
Breaking the Restriction Barriers and Applying CRISPRi as a Gene Silencing Tool in Pseudoclostridium thermosuccinogenes

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Preparation and Purification of P. thermosuccinogenes DSM 5809 Spores
2.3. Plasmid Construction
2.4. Electroporation of P. thermosuccinogenes DSM 5809
2.5. Prediction of P. thermosuccinogenes DSM 5809 Methylome Based on PacBio Sequence Information
2.6. Generating E. coli HST04 Strains with Methyltransferases Genes of P. thermosuccinogenes
2.7. RNA Isolation and First Strand cDNA Synthesis
2.8. Quantitative Real Time PCR
2.9. High-Performance Liquid Chromatography
3. Results
3.1. Initial Transformation Steps for P. thermosuccinogenes DSM 5809
3.2. Bioinformatics Analysis of Restriction Modification Systems in P. thermosuccinogenes
3.3. Strategies to Overcome Restriction Barrier to Improve Transformation Efficiency
3.4. CRISPRi as a Silencing Tool for P. thermosuccinogenes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Czajka, J.; Wang, Q.; Wang, Y.; Tang, Y.J. Synthetic Biology for Manufacturing Chemicals: Constraints Drive the Use of Non-Conventional Microbial Platforms. Appl. Microbiol. Biotechnol. 2017, 101, 7427–7434. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yan, Q.; Jones, J.A.; Tang, Y.J.; Fong, S.S.; Koffas, M.A.G. Metabolic Burden: Cornerstones in Synthetic Biology and Metabolic Engineering Applications. Trends Biotechnol. 2016, 34, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.C.; Kim, N.M.; Sandoval, N.R. Recent Developments of the Synthetic Biology Toolkit for Clostridium. Front. Microbiol. 2018, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Weusthuis, R.A.; Lamot, I.; van der Oost, J.; Sanders, J.P.M. Microbial Production of Bulk Chemicals: Development of Anaerobic Processes. Trends Biotechnol. 2011, 29, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Bruder, M.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Technical Guide for Genetic Advancement of Underdeveloped and Intractable Clostridium. Biotechnol. Adv. 2014, 32, 623–641. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A. CRISPR-Cas Immunity in Prokaryotes. Nature 2015, 526, 55–61. [Google Scholar] [CrossRef]
- Mohanraju, P.; Makarova, K.S.; Zetsche, B.; Zhang, F.; Koonin, E.V.; van der Oost, J. Diverse Evolutionary Roots and Mechanistic Variations of the CRISPR-Cas Systems. Science 2016, 353, aad5147. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S. Origins and Evolution of CRISPR-Cas Systems. Philos. Trans. Royal Soc. B: Biol. Sci. 2019, 374, 20180087. [Google Scholar] [CrossRef]
- Ofir, G.; Melamed, S.; Sberro, H.; Mukamel, Z.; Silverman, S.; Yaakov, G.; Doron, S.; Sorek, R. DISARM Is a Widespread Bacterial Defence System with Broad Anti-Phage Activities. Nat. Microbiol. 2018, 3, 90–98. [Google Scholar]
