
Development of Droplet Digital PCR Assay for Detection of Seed-Borne Burkholderia glumae and B. gladioli Causing Bacterial Panicle Blight Disease of Rice

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacteria Strains
2.2. Preparation of Bacterial Suspension and Rice Seeds Carrying Bacteria
2.3. Preparation of Bacterial Genomic DNA
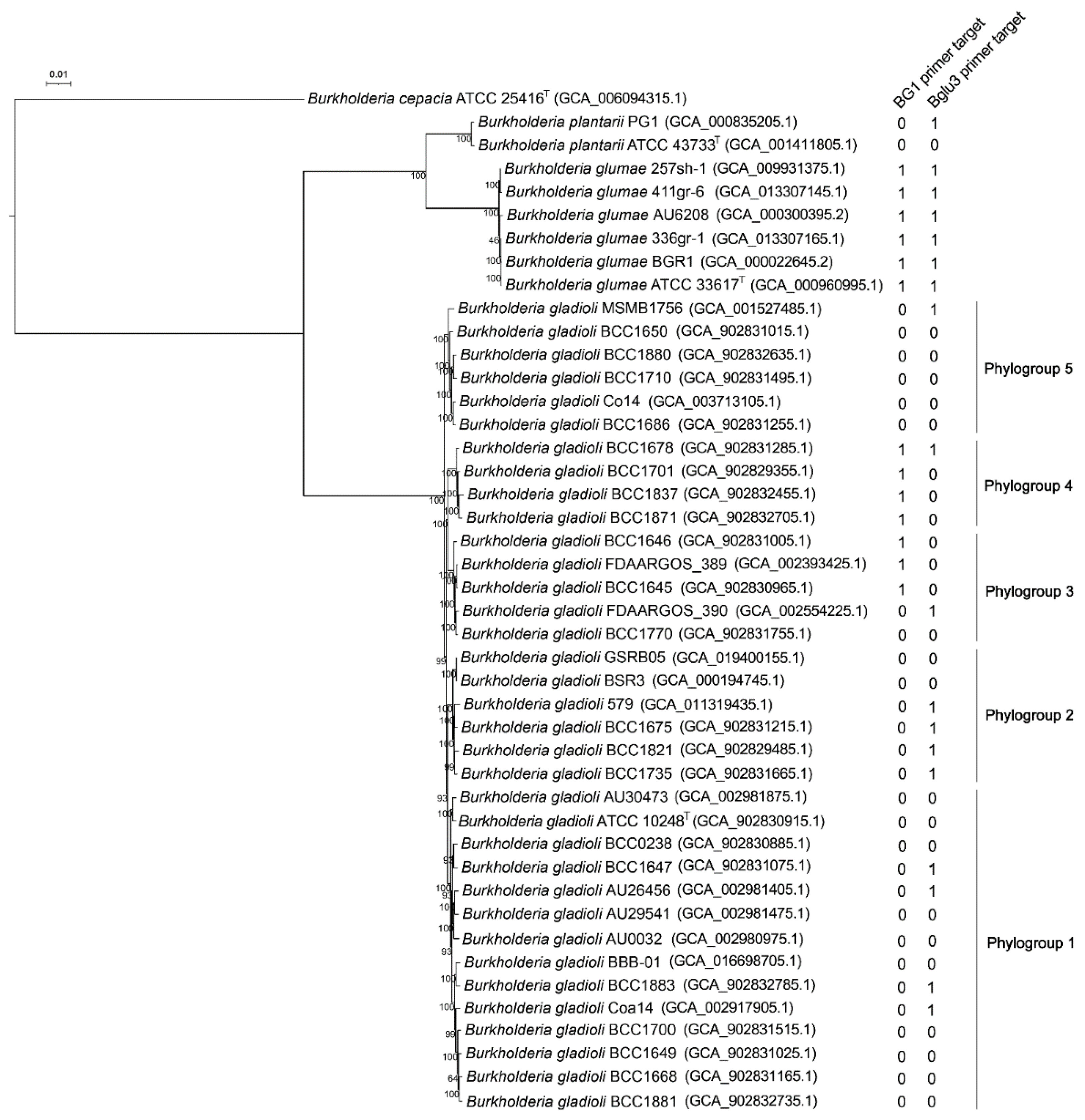
2.4. Genome Relatedness Analysis of Burkholderia gladioli
2.5. Phylogenomic Analysis
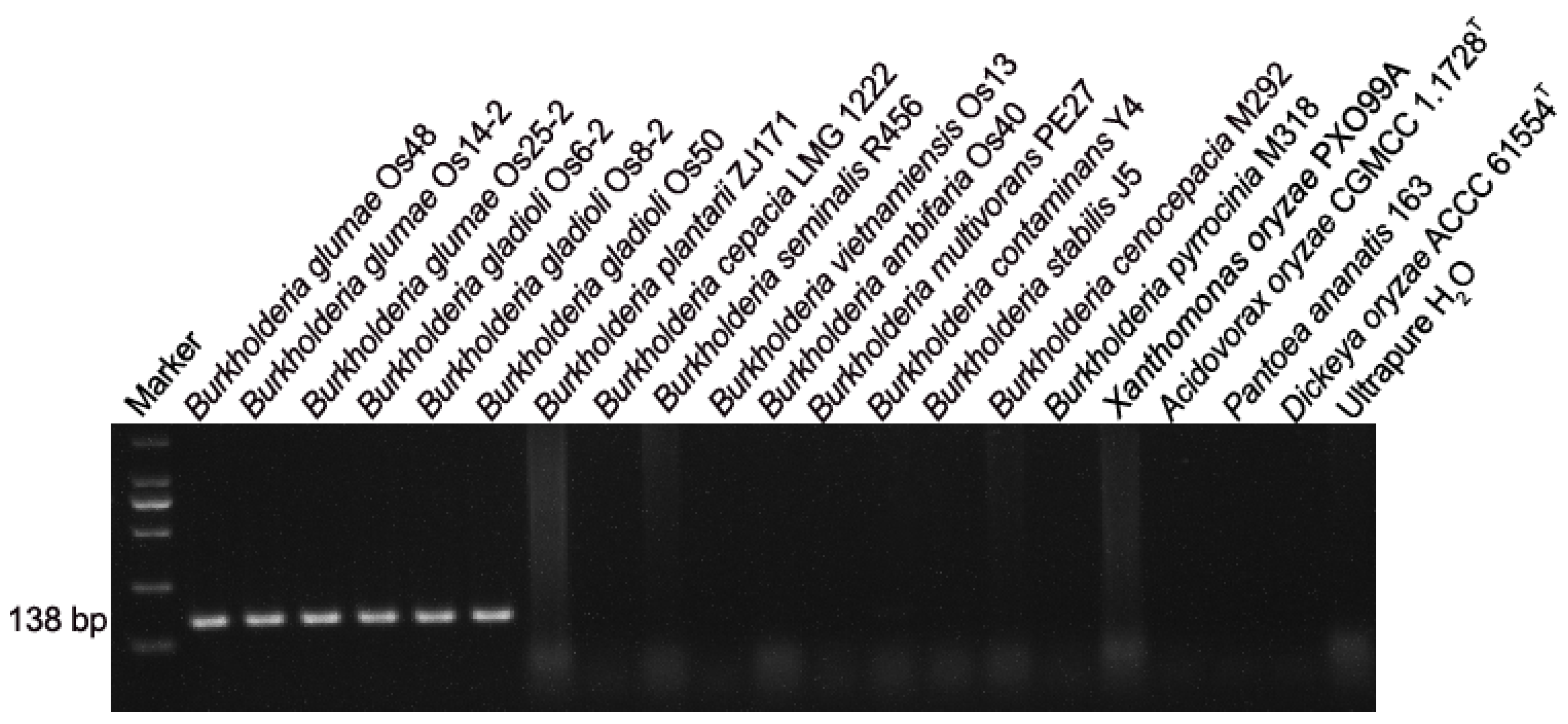
2.6. Evaluation of Previously Designed B. glumae-Specific Primers
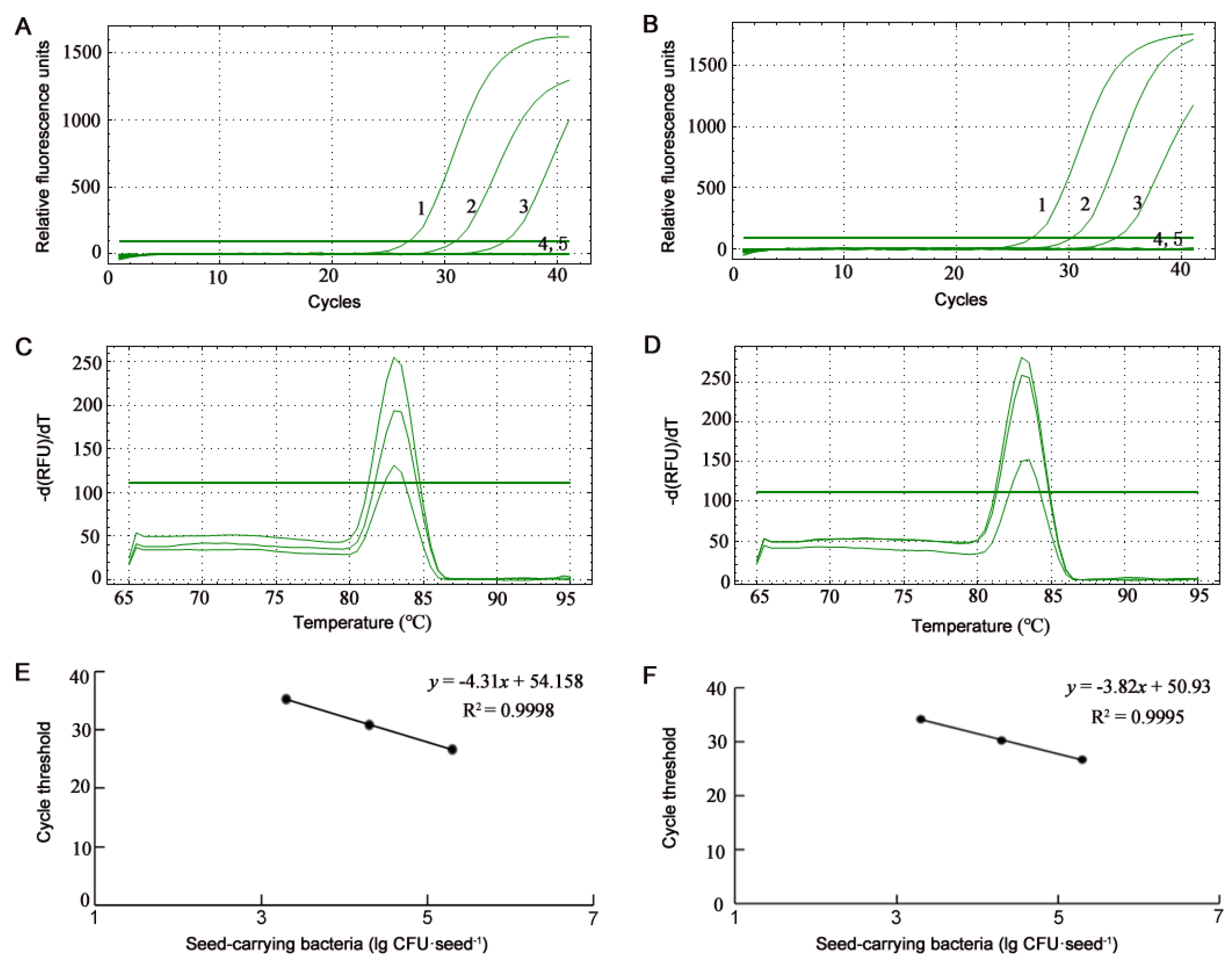
2.7. qPCR
2.8. dPCR
3. Results and Discussion
3.1. BG1 Primers Are Specific to B. glumae and Two Phylogroups within B. gladioli
3.2. Detection of B. glumae and B. gladioli Using SYBR Green qPCR
3.3. Detection of B. glumae and B. gladioli Using Droplet EvaGreen dPCR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ham, J.H.; Melanson, R.A.; Rush, M.C. Burkholderia glumae: Next major pathogen of rice? Mol. Plant Pathol. 2011, 12, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Ura, H.; Furuya, N.; Iiyama, K.; Hidaka, M.; Tsuchiya, K.; Matsuyama, N. Burkholderia gladioli associated with symptoms of bacterial grain rot and leaf-sheath browning of rice plants. J. Gen. Plant Pathol. 2006, 72, 98–103. [Google Scholar] [CrossRef]
- Nandakumar, R.; Rush, M.C.; Correa, F. Association of Burkholderia glumae and B. gladioli with panicle blight symptoms on rice in Panama. Plant Dis. 2007, 91, 767. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, R.; Bollich, P.A.; Shahjahan, A.K.M.; Groth, D.E.; Rush, M.C. Evidence for the soilborne nature of the rice sheath rot and panicle blight pathogen, Burkholderia gladioli. Can. J. Plant Pathol. 2008, 30, 148–154. [Google Scholar] [CrossRef]
- Sayler, R.J.; Cartwright, R.D.; Yang, Y. Genetic characterization and real-time PCR detection of Burkholderia glumae, a newly emerging bacterial pathogen of rice in the United States. Plant Dis. 2006, 90, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandakumar, R.; Shahjahan, A.K.M.; Yuan, X.L.; Dickstein, E.R.; Groth, D.E.; Clark, C.A.; Cartwright, R.D.; Rush, M.C. Burkholderia glumae and B. gladioli cause bacterial panicle blight in rice in the Southern United States. Plant Dis. 2009, 93, 896–905. [Google Scholar] [CrossRef] [Green Version]
