
Trans-Translation Is an Appealing Target for the Development of New Antimicrobial Compounds

 , ,
, , 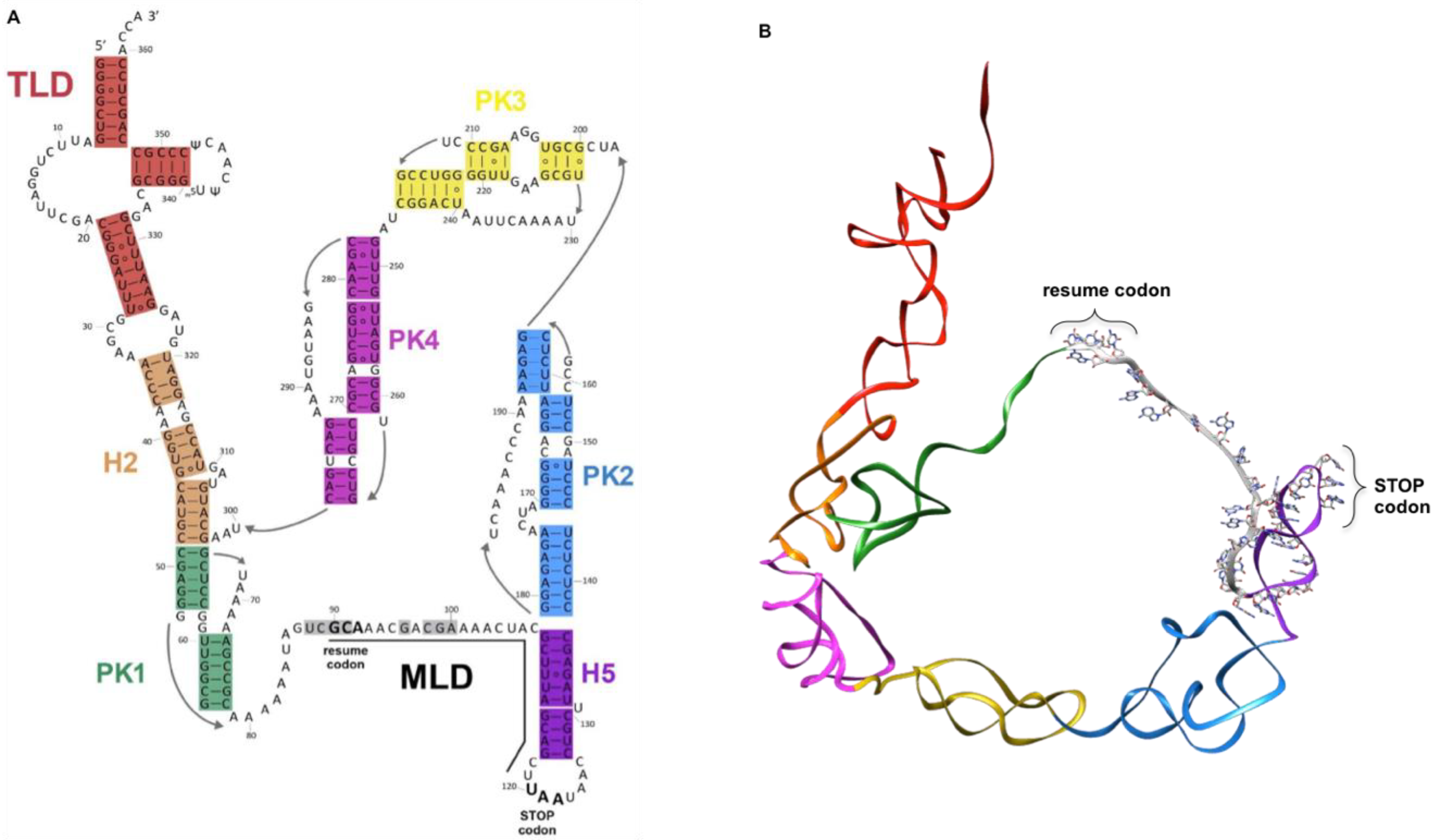{kind=link}
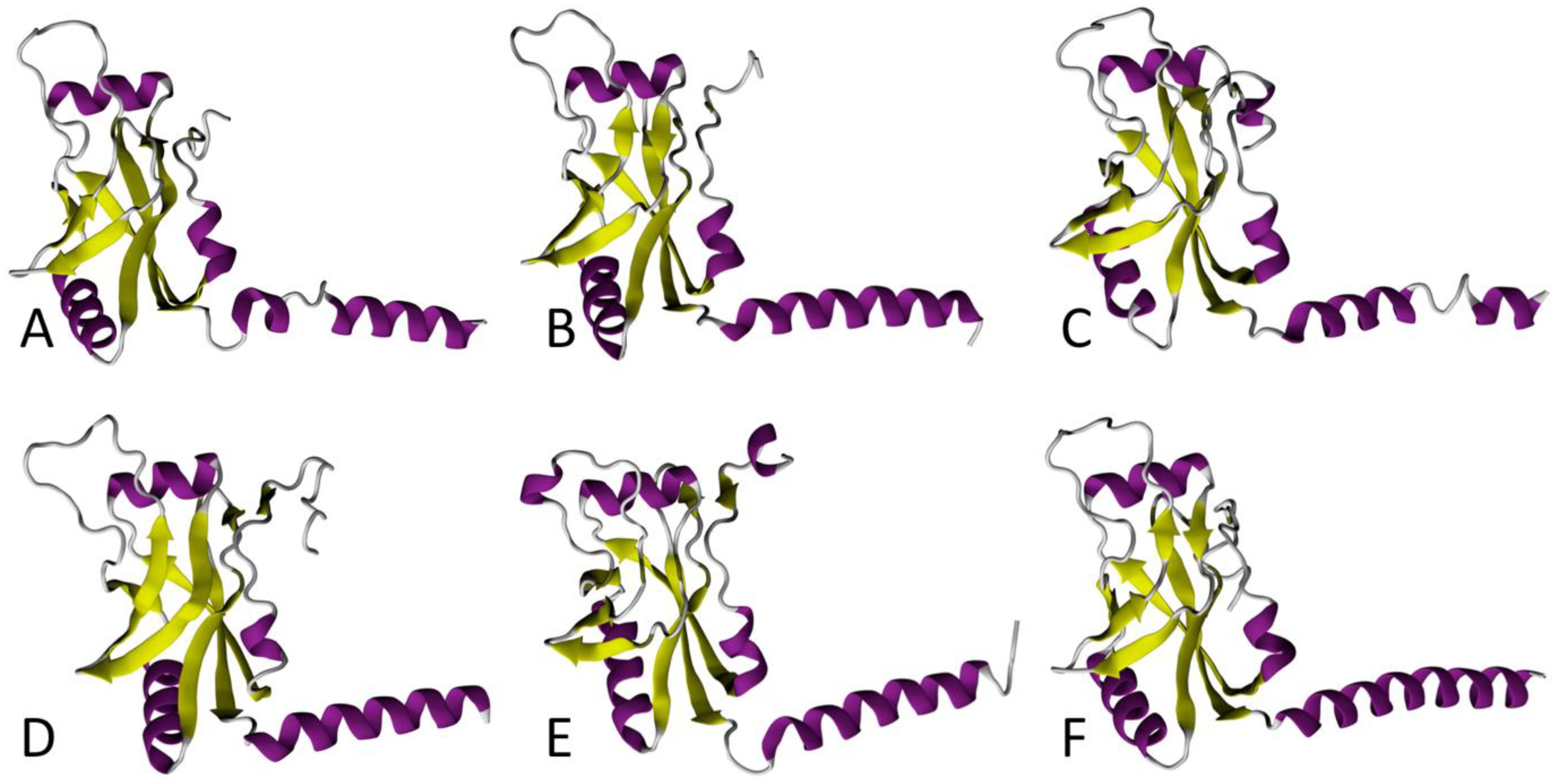{kind=link}
{kind=link}
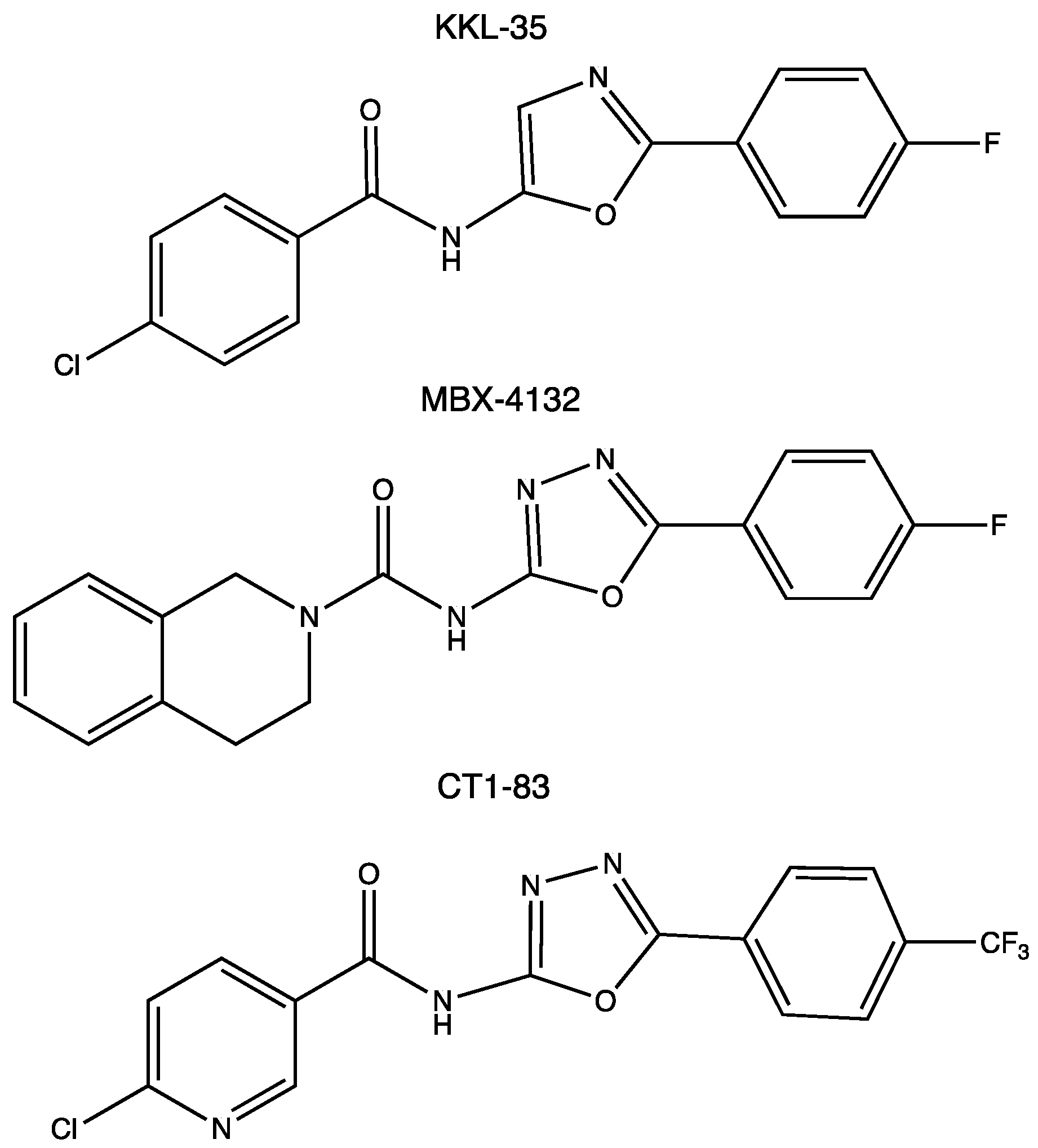{kind=link}
{kind=link}
Abstract
1. Introduction
2. Ribosomal Stalling: From No-Go to Non-Stop
3. Trans-Translation Components Are Major Targets for Interference
3.1. Transfer-Messenger RNA (tmRNA)
3.2. SmpB
4. The Molecular Process of Trans-Translation
5. Trans-Translation as a Target for New Antimicrobial Compounds
6. Antibiotics Targeting Trans-Translation: Are We There Yet?
6.1. Oxadiazole Compounds
6.2. Pyrazinamide
6.3. Peptides and Oligonucleotides
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vázquez-Laslop, N.; Mankin, A.S. Context-specific action of ribosomal antibiotics. Annu. Rev. Microbiol. 2018, 72, 185–207. [Google Scholar] [CrossRef]
- Ero, R.; Yan, X.F.; Gao, Y.G. Ribosome protection proteins—“New” players in the global arms race with antibiotic-resistant pathogens. Int. J. Mol. Sci. 2021, 22, 5356. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Santajit, S.; Indrawattana, N. Mechanisms of antimicrobial resistance in ESKAPE pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef]
- Kakkar, A.K.; Shafiq, N.; Singh, G.; Ray, P.; Gautam, V.; Agarwal, R.; Muralidharan, J.; Arora, P. Antimicrobial stewardship programs in resource constrained environments: Understanding and addressing the need of the systems. Front. Public Health 2020, 8, 140. [Google Scholar] [CrossRef]
- Tyers, M.; Wright, G.D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 2019, 17, 141–155. [Google Scholar] [CrossRef]
- Janssen, B.D.; Diner, E.J.; Hayes, C.S. Analysis of aminoacyl- and peptidyl-tRNAs by gel electrophoresis. Bact. Regul. RNA 2012, 905, 291–309. [Google Scholar] [CrossRef][Green Version]
