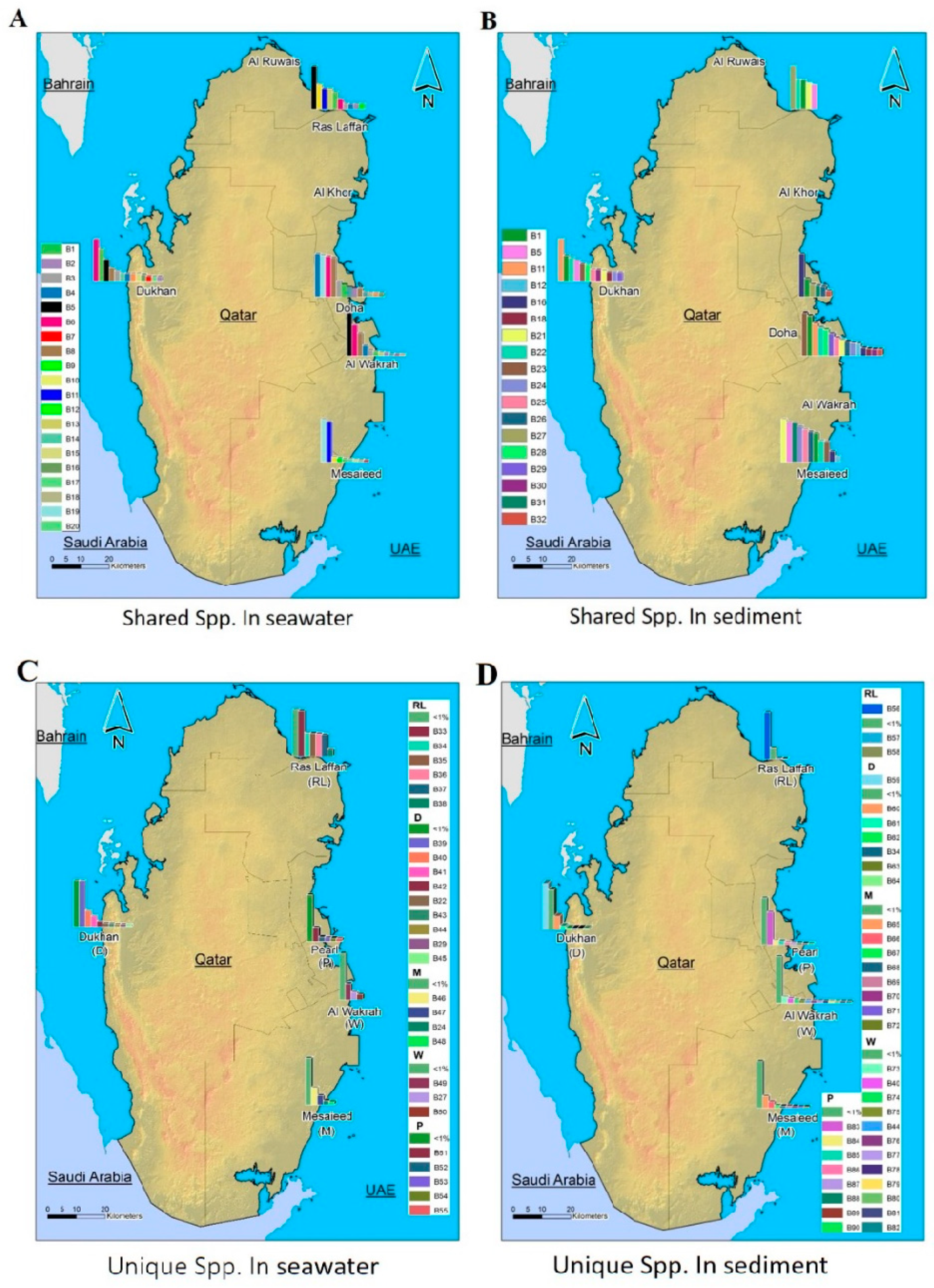
3.5. Microbial Composition and Distribution in Seawater and Sediment Samples
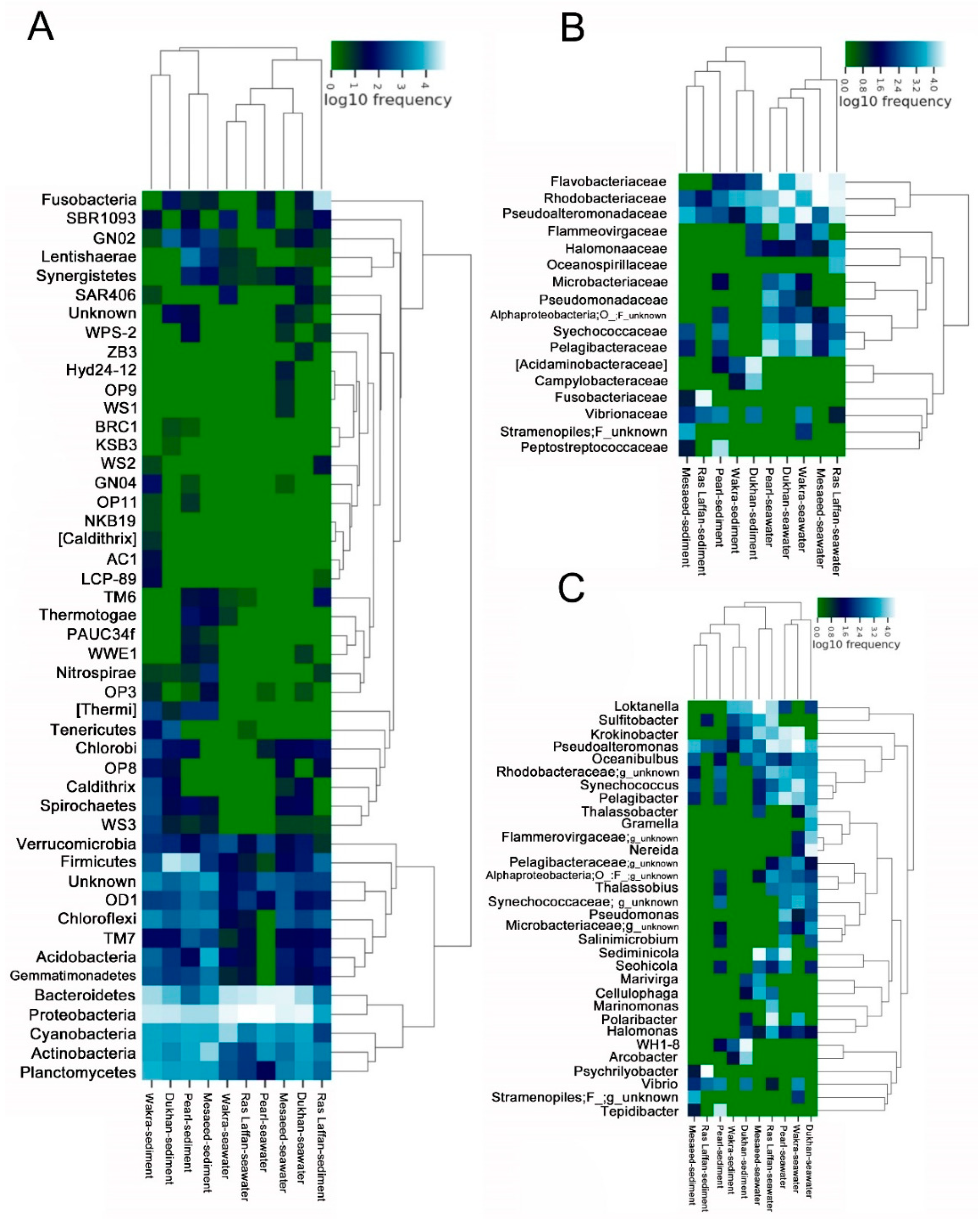
3.6. Structural Analysis of the Microbial Communities in the Samples
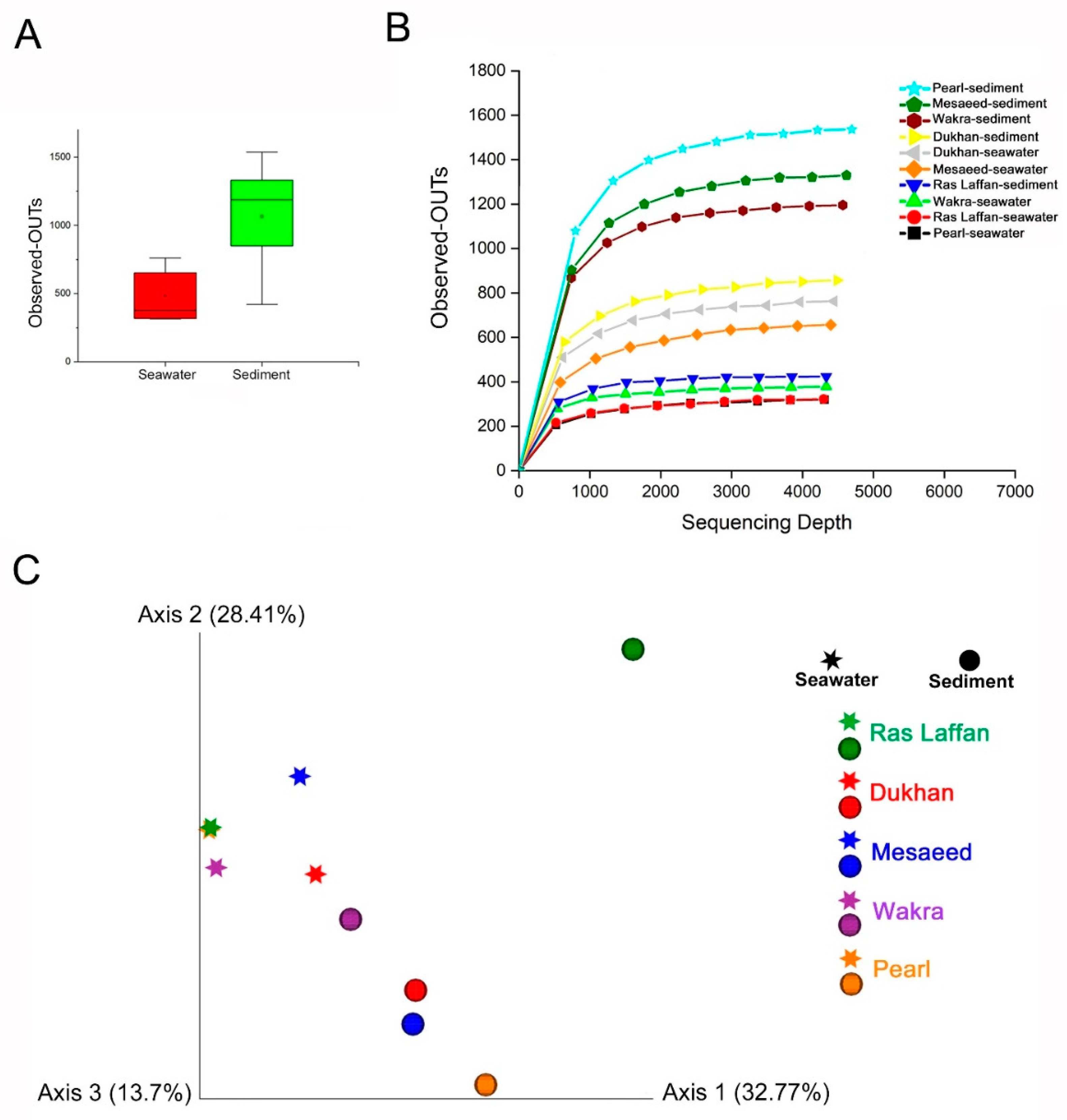
3.7. Alpha and Beta Diversity of the Microbial Communities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nair, M.; Kumar, D. Water desalination and challenges: The Middle East perspective: A review. Desalination Water Treat. 2013, 51, 2030–2040. [Google Scholar] [CrossRef]
- Belila, A.; El-Chakhtoura, J.; Otaibi, N.; Muyzer, G.; Gonzalez-Gil, G.; Saikaly, P.E.; van Loosdrecht, M.C.M.; Vrouwenvelder, J.S. Bacterial community structure and variation in a full-scale seawater desalination plant for drinking water production. Water Res. 2016, 94, 62–72. [Google Scholar] [CrossRef]
- Kormas, K.A.; Neofitou, C.; Pachiadaki, M.; Koufostathi, E. Changes of the bacterial assemblages throughout an urban drinking water distribution system. Environ. Monit. Assess. 2010, 165, 27–38. [Google Scholar] [CrossRef]