- Cottyn, B.; Regalado, E.; Lanoot, B.; De Cleene, M.; Mew, T.W.; Swings, J. Bacterial populations associated with rice seed in the tropical environment. Phytopathology 2001, 91, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Cottyn, B.; Debode, J.; Regalado, E.; Mew, T.W.; Swings, J. Phenotypic and genetic diversity of rice seed-associated bacteria and their role in pathogenicity and biological control. J. Appl. Microbiol. 2009, 107, 885–897. [Google Scholar] [CrossRef]
- Morcia, C.; Ghizzoni, R.; Delogu, C.; Andreani, L.; Carnevali, P.; Terzi, V. Digital PCR: What relevance to plant studies? Biology 2020, 9, 433. [Google Scholar] [CrossRef]
- Salipante, S.J.; Jerome, K.R. Digital PCR—An emerging technology with broad applications in microbiology. Clin. Chem. 2020, 66, 117–123. [Google Scholar] [CrossRef]
- Dreo, T.; Pirc, M.; Ramšak, Ž.; Pavšič, J.; Milavec, M.; Zel, J.; Gruden, K. Optimising droplet digital PCR analysis approaches for detection and quantification of bacteria: A case study of fire blight and potato brown rot. Anal. Bioanal. Chem. 2014, 406, 6513–6528. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xia, Q.; Yin, Y.; Wang, Z. Comparison of droplet digital PCR and quantitative PCR assays for quantitative detection of Xanthomonas citri subsp. citri. PLoS ONE 2016, 11, e0159004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maheshwari, Y.; Selvaraj, V.; Hajeri, S.; Yokomi, R. Application of droplet digital PCR for quantitative detection of Spiroplasma citri in comparison with real time PCR. PLoS ONE 2017, 12, e0184751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maheshwari, Y.; Selvaraj, V.; Godfrey, K.; Hajeri, S.; Yokomi, R. Multiplex detection of “Candidatus Liberibacter asiaticus” and Spiroplasma citri by qPCR and droplet digital PCR. PLoS ONE 2021, 16, e0242392. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liu, X.L.; Lou, B.H.; Zhou, C.Y.; Wang, X.F. Development of a sensitive and reliable droplet digital PCR assay for the detection of ‘Candidatus Liberibacter asiaticus’. J. Integr. Agric. 