- Doma, M.K.; Parker, R. RNA quality control in eukaryotes. Cell 2007, 131, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.; Binet, E.; Bouloc, P. Competition between SsrA tagging and translational termination at weak stop codons in Escherichia coli. Mol. Microbiol. 2002, 45, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Yamamoto, Y.; Ogawa, K.; Abo, T.; Inokuchi, H.; Aiba, H. Bacterial SsrA system plays a role in coping with unwanted translational readthrough caused by suppressor tRNAs. Genes Cells 2002, 7, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.S.; Sauer, R.T. Cleavage of the a site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol. Cell 2003, 12, 903–911. [Google Scholar] [CrossRef]
- Abo, T.; Ueda, K.; Sunohara, T.; Ogawa, K.; Aiba, H. SsrA-mediated protein tagging in the presence of miscoding drugs and its physiological role in Escherichia coli. Genes Cells 2002, 7, 629–638. [Google Scholar] [CrossRef]
- Laursen, B.S.; Sørensen, H.P.; Mortensen, K.K.; Sperling-Petersen, H.U. Initiation of protein synthesis in bacteria. Microbiol. Mol. Biol. Rev. 2005, 69, 101–123. [Google Scholar] [CrossRef]
- Pedersen, K.; Zavialov, A.V.; Pavlov, M.Y.; Elf, J.; Gerdes, K.; Ehrenberg, M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 2003, 112, 131–140. [Google Scholar] [CrossRef]
- Keiler, K.C.; Feaga, H.A. Resolving nonstop translation complexes is a matter of life or death. J. Bacteriol. 2014, 196, 2123–2130. [Google Scholar] [CrossRef]
- Müller, C.; Crowe-McAuliffe, C.; Wilson, D.N. Ribosome rescue pathways in bacteria. Front. Microbiol. 2021, 12, 652980. [Google Scholar] [CrossRef]
- Gueneau de Novoa, P.; Williams, K.P. The tmRNA website: Reductive evolution of tmRNA in plastids and other endosymbionts. Nucleic Acids Res. 2004, 32, D104–D108. [Google Scholar] [CrossRef]
- Komine, Y.; Kitabatake, M.; Yokogawa, T.; Nishikawa, K.; Inokuchi, H. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc. Natl. Acad. Sci. USA 1994, 91, 9223–9227. [Google Scholar] [CrossRef]
- Li, Z.; Pandit, S.; Deutscher, M.P. 3′ Exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli. Proc. Natl. Acad. Sci. USA 1998, 95, 2856–2861. [Google Scholar] [CrossRef] [PubMed]
- Lin-Chao, S.; Wei, C.L.; Lin, Y.T. RNase E is required for the maturation of ssrA RNA and normal ssrA RNA peptide-tagging activity. Proc. Natl. Acad. Sci. USA 1999, 96, 12406–12411. [Google Scholar] [CrossRef] [PubMed]
- Gimple, O.; Schön, A. In vitro and in vivo processing of Cyanelle tmRNA by RNase P. Biol. Chem. 2001, 382, 1421–1429. [Google Scholar] [CrossRef]