- El-Chakhtoura, J.; Prest, E.; Saikaly, P.; van Loosdrecht, M.; Hammes, F.; Vrouwenvelder, H. Dynamics of bacterial communities before and after distribution in a full-scale drinking water network. Water Res. 2015, 74, 180–190. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, S.; Tanuwidjaja, D.; Voutchkov, N.; Hoek, E.M.; Cai, B. Composition and variability of biofouling organisms in seawater reverse osmosis desalination plants. Appl. Environ. Microbiol. 2011, 77, 4390–4398. [Google Scholar] [CrossRef] [PubMed]
- Chiellini, C.; Iannelli, R.; Modeo, L.; Bianchi, V.; Petroni, G. Biofouling of reverse osmosis membranes used in river water purification for drinking purposes: Analysis of microbial populations. Biofouling 2012, 28, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Bereschenko, L.A.; Heilig, G.H.; Nederlof, M.M.; van Loosdrecht, M.C.; Stams, A.J.; Euverink, G.J. Molecular characterization of the bacterial communities in the different compartments of a full-scale reverse-osmosis water purification plant. Appl. Environ. Microbiol. 2008, 74, 5297–5304. [Google Scholar] [CrossRef] [PubMed]
- Eichler, S.; Christen, R.; Höltje, C.; Westphal, P.; Bötel, J.; Brettar, I.; Mehling, A.; Höfle, M.G. Composition and dynamics of bacterial communities of a drinking water supply system as assessed by RNA- and DNA-based 16S rRNA gene fingerprinting. Appl. Environ. Microbiol. 2006, 72, 1858–1872. [Google Scholar] [CrossRef]