2018, 17, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, H.J.; Zhao, Z.J.; Wen, C.L.; Ping, W.U.; Song, S.H.; Yu, S.C.; Luo, L.X.; Xu, X.L. Application of droplet digital PCR in detection of seed-transmitted pathogen Acidovorax citrulli. J. Integr. Agric. 2020, 19, 561–569. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Q.; Zhou, P.; Zhao, W.; Sun, X. Evaluation of droplet digital PCR for the detection of black canker disease in tomato. Plant Dis. 2022, 106, 395–405. [Google Scholar] [CrossRef]
- Thornton, B.; Basu, C. Real-time PCR (qPCR) primer design using free online software. Biochem. Mol. Biol. Educ. 2011, 39, 145–154. [Google Scholar] [CrossRef]
- Voegel, T.M.; Nelson, L.M. Quantification of Agrobacterium vitis from grapevine nursery stock and vineyard soil using droplet digital PCR. Plant Dis. 2018, 102, 2136–2141. [Google Scholar] [CrossRef] [Green Version]
- Barrett-Manako, K.; Andersen, M.; Martínez-Sánchez, M.; Jenkins, H.; Hunter, S.; Reese-George, J.; Montefiori, M.; Wohlers, M.; Rikkerink, E.; Templeton, M.; et al. Real-time PCR and droplet digital PCR are accurate and reliable methods to quantify Pseudomonas syringae pv. actinidiae biovar 3 in kiwifruit infected plantlets. Plant Dis. 2021, 105, 1748–1757. [Google Scholar] [CrossRef]
- Mao, F.; Leung, W.Y.; Xin, X. Characterization of EvaGreen and the implication of its physicochemical properties for qPCR applications. BMC Biotechnol. 2007, 7, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eischeid, A.C. SYTO dyes and EvaGreen outperform SYBR Green in real-time PCR. BMC Res. Notes 2011, 4, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.K.; Cho, M.S.; Kim, M.H.; Choi, H.J.; Kang, M.J.; Shim, H.S.; Ahn, T.Y.; Kim, J.; Park, D.S. Rapid and specific detection of Burkholderia glumae in rice seed by real-time Bio-PCR using species-specific primers based on an rhs family gene. Plant Dis. 2012, 96, 577–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Lee, H.H.; Mannaa, M.; Kim, N.; Park, J.; Kim, J.; Seo, Y.S. Genomics-based sensitive and specific novel primers for simultaneous detection of Burkholderia glumae and Burkholderia gladioli in rice seeds. Plant Pathol. J. 2018, 34, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chang, S.; Lin, L.; Li, Y.; An, Q. A colorimetric assay of 1-aminocyclopropane-1-carboxylate (ACC) based on ninhydrin reaction for rapid screening of bacteria containing ACC deaminase. Lett. Appl. Microbiol. 