- Lee, S.Y.; Bailey, S.C.; Apirion, D. Small stable RNAs from Escherichia coli: Evidence for the existence of new molecules and for a new ribonucleoprotein particle containing 6S RNA. J. Bacteriol. 1978, 133, 1015–1023. [Google Scholar] [CrossRef]
- Moore, S.D.; Sauer, R.T. Ribosome rescue: tmRNA tagging activity and capacity in Escherichia coli. Mol. Microbiol. 2005, 58, 456–466. [Google Scholar] [CrossRef]
- Felden, B.; Hanawa, K.; Atkins, J.F.; Himeno, H.; Muto, A.; Gesteland, R.F.; McCloskey, J.A.; Crain, P.F. Presence and location of modified nucleotides in Escherichia coli tmRNA: Structural mimicry with tRNA acceptor branches. EMBO J. 1998, 17, 3188–3196. [Google Scholar] [CrossRef]
- Ranaei-Siadat, E.; Fabret, C.; Seijo, B.; Dardel, F.; Grosjean, H.; Nonin-Lecomte, S. RNA-methyltransferase TrmA is a dual-specific enzyme responsible for C5-methylation of uridine in both tmRNA and tRNA. RNA Biol. 2013, 10, 572–578. [Google Scholar] [CrossRef]
- Hou, Y.M.; Schimmel, P. A simple structural feature is a major determinant of the identity of a transfer RNA. Nat. Cell Biol. 1988, 333, 140–145. [Google Scholar] [CrossRef]
- Schimmel, P.; Ribas de Pouplana, L. Footprints of aminoacyl-tRNA synthetases are everywhere. Trends Biochem. Sci. 2000, 25, 207–209. [Google Scholar] [CrossRef]
- Ribas de Pouplana, L.; Schimmel, P. Aminoacyl-tRNA synthetases: Potential markers of genetic code development. Trends Biochem. Sci. 2001, 26, 591–596. [Google Scholar] [CrossRef]
- Nameki, N.; Tadaki, T.; Himeno, H.; Muto, A. Three of four pseudoknots in tmRNA are interchangeable and are substitutable with single-stranded RNAs. FEBS Lett. 2000, 470, 345–349. [Google Scholar] [CrossRef]
- Williams, K.P.; Martindale, K.A.; Bartel, D.P. Resuming translation on tmRNA: A unique mode of determining a reading frame. EMBO J. 1999, 18, 5423–5433. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ishii, M.; Tadaki, T.; Muto, A.; Himeno, H. Determinants on tmRNA for initiating efficient and precise trans-translation: Some mutations upstream of the tag-encoding sequence of Escherichia coli tmRNA shift the initiation point of trans-translation in vitro. RNA 2001, 7, 999–1012. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, M.R.; Healey, D.W.; Robison, S.G.; Dewey, J.D.; Buskirk, A.R. The role of upstream sequences in selecting the reading frame on tmRNA. BMC Biol. 2008, 6, 29, Published 30 June 2008. [Google Scholar] [CrossRef]