- Pinto, A.J.; Xi, C.; Raskin, L. Bacterial Community Structure in the Drinking Water Microbiome Is Governed by Filtration Processes. Environ. Sci. Technol. 2012, 46, 8851–8859. [Google Scholar] [CrossRef]
- Salazar, G.; Sunagawa, S. Marine microbial diversity. Curr. Biol. 2017, 27, R489–R494. [Google Scholar] [CrossRef] [PubMed]
- Halpern, B.S.; Walbridge, S.; Selkoe, K.A.; Kappel, C.V.; Micheli, F.; D’agrosa, C.; Bruno, J.F.; Casey, K.S.; Ebert, C.; Fox, H.E. A global map of human impact on marine ecosystems. Science 2008, 319, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Gevao, B.; Al-Ghadban, A.; Nithyanandan, M.; Al-Shamroukh, D. Acidification in Arabian Gulf–Insights from pH and temperature measurements. J. Environ. Monit. 2012, 14, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Lattemann, S.; Höpner, T. Environmental impact and impact assessment of seawater desalination. Desalination 2008, 220, 1–15. [Google Scholar] [CrossRef]
- Naser, H.A. Effects of multi-stage flash and reverse osmosis desalinations on benthic assemblages in Bahrain, Arabian Gulf. J. Environ. Prot. 2013, 4, 180. [Google Scholar] [CrossRef]
- Sheppard, C.; Al-Husiani, M.; Al-Jamali, F.; Al-Yamani, F.; Baldwin, R.; Bishop, J.; Benzoni, F.; Dutrieux, E.; Dulvy, N.K.; Durvasula, S.R.V. The Gulf: A young sea in decline. Mar. Pollut. Bull. 2010, 60, 13–38. [Google Scholar] [CrossRef]
- Naser, H. Human impacts on marine biodiversity: Macrobenthos in Bahrain, Arabian Gulf. In The Importance of Biological Interactions in the Study of Biodiversity; Lopez-Pujol, J., Ed.; InTech Publishing: Rijeka, Croatia, 2011; pp. 109–126. [Google Scholar]
- Karbasdehi, V.N.; Dobaradaran, S.; Nabipour, I.; Ostovar, A.; Arfaeinia, H.; Vazirizadeh, A.; Mirahmadi, R.; Keshtkar, M.; Ghasemi, F.F.; Khalifei, F. Indicator bacteria community in seawater and coastal sediment: The Persian Gulf as a case. J. Environ. Health Sci. Eng. 2017, 15, 6. [Google Scholar] [CrossRef]
- ASTM E1391-03; Standard Guide for Collection, Storage, Characterization, and Manipulation of Sediments for Toxicological Testing and for Selection of Samplers Used to Collect Benthic Invertebrates. American Society for Testing and Materials: Philadelphia, PA, USA, 2014.