- Suzuki, H.; Yoshida, K. Genetic Transformation of Geobacillus Kaustophilus HTA426 by Conjugative Transfer of Host-Mimicking Plasmids. J. Microbiol. Biotechnol. 2012, 22, 1279–1287. [Google Scholar] [CrossRef]
- Jensen, T.Ø.; Tellgren-Roth, C.; Redl, S.; Maury, J.; Jacobsen, S.A.B.; Pedersen, L.E.; Nielsen, A.T. Genome-Wide Systematic Identification of Methyltransferase Recognition and Modification Patterns. Nat. Commun. 2019, 10, 3311. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, Y.; Dai, Z.; Li, Y. Engineering Clostridium Strain to Accept Unmethylated DNA. PLoS ONE 2010, 5, e9038. [Google Scholar] [CrossRef]
- Riley, L.A.; Ji, L.; Schmitz, R.J.; Westpheling, J.; Guss, A.M. Rational Development of Transformation in Clostridium Thermocellum ATCC 27405 via Complete Methylome Analysis and Evasion of Native Restriction–Modification Systems. J. Ind. Microbiol. Biotechnol. 2019, 46, 1435–1443. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, W.; Deng, A.; Sun, Z.; Zhang, Y.; Liang, Y.; Che, Y.; Wen, T. A Mimicking-of-DNA-Methylation-Patterns Pipeline for Overcoming the Restriction Barrier of Bacteria. PLoS Genet. 2012, 8, e1002987. [Google Scholar] [CrossRef]
- Sridhar, J.; Eiteman, M.A. Influence of Redox Potential on Product Distribution in Clostridium Thermosuccinogenes. Appl. Biochem. Biotechnol. 1999, 82, 91–101. [Google Scholar] [CrossRef]
- Sridhar, J.; Eiteman, M.A.; Wiegel, J.W. Elucidation of Enzymes in Fermentation Pathways Used by Clostridium Thermosuccinogenes Growing on Inulin. Appl. Environ. Microbiol. 2000, 66, 246–251. [Google Scholar] [CrossRef]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The Advantages of SMRT Sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef]
- Beaulaurier, J.; Schadt, E.E.; Fang, G. Deciphering Bacterial Epigenomes Using Modern Sequencing Technologies. Nat. Rev. Genet. 2019, 20, 157–172. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
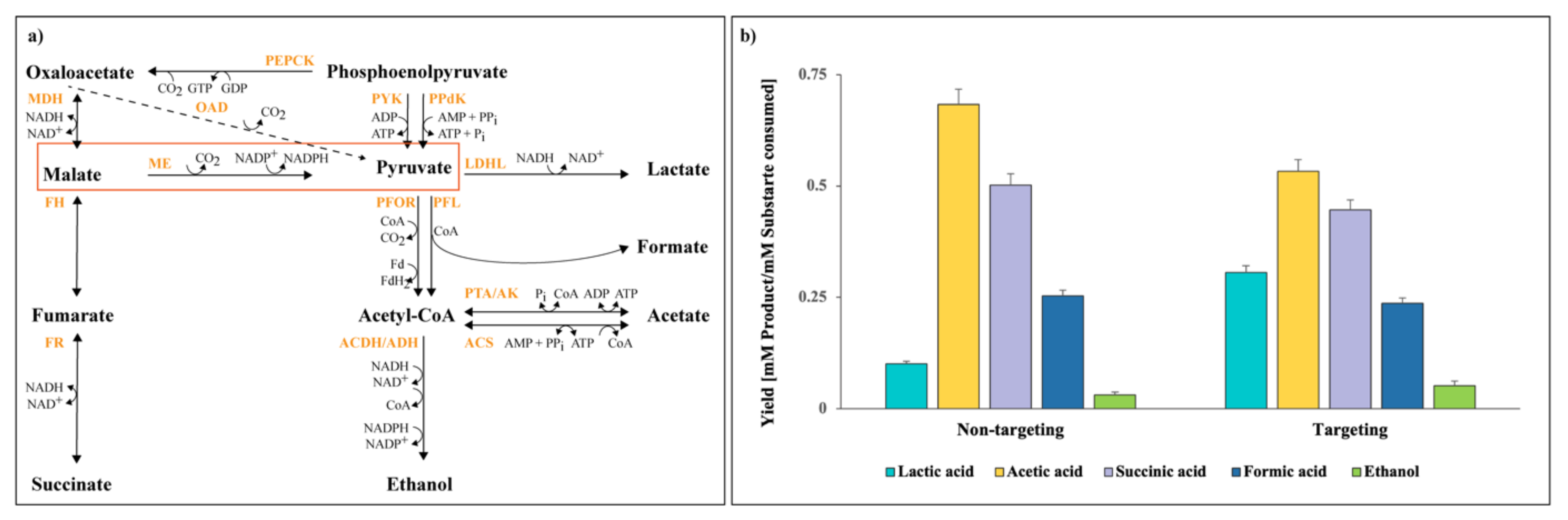
- Koendjbiharie, J.G.; Wiersma, K.; Kranenburg, R. van Investigating the Central Metabolism of Clostridium Thermosuccinogenes. Appl. Environ. Microbiol. 2018, 84, e00363. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9–CrRNA Ribonucleoprotein Complex Mediates Specific DNA Cleavage for Adaptive Immunity in Bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-Guided Editing of Bacterial Genomes Using CRISPR-Cas Systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef]
- Harrington, L.B.; Paez-Espino, D.; Staahl, B.T.; Chen, J.S.; Ma, E.; Kyrpides, N.C.; Doudna, J.A. A Thermostable Cas9 with Increased Lifetime in Human Plasma. Nat. Commun. 2017, 8, 1424. [Google Scholar] [CrossRef]
- Mougiakos, I.; Mohanraju, P.; Bosma, E.F.; Vrouwe, V.; Finger Bou, M.; Naduthodi, M.I.S.; Gussak, A.; Brinkman, R.B.L.; van Kranenburg, R.; van der Oost, J. Characterizing a Thermostable Cas9 for Bacterial Genome Editing and Silencing. Nat. Commun. 2017, 8, 1647. [Google Scholar] [CrossRef]