- Thibonnier, M.; Thiberge, J.M.; De Reuse, H. Trans-translation in Helicobacter pylori: Essentiality of ribosome rescue and requirement of protein tagging for stress resistance and competence. PLoS ONE 2008, 3, e3810. [Google Scholar] [CrossRef] [PubMed]
- Kieft, J.S. Viral IRES RNA structures and ribosome interactions. Trends Biochem. Sci. 2008, 33, 274–283. [Google Scholar] [CrossRef]
- Yang, C.; Glover, J.R. The SmpB-tmRNA tagging system plays important roles in Streptomyces coelicolor growth and development. PLoS ONE 2009, 4, e4459. [Google Scholar] [CrossRef] [PubMed]
- Barends, S.; Zehl, M.; Bialek, S.; De Waal, E.; A Traag, B.; Willemse, J.; Jensen, O.N.; Vijgenboom, E.; Van Wezel, G.P. Transfer–messenger RNA controls the translation of cell-cycle and stress proteins in Streptomyces. EMBO Rep. 2010, 11, 119–125. [Google Scholar] [CrossRef]
- Katz, A.; Elgamal, S.; Rajkovic, A.; Ibba, M. Non-canonical roles of tRNAs and tRNA mimics in bacterial cell biology. Mol. Microbiol. 2016, 101, 545–558. [Google Scholar] [CrossRef]
- Keiler, K.C.; Shapiro, L.; Williams, K.P. tmRNAs that encode proteolysis-inducing tags are found in all known bacterial genomes: A two-piece tmRNA functions in Caulobacter. Proc. Natl. Acad. Sci. USA 2000, 97, 7778–7783. [Google Scholar] [CrossRef] [PubMed]
- Sharkady, S.M.; Williams, K.P. A third lineage with two-piece tmRNA. Nucleic Acids Res. 2004, 32, 4531–4538. [Google Scholar] [CrossRef]
- Gaudin, C.; Zhou, X.; Williams, K.P.; Felden, B. Two-piece tmRNA in cyanobacteria and its structural analysis. Nucleic Acids Res. 2002, 30, 2018–2024. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.D.; Sauer, R.T. The tmRNA system for translational surveillance and ribosome rescue. Annu. Rev. Biochem. 2007, 76, 101–124. [Google Scholar] [CrossRef]
- Karzai, A.W.; Susskind, M.M.; Sauer, R. SmpB, a unique RNA-binding protein essential for the peptide-tagging activity of SsrA (tmRNA). EMBO J. 1999, 18, 3793–3799. [Google Scholar] [CrossRef]