- Turturice, B.A.; Gold, D.R.; Litonjua, A.A.; Oken, E.; Rifas-Shiman, S.; Perkins, D.L.; Finn, P.W. Lower perinatal exposure to Proteobacteria is an independent predictor of early childhood wheezing. J. Allergy Clin. Immunol. 2019, 143, 419–421.e5. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef]
- Kumar, R.; Mishra, A.; Jha, B. Bacterial community structure and functional diversity in subsurface seawater from the western coastal ecosystem of the Arabian Sea, India. Gene 2019, 701, 55–64. [Google Scholar] [CrossRef]
- Keshri, J.; Mishra, A.; Jha, B. Microbial population index and community structure in saline–alkaline soil using gene targeted metagenomics. Microbiol. Res. 2013, 168, 165–173. [Google Scholar] [CrossRef]
- Oberbeckmann, S.; Osborn, A.M.; Duhaime, M.B. Microbes on a bottle: Substrate, season and geography influence community composition of microbes colonizing marine plastic debris. PLoS ONE 2016, 11, e0159289. [Google Scholar] [CrossRef] [PubMed]
- Giani, M.; Djakovac, T.; Degobbis, D.; Cozzi, S.; Solidoro, C.; Umani, S.F. Recent changes in the marine ecosystems of the northern Adriatic Sea. Estuar. Coast. Shelf Sci. 2012, 115, 1–13. [Google Scholar] [CrossRef]
- Goyal, S.M.; Gerba, C.P.; Melnick, J.L. Occurrence and distribution of bacterial indicators and pathogens in canal communities along the Texas coast. Appl. Environ. Microbiol. 1977, 34, 139–149. [Google Scholar] [CrossRef]
- Placha, I.; Venglovský, J.; Sasakova, N.; Svoboda, I. The effect of summer and winter seasons on the survival of Salmonella typhimurium and indicator micro-organisms during the storage of solid fraction of pig slurry. J. Appl. Microbiol. 2001, 91, 1036–1043. [Google Scholar] [CrossRef]
- Pedrós-Alió, C. Trophic ecology of solar salterns. In Halophilic Microorganisms; Springer: Berlin/Heidelberg, Germany, 2004; pp. 33–48. [Google Scholar]
- Pachepsky, Y.; Shelton, D. Escherichia coli and fecal coliforms in freshwater and estuarine sediments. Crit. Rev. Environ. Sci. Technol. 2011, 41, 1067–1110. [Google Scholar] [CrossRef]
- Brinkmeyer, R.; Amon, R.M.; Schwarz, J.R.; Saxton, T.; Roberts, D.; Harrison, S.; Ellis, N.; Fox, J.; DiGuardi, K.; Hochman, M. Distribution and persistence of Escherichia coli and Enterococci in stream bed and bank sediments from two urban streams in Houston, TX. Sci. Total Environ. 2015, 502, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Weagant, S.D.; Grant, M.A.; Burkhardt, W.; Shellfish, M.; Water, B. BAM: Enumeration of Escherichia coli and the Coliform Bacteria. Bacteriol. Anal. Man. 2002, 13, 1–13. [Google Scholar]
- Donovan, E.; Unice, K.; Roberts, J.D.; Harris, M.; Finley, B. Risk of gastrointestinal disease associated with exposure to pathogens in the water of the Lower Passaic River. Appl. Environ. Microbiol. 2008, 74, 994–1003. [Google Scholar] [CrossRef]
- Droppo, I.; Krishnappan, B.; Liss, S.; Marvin, C.; Biberhofer, J. Modelling sediment-microbial dynamics in the South Nation River, Ontario, Canada: Towards the prediction of aquatic and human health risk. Water Res. 2011, 45, 3797–3809. [Google Scholar] [CrossRef]
- Lee, J.; Kim, I.S. Microbial community in seawater reverse osmosis and rapid diagnosis of membrane biofouling. Desalination 2011, 273, 118–126. [Google Scholar] [CrossRef]
- Jeong, D.; Lee, C.-H.; Lee, S.; Bae, H. Intermittent chlorination shifts the marine biofilm population on reverse osmosis membranes. Membr. Water Treat. 2019, 10, 395–404. [Google Scholar]