- Ganguly, J.; Martin-Pascual, M.; van Kranenburg, R. CRISPR Interference (CRISPRi) as Transcriptional Repression Tool for Hungateiclostridium Thermocellum DSM 1313. Microb. Biotechnol. 2020, 13, 339–349. [Google Scholar] [CrossRef]
- Walker, J.E.; Lanahan, A.A.; Zheng, T.; Toruno, C.; Lynd, L.R.; Cameron, J.C.; Olson, D.G.; Eckert, C.A. Development of Both Type I-B and Type II CRISPR/Cas Genome Editing Systems in the Cellulolytic Bacterium Clostridium Thermocellum. Metab. Eng. Commun. 2020, 10, e00116. [Google Scholar] [CrossRef]
- Mougiakos, I.; Bosma, E.F.; Ganguly, J.; van der Oost, J.; van Kranenburg, R. Hijacking CRISPR-Cas for High-Throughput Bacterial Metabolic Engineering: Advances and Prospects. Curr. Opin. Biotechnol. 2018, 50, 146–157. [Google Scholar] [CrossRef]
- Mougiakos, I.; Bosma, E.F.; Weenink, K.; Vossen, E.; Goijvaerts, K.; van der Oost, J.; van Kranenburg, R. Efficient Genome Editing of a Facultative Thermophile Using Mesophilic SpCas9. ACS Synth. Biol. 2017, 6, 849–861. [Google Scholar] [CrossRef]
- Schultenkämper, K.; Brito, L.F.; Wendisch, V.F. Impact of CRISPR Interference on Strain Development in Biotechnology. Biotechnol. Appl. Biochem. 2020, 67, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Guo, F.; Yan, W.; Dai, Z.; Dong, W.; Zhou, J.; Zhang, W.; Xin, F.; Jiang, M. Recent Advances of CRISPR/Cas9-Based Genetic Engineering and Transcriptional Regulation in Industrial Biology. Front. Bioeng. Biotechnol. 2020, 7, 459. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Shen, X.; Sun, X.; Yan, Y.; Wang, J.; Yuan, Q. CRISPR-Based Metabolic Engineering in Non-Model Microorganisms. Curr. Opin. Biotechnol. 2022, 75, 102698. [Google Scholar] [CrossRef] [PubMed]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 Phage Cloning Vectors and Host Strains: Nucleotide Sequences of the M13mp18 and PUC19 Vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Plugge, C.M. Anoxic Media Design, Preparation, and Considerations. Methods Enzymol. 2005, 397, 3–16. [Google Scholar] [PubMed]
- Olson, D.G.; Lynd, L.R. Transformation of Clostridium Thermocellum by Electroporation. Methods Enzymol. 2012, 510, 317–330. [Google Scholar] [PubMed]
- Mearls, E.B.; Izquierdo, J.A.; Lynd, L.R. Formation and Characterization of Non-Growth States in Clostridium Thermocellum: Spores and L-Forms. BMC Microbiol. 2012, 12, 180. [Google Scholar] [CrossRef]
- Yang, W.-W.; Crow-Willard, E.N.; Ponce, A. Production and Characterization of Pure Clostridium Spore Suspensions. J. Appl. Microbiol. 2009, 106, 27–33. [Google Scholar] [CrossRef]