- Hanawa-Suetsugu, K.; Takagi, M.; Inokuchi, H.; Himeno, H.; Muto, A. SmpB functions in various steps of trans-translation. Nucleic Acids Res. 2002, 30, 1620–1629. [Google Scholar] [CrossRef]
- Shimizu, Y.; Ueda, T. The role of SmpB protein in trans-translation. FEBS Lett. 2002, 514, 74–77. [Google Scholar] [CrossRef]
- Wower, J.; Zwieb, C.W.; Hoffman, D.W.; Wower, I.K. SmpB: A protein that binds to double-stranded segments in tmRNA and tRNA. Biochemistry 2002, 41, 8826–8836. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, M.; Hubbard, T.J.; Proctor, M.; Freund, S.M.; Murzin, A.G. The solution structure of the S1 RNA binding domain: A member of an ancient nucleic acid–binding fold. Cell 1997, 88, 235–242. [Google Scholar] [CrossRef]
- Dong, G.; Nowakowski, J.; Hoffman, D.W. Structure of small protein B: The protein component of the tmRNA–SmpB system for ribosome rescue. EMBO J. 2002, 21, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Someya, T.; Nameki, N.; Hosoi, H.; Suzuki, S.; Hatanaka, H.; Fujii, M.; Terada, T.; Shirouzu, M.; Inoue, Y.; Shibata, T.; et al. Solution structure of a tmRNA-binding protein, SmpB, from Thermus thermophilus. FEBS Lett. 2003, 535, 94–100. [Google Scholar] [CrossRef]
- Gutmann, S.; Haebel, P.W.; Metzinger, L.; Sutter, M.; Felden, B.; Ban, N. Crystal structure of the transfer-RNA domain of transfer-messenger RNA in complex with SmpB. Nature 2003, 424, 699–703. [Google Scholar] [CrossRef]
- Bessho, Y.; Shibata, R.; Sekine, S.; Murayama, K.; Higashijima, K.; Hori-Takemoto, C.; Shirouzu, M.; Kuramitsu, S.; Yokoyama, S. Structural basis for functional mimicry of long-variable-arm tRNA by transfer-messenger RNA. Proc. Natl. Acad. Sci. USA 2007, 104, 8293–8298. [Google Scholar] [CrossRef]
- Kaur, S.; Gillet, R.; Li, W.; Gursky, R.; Frank, J. Cryo-EM visualization of transfer messenger RNA with two SmpBs in a stalled ribosome. Proc. Natl. Acad. Sci. USA 2006, 103, 16484–16489. [Google Scholar] [CrossRef] [PubMed]