2011, 53, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Palleroni, N.J.; Holmes, B. Pseudomonas cepacia sp. nov., nom. rev. Int. J. Syst. Bacteriol. 1981, 31, 479–481. [Google Scholar] [CrossRef]
- Wang, M.; Wei, P.; Cao, M.; Zhu, L.; Lu, Y. First report of rice seedling blight caused by Burkholderia plantarii in North and Southeast China. Plant Dis. 2016, 100, 645. [Google Scholar] [CrossRef]
- Zhang, L.; Xie, G. Diversity and distribution of Burkholderia cepacia complex in the rhizosphere of rice and maize. FEMS Microbiol. Lett. 2007, 266, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, M.; Loh, B.; Leptihn, S.; Ahmed, T.; Li, B. A novel NRPS cluster, acquired by horizontal gene transfer from algae, regulates siderophore iron metabolism in Burkholderia seminalis R456. Int. J. Biol. Macromol. 2021, 182, 838–848. [Google Scholar] [CrossRef]
- Fang, Y.; Xie, G.L.; Lou, M.M.; Li, B.; Muhammad, I. Diversity analysis of Burkholderia cepacia complex in the water bodies of West Lake, Hangzhou, China. J. Microbiol. 2011, 49, 309–314. [Google Scholar] [CrossRef]
- Fang, Y.; Lou, M.M.; Li, B.; Xie, G.L.; Wang, F.; Zhang, L.X.; Luo, Y.C. Characterization of Burkholderia cepacia complex from cystic fibrosis patients in China and their chitosan susceptibility. World J. Microbiol. Biotechnol. 2010, 26, 443–450. [Google Scholar] [CrossRef]
- Mew, T.W.; Vera Cruz, C.M.; Medalla, E.S. Changes in race frequency of Xanthomonas oryzae pv. oryzae in response to rice cultivars planted in the Philippines. Plant Dis. 1992, 76, 1029–1032. [Google Scholar] [CrossRef]