- Wang, Y.; Yau, Y.-Y.; Perkins-Balding, D.; Thomson, J.G. Recombinase Technology: Applications and Possibilities. Technology 2011, 30, 267–285. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-Step Inactivation of Chromosomal Genes in Escherichia Coli K-12 Using PCR Products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Stothard, P. The Sequence Manipulation Suite: JavaScript Programs for Analyzing and Formatting Protein and DNA Sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef]
- Pyne, M.E.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Development of an Electrotransformation Protocol for Genetic Manipulation of Clostridium Pasteurianum. Biotechnol. Biofuels 2013, 6, 50. [Google Scholar] [CrossRef]
- Purdy, D.; O’Keeffe, T.A.T.; Elmore, M.; Herbert, M.; McLeod, A.; Bokori-Brown, M.; Ostrowski, A.; Minton, N.P. Conjugative Transfer of Clostridial Shuttle Vectors from Escherichia Coli to Clostridium Difficile through Circumvention of the Restriction Barrier. Mol. Microbiol. 2002, 46, 439–452. [Google Scholar] [CrossRef]
- Herman, N.A.; Li, J.; Bedi, R.; Turchi, B.; Liu, X.; Miller, M.J.; Zhang, W. Development of a High-Efficiency Transformation Method and Implementation of Rational Metabolic Engineering for the Industrial Butanol Hyperproducer Clostridium Saccharoperbutylacetonicum Strain N1-4. Appl. Environ. Microbiol. 2017, 83, e02942-16. [Google Scholar] [CrossRef]
- Tyurin, M.V.; Desai, S.G.; Lynd, L.R. Electrotransformation of Clostridium Thermocellum. Appl. Environ. Microbiol. 2004, 70, 883–890. [Google Scholar] [CrossRef]
- Song, Y.; Hahn, T.; Thompson, I.P.; Mason, T.J.; Preston, G.M.; Li, G.; Paniwnyk, L.; Huang, W.E. Ultrasound-Mediated DNA Transfer for Bacteria. Nucleic Acids Res. 2007, 35, e129. [Google Scholar] [CrossRef]
- Guss, A.M.; Olson, D.G.; Caiazza, N.C.; Lynd, L.R. Dcm Methylation Is Detrimental to Plasmid Transformation in Clostridium Thermocellum. Biotechnol. Biofuels 2012, 5, 30. [Google Scholar] [CrossRef]
- Kolek, J.; Sedlar, K.; Provaznik, I.; Patakova, P. Dam and Dcm Methylations Prevent Gene Transfer into Clostridium Pasteurianum NRRL B-598: Development of Methods for Electrotransformation, Conjugation, and Sonoporation. Biotechnol. Biofuels 2016, 9, 14. [Google Scholar] [CrossRef]
- Bartosiak-Jentys, J.; Hussein, A.H.; Lewis, C.J.; Leak, D.J. Modular System for Assessment of Glycosyl Hydrolase Secretion in Geobacillus Thermoglucosidasius. Microbiology (Reading) 2013, 159, 1267–1275. [Google Scholar] [CrossRef]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond Editing: Repurposing CRISPR-Cas9 for Precision Genome Regulation and Interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Colavin, A.; Shi, H.; Czarny, T.L.; Larson, M.H.; Wong, S.; Hawkins, J.S.; Lu, C.H.S.; Koo, B.-M.; Marta, E.; et al. A Comprehensive, CRISPR-Based Functional Analysis of Essential Genes in Bacteria. Cell 2016, 165, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Koendjbiharie, J.G.; Wevers, K.; van Kranenburg, R. Assessing Cofactor Usage in Pseudoclostridium Thermosuccinogenes via Heterologous Expression of Central Metabolic Enzymes. Front. Microbiol. 