- Kurita, D.; Sasaki, R.; Muto, A.; Himeno, H. Interaction of SmpB with ribosome from directed hydroxyl radical probing. Nucleic Acids Res. 2007, 35, 7248–7255. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nonin-Lecomte, S.; Germain-Amiot, N.; Gillet, R.; Hallier, M.; Ponchon, L.; Dardel, F.; Felden, B. Ribosome hijacking: A role for small protein B during trans -translation. EMBO Rep. 2009, 10, 160–165. [Google Scholar] [CrossRef]
- Rae, C.D.; Gordiyenko, Y.; Ramakrishnan, V. How a circularized tmRNA moves through the ribosome. Science 2019, 363, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Guyomar, C.; D’Urso, G.; Chat, S.; Giudice, E.; Gillet, R. Structures of tmRNA and SmpB as they transit through the ribosome. Nat. Commun. 2021, 12, 4909. [Google Scholar] [CrossRef]
- Neubauer, C.; Gillet, R.; Kelley, A.C.; Ramakrishnan, V. Decoding in the absence of a codon by tmRNA and SmpB in the Ribosome. Science 2012, 335, 1366–1369. [Google Scholar] [CrossRef]
- Kurita, D.; Muto, A.; Himeno, H. Role of the C-terminal tail of SmpB in the early stage of trans-translation. RNA 2010, 16, 980–990. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, G.; Guyomar, C.; Chat, S.; Giudice, E.; Gillet, R. Insights into the ribosomal trans-translation rescue system: Lessons from recent structural studies. FEBS J. 2022; in press. [Google Scholar]
- Fei, X.; Bell, T.A.; Barkow, S.R.; Baker, T.A.; Sauer, R.T. Structural basis of ClpXP recognition and unfolding of ssrA-tagged substrates. eLife 2020, 9, e61496. [Google Scholar] [CrossRef] [PubMed]
- Karzai, A.W.; Roche, E.D.; Sauer, R.T. The SsrA–SmpB system for protein tagging, directed degradation and ribosome rescue. Nat. Struct. Biol. 2000, 7, 449–455. [Google Scholar] [CrossRef]
- Richards, J.; Mehta, P.; Karzai, A.W. RNase R degrades non-stop mRNAs selectively in an SmpB-tmRNA-dependent manner. Mol. Microbiol. 2006, 62, 1700–1712. [Google Scholar] [CrossRef]
- Cheng, Z.-F.; Deutscher, M.P. Purification and characterization of the Escherichia coli exoribonuclease RNase R. Comparison with RNase II. J. Biol. Chem. 2002, 277, 21624–21629. [Google Scholar] [CrossRef] [PubMed]
- Matos, R.G.; Pobre, V.; Reis, F.P.; Malecki, M.; Andrade, J.M.; Arraiano, C.M. Structure and degradation mechanisms of 3′ to 5′ exoribonucleases. In Ribonucleases. Nucleic Acids and Molecular Biology; Nicholson, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 192–222. [Google Scholar] [CrossRef]