- Levi, A.; Bar-Zeev, E.; Elifantz, H.; Berman, T.; Berman-Frank, I. Characterization of microbial communities in water and biofilms along a large scale SWRO desalination facility: Site-specific prerequisite for biofouling treatments. Desalination 2016, 378, 44–52. [Google Scholar] [CrossRef]
- Ghiglione, J.-F.; Galand, P.E.; Pommier, T.; Pedrós-Alió, C.; Maas, E.W.; Bakker, K.; Bertilson, S.; Kirchman, D.L.; Lovejoy, C.; Yager, P.L. Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl. Acad. Sci. USA 2012, 109, 17633–17638. [Google Scholar] [CrossRef] [PubMed]
- Soininen, J.; Lennon, J.J.; Hillebrand, H. A multivariate analysis of beta diversity across organisms and environments. Ecology 2007, 88, 2830–2838. [Google Scholar] [CrossRef] [PubMed]
- Turley, C. The importance of ‘marine snow’. Microbiol. Today 2002, 29, 177–179. [Google Scholar]
- Treusch, A.H.; Vergin, K.L.; Finlay, L.A.; Donatz, M.G.; Burton, R.M.; Carlson, C.A.; Giovannoni, S.J. Seasonality and vertical structure of microbial communities in an ocean gyre. ISME J. 2009, 3, 1148–1163. [Google Scholar] [CrossRef]
- Dang, H.; Chen, R.; Wang, L.; Shao, S.; Dai, L.; Ye, Y.; Guo, L.; Huang, G.; Klotz, M.G. Molecular characterization of putative biocorroding microbiota with a novel niche detection of Epsilon- and Zetaproteobacteria in Pacific Ocean coastal seawaters. Environ. Microbiol. 2011, 13, 3059–3074. [Google Scholar] [CrossRef]
- Techtmann, S.M.; Fortney, J.L.; Ayers, K.A.; Joyner, D.C.; Linley, T.D.; Pfiffner, S.M.; Hazen, T.C. The Unique Chemistry of Eastern Mediterranean Water Masses Selects for Distinct Microbial Communities by Depth. PLoS ONE 2015, 10, e0120605. [Google Scholar] [CrossRef]
- Kirchman, D.L.; Kirchman, D.L. 195Processes in anoxic environments. In Processes in Microbial Ecology; Oxford University Press: Oxford, UK, 2011; pp. 195–216. [Google Scholar]
- Nagaraj, V.; Skillman, L.; Ho, G.; Li, D.; Gofton, A. Characterisation and comparison of bacterial communities on reverse osmosis membranes of a full-scale desalination plant by bacterial 16S rRNA gene metabarcoding. NPJ Biofilms Microbiomes 2017, 3, 13. [Google Scholar] [CrossRef]
- Selje, N.; Simon, M.; Brinkhoff, T. A newly discovered Roseobacter cluster in temperate and polar oceans. Nature 2004, 427, 445–448. [Google Scholar] [CrossRef]
- Dang, H.; Lovell, C.R. Microbial surface colonization and biofilm development in marine environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Keats, K.F.; Rivkin, R.B.; Lang, A.S. Water mass and depth determine the distribution and diversity of Rhodobacterales in an Arctic marine system. FEMS Microbiol. Ecol. 2013, 84, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Kirchman, D.L. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Ul-Hasan, S.; Bowers, R.M.; Figueroa-Montiel, A.; Licea, A.F.; Beman, J.M.; Woyke, T.; Nobile, C.J. Archaeal, bacterial, and eukaryal microbial community structure of sediment and seawater in a coastal region near Puerto Nuevo, Baja California. bioRxiv 2018, 324442. [Google Scholar]
- Hao, Y.Q.; Zhao, X.F.; Zhang, D.Y. Field experimental evidence that stochastic processes predominate in the initial assembly of bacterial communities. Environ. Microbiol. 2016, 18, 1730–1739. [Google Scholar] [CrossRef]
- Langenheder, S.; Ragnarsson, H. The role of environmental and spatial factors for the composition of aquatic bacterial communities. Ecology 2007, 88, 2154–2161. [Google Scholar] [CrossRef]
- Hamdan, L.J.; Coffin, R.B.; Sikaroodi, M.; Greinert, J.; Treude, T.; Gillevet, P.M. Ocean currents shape the microbiome of Arctic marine sediments. ISME J. 2013, 7, 685–696. [Google Scholar] [CrossRef]