2019, 10, 1162. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Olson, D.G.; Zhou, J.; Herring, C.D.; Joe Shaw, A.; Lynd, L.R. Redirecting Carbon Flux through Exogenous Pyruvate Kinase to Achieve High Ethanol Yields in Clostridium Thermocellum. Metab. Eng. 2013, 15, 151–158. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Deepbinner: Demultiplexing barcoded Oxford Nanopore reads with deep convolutional neural networks. PLoS Comput. Biol. 2018, 14, e1006583. [Google Scholar] [CrossRef]
- Lanfear, R.; Schalamun, M.; Kainer, D.; Wang, W.; Schwessinger, B. MinIONQC: Fast and simple quality control for MinION sequencing data. Bioinformatics 2019, 35, 523–525. [Google Scholar] [CrossRef]
- Stoiber, M.; Egan, R.; Lee, J.E.; Celniker, S.; Neely, R.K.; Loman, N.; Pennacchio, L.A.; Brown, J. De novo Identification of DNA Modifications Enabled by Genome-Guided Nanopore Signal Processing. BioRxiv 2017. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RM System | RM Type | Motifs | Modification Type | % Modified | Motifs in Genome |
|---|---|---|---|---|---|
| 1 | I | CACNNNNNNNTNGC/GCNANNNNNNNGTG | m6 A | 95.09/88.47 | 876 |
| 2 | I | GATNNNNCTC/GAGNNNNATC | m6 A | 94.29/82.99 | 1630 |
| 3 | I | TCABNNNNNNTARG/CYTANNNNNNVTGA | m6 A | 90.24/85.79 | 697 |
| 4 | III | CGAG | m6 A | 66.04 | 10,352 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganguly, J.; Martin-Pascual, M.; Montiel González, D.; Bulut, A.; Vermeulen, B.; Tjalma, I.; Vidaki, A.; van Kranenburg, R. Breaking the Restriction Barriers and Applying CRISPRi as a Gene Silencing Tool in Pseudoclostridium thermosuccinogenes. Microorganisms 2022, 10, 698. https://doi.org/10.3390/microorganisms10040698
Ganguly J, Martin-Pascual M, Montiel González D, Bulut A, Vermeulen B, Tjalma I, Vidaki A, van Kranenburg R. Breaking the Restriction Barriers and Applying CRISPRi as a Gene Silencing Tool in Pseudoclostridium thermosuccinogenes. Microorganisms. 2022; 10(4):698. https://doi.org/10.3390/microorganisms10040698
Chicago/Turabian StyleGanguly, Joyshree, Maria Martin-Pascual, Diego Montiel González, Alkan Bulut, Bram Vermeulen, Ivo Tjalma, Athina Vidaki, and Richard van Kranenburg. 2022. "Breaking the Restriction Barriers and Applying CRISPRi as a Gene Silencing Tool in Pseudoclostridium thermosuccinogenes" Microorganisms 10, no. 4: 698. https://doi.org/10.3390/microorganisms10040698
APA StyleGanguly, J., Martin-Pascual, M., Montiel González, D., Bulut, A., Vermeulen, B., Tjalma, I., Vidaki, A., & van Kranenburg, R. (2022). Breaking the Restriction Barriers and Applying CRISPRi as a Gene Silencing Tool in Pseudoclostridium thermosuccinogenes. Microorganisms, 10(4), 698. https://doi.org/10.3390/microorganisms10040698