- Schaad, N.W.; Postnikova, E.; Sechler, A.; Claflin, L.E.; Vidaver, A.K.; Jones, J.B.; Agarkova, I.; Ignatov, A.; Dickstein, E.; Ramundo, B.A. Reclassification of subspecies of Acidovorax avenae as A. avenae (Manns 1905) emend., A. cattleyae (Pavarino, 1911) comb. nov., A. citrulli (Schaad et al., 1978) comb. nov., and proposal of A. oryzae sp. nov. Syst. Appl. Microbiol. 2008, 31, 434–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; He, S.W.; Guo, H.B.; Han, J.G.; Thin, K.K.; Gao, J.S.; Wang, Y.; Zhang, X.X. Dickeya oryzae sp. nov., isolated from the roots of rice. Int. J. Syst. Evol. Microbiol. 2020, 70, 4171–4178. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Kuma, K.I.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Lin, L.; Li, Z.; Hu, C.; Zhang, X.; Chang, S.; Yang, L.; Li, Y.; An, Q. Plant growth-promoting nitrogen-fixing enterobacteria are in association with sugarcane plants growing in Guangxi, China. Microbes Environ. 2012, 27, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.; Webster, G.; Mullins, A.J.; Jenner, M.; Bull, M.J.; Dashti, Y.; Spilker, T.; Parkhill, J.; Connor, T.R.; LiPuma, J.J.; et al. Kill and cure: Genomic phylogeny and bioactivity of Burkholderia gladioli bacteria capable of pathogenic and beneficial lifestyles. Microb. Genom. 2021, 7, mgen000515. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Lee, C.C.; Leu, W.M.; Wu, J.J.; Huang, Y.C.; Meng, M. Fungicidal activity of volatile organic compounds emitted by Burkholderia gladioli strain BBB-01. Molecules 2021, 26, 745. [Google Scholar] [CrossRef]
- Peng, Z.; Dottorini, T.; Hu, Y.; Li, M.; Yan, S.; Fanning, S.; Baker, M.; Xu, J.; Li, F. Comparative genomic analysis of the foodborne pathogen Burkholderia gladioli pv. cocovenenans harboring a bongkrekic acid biosynthesis gene cluster. Front. Microbiol. 2021, 12, 628538. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strains | Isolation Source | Reference |
|---|---|---|
| Burkholderia glumae Os48 | Rice | [25] |
| Burkholderia glumae Os14-2 | Rice | This study |
| Burkholderia glumae Os25-2 | Rice | This study |
| Burkholderia gladioli Os6-2 | Rice | This study |
| Burkholderia gladioli Os8-2 | Rice | This study |
| Burkholderia gladioli Os50 | Rice | [25] |
| Burkholderia cepacia LMG 1222T | Onion | [26] |
| Burkholderia plantarii ZJ171 | Rice paddy | [27] |
| Burkholderia cenocepacia M292 | Maize rhizosphere | [28] |
| Burkholderia pyrrocinia M318 | Maize rhizosphere | [28] |