- Caruso, G. Microbial Colonization in Marine Environments: Overview of Current Knowledge and Emerging Research Topics. J. Mar. Sci. Eng. 2020, 8, 78. [Google Scholar] [CrossRef]
- Dang, H.; Lovell, C.R. Numerical dominance and phylotype diversity of marine Rhodobacter species during early colonization of submerged surfaces in coastal marine waters as determined by 16S ribosomal DNA sequence analysis and fluorescence in situ hybridization. Appl. Environ. Microbiol. 2002, 68, 496–504. [Google Scholar] [CrossRef]
- Jones, P.R.; Cottrell, M.T.; Kirchman, D.L.; Dexter, S.C. Bacterial community structure of biofilms on artificial surfaces in an estuary. Microb. Ecol. 2007, 53, 153–162. [Google Scholar] [CrossRef]
- Hanson, C.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Martiny, J.B. Beyond biogeographic patterns: Processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012, 10, 497. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, N.; Scheu, S.; Jousset, A. Bacterial diversity stabilizes community productivity. PLoS ONE 2012, 7, e34517. [Google Scholar] [CrossRef] [PubMed]
- Zinger, L.; Amaral-Zettler, L.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Huse, S.M.; Welch, D.B.M.; Martiny, J.B.; Sogin, M.; Boetius, A.; Ramette, A. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE 2011, 6, e24570. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Geographical Location | |
|---|---|---|
| Latitude | Longitude | |
| Ras Laffan (RL) | 25°57′15.7″ N | 51°29′23.5″ E |
| Dukhan (D) | 25°24′57.6″ N | 50°45′30.2″ E |
| Mesaeed (M) | 24°51′17.4″ N | 51°30′43.5″ E |
| Wakra (W) | 25°11′07.3″ N | 51°37′12.7″ E |
| Pearl (P) | 25°22′12.5″ N | 51°31′44.3″ E |
| Sampling Time | Location | Temp. (°C) | pH | Conductivity (mS/cm) |
|---|---|---|---|---|
| July | Ras Laffan | 37 ± 0.19 | 8.27 ± 0.1 | 60.9 ±0.36 |
| Dukhan | 31 ± 0.18 | 8.24 ± 0.1 | 76.9 ± 0.36 | |
| Mesaeed | 34 ± 0.18 | 8.28 ± 0.1 | 65.9 ± 0.36 | |
| Wakra | 37 ± 0.19 | 8.60 ± 0.1 | 63.2 ± 0.36 | |
| Pearl | 36 ± 0.19 | 8.24 ± 0.1 | 60.0 ± 0.36 | |
| September | Ras Laffan | 21.4 ± 0.18 | 8.26 ± 0.1 | 63.3 ± 0.36 |
| Dukhan | 22.1 ± 0.18 | 8.23 ± 0.1 | 80.4 ± 0.36 | |
| Mesaeed | 22.5 ± 0.18 | 8.40 ± 0.1 | 68.3 ± 0.36 | |
| Wakra | 24 ± 0.19 | 8.60 ± 0.1 | 68.2 ± 0.36 | |
| Pearl | 22 ± 0.18 | 8.31 ± 0.1 | 63.0 ± 0.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Malah, S.S.; Rasool, K.; Jabbar, K.A.; Sohail, M.U.; Baalousha, H.M.; Mahmoud, K.A. Marine Bacterial Community Structures of Selected Coastal Seawater and Sediment Sites in Qatar. Microorganisms 2023, 11, 2827. https://doi.org/10.3390/microorganisms11122827
El-Malah SS, Rasool K, Jabbar KA, Sohail MU, Baalousha HM, Mahmoud KA. Marine Bacterial Community Structures of Selected Coastal Seawater and Sediment Sites in Qatar. Microorganisms. 2023; 11(12):2827. https://doi.org/10.3390/microorganisms11122827
Chicago/Turabian StyleEl-Malah, Shimaa S., Kashif Rasool, Khadeeja Abdul Jabbar, Muhammad Umar Sohail, Husam Musa Baalousha, and Khaled A. Mahmoud. 2023. "Marine Bacterial Community Structures of Selected Coastal Seawater and Sediment Sites in Qatar" Microorganisms 11, no. 12: 2827. https://doi.org/10.3390/microorganisms11122827
APA StyleEl-Malah, S. S., Rasool, K., Jabbar, K. A., Sohail, M. U., Baalousha, H. M., & Mahmoud, K. A. (2023). Marine Bacterial Community Structures of Selected Coastal Seawater and Sediment Sites in Qatar. Microorganisms, 11(12), 2827. https://doi.org/10.3390/microorganisms11122827