| Burkholderia vietnamiensis Os13 | Rice | [25] |
| Burkholderia ambifaria Os40 | Rice | [25] |
| Burkholderia seminalis R456 | Rice rhizosphere | [29] |
| Burkholderia multivorans PE27 | Lake water | [30] |
| Burkholderia stabilis J5 | Lake water | [30] |
| Burkholderia contaminans Y4 | Human sputum | [31] |
| Xanthomonas oryzae pv. oryzae PXO99A | Rice | [32] |
| Acidovorax oryzae CGMCC 1.1728T | Rice | [33] |
| Dickeya oryzae ACCC 61554T | Rice | [34] |
| Pantoea ananatis F163 | Rice | This study |
| Primer Name | Primer Sequence (5′–3′) | Target Sequence Length (bp) | Target Protein |
|---|---|---|---|
| BG1F | CCGCGCTGTTCATGAGGGATAA | 138 | Rhs family protein |
| BG1R | CGGGCGGAACGACGGTAAGT | ||
| Bglu3F | TGTCGAGGGCGTATGATCAG | 174 | Hypothetical protein |
| Bglu3R | AAATGATGGTGATTTCCCTGGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Luo, J.; Chen, L.; Ahmed, T.; Alotaibi, S.S.; Wang, Y.; Sun, G.; Li, B.; An, Q. Development of Droplet Digital PCR Assay for Detection of Seed-Borne Burkholderia glumae and B. gladioli Causing Bacterial Panicle Blight Disease of Rice. Microorganisms 2022, 10, 1223. https://doi.org/10.3390/microorganisms10061223
Zhang J, Luo J, Chen L, Ahmed T, Alotaibi SS, Wang Y, Sun G, Li B, An Q. Development of Droplet Digital PCR Assay for Detection of Seed-Borne Burkholderia glumae and B. gladioli Causing Bacterial Panicle Blight Disease of Rice. Microorganisms. 2022; 10(6):1223. https://doi.org/10.3390/microorganisms10061223
Chicago/Turabian StyleZhang, Jiannan, Jinyan Luo, Lei Chen, Temoor Ahmed, Saqer S. Alotaibi, Yanli Wang, Guochang Sun, Bin Li, and Qianli An. 2022. "Development of Droplet Digital PCR Assay for Detection of Seed-Borne Burkholderia glumae and B. gladioli Causing Bacterial Panicle Blight Disease of Rice" Microorganisms 10, no. 6: 1223. https://doi.org/10.3390/microorganisms10061223
APA StyleZhang, J., Luo, J., Chen, L., Ahmed, T., Alotaibi, S. S., Wang, Y., Sun, G., Li, B., & An, Q. (2022). Development of Droplet Digital PCR Assay for Detection of Seed-Borne Burkholderia glumae and B. gladioli Causing Bacterial Panicle Blight Disease of Rice. Microorganisms, 10(6), 1223. https://doi.org/10.3390/microorganisms10061223