- Saramago, M.; Bárria, C.; Dos Santos, R.F.; Silva, I.; Pobre, V.; Domingues, S.; Andrade, J.; Viegas, S.; Arraiano, C.M. The role of RNases in the regulation of small RNAs. Curr. Opin. Microbiol. 2014, 18, 105–115. [Google Scholar] [CrossRef]
- Ramadoss, N.S.; Alumasa, J.N.; Cheng, L.; Wang, Y.; Li, S.; Chambers, B.S.; Chang, H.; Chatterjee, A.K.; Brinker, A.; Engels, I.H.; et al. Small molecule inhibitors of trans-translation have broad-spectrum antibiotic activity. Proc. Natl. Acad. Sci. USA 2013, 110, 10282–10287. [Google Scholar] [CrossRef]
- Huang, Y.; Alumasa, J.N.; Callaghan, L.T.; Baugh, R.S.; Rae, C.D.; Keiler, K.C.; McGillivray, S.M. A Small-molecule inhibitor of trans-translation synergistically interacts with cathelicidin antimicrobial peptides to impair survival of Staphylococcus aureus. Antimicrob. Agents Chemother. 2019, 63, e02362-18. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz, J.; Vioque, A. Increased sensitivity to protein synthesis inhibitors in cells lacking tmRNA. RNA 2001, 7, 1708–1716. [Google Scholar]
- Vioque, A.; de la Cruz, J. Trans-translation and protein synthesis inhibitors. FEMS Microbiol. Lett. 2003, 218, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Feaga, H.A.; Viollier, P.; Keiler, K.C. Release of nonstop ribosomes is essential. mBio 2014, 5, e01916-14. [Google Scholar] [CrossRef]
- Giudice, E.; Macé, K.; Gillet, R. Trans-translation exposed: Understanding the structures and functions of tmRNA-SmpB. Front. Microbiol. 2014, 5, 113. [Google Scholar] [CrossRef]
- Andini, N.; Nash, K.A. Expression of tmRNA in mycobacteria is increased by antimicrobial agents that target the ribosome. FEMS Microbiol. Lett. 2011, 322, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Luidalepp, H.; Hallier, M.; Felden, B.; Tenson, T. tmRNA decreases the bactericidal activity of aminoglycosides and the susceptibility to inhibitors of cell wall synthesis. RNA Biol. 2005, 2, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ji, L.; Shi, W.; Xie, J.; Zhang, Y. Trans-translation mediates tolerance to multiple antibiotics and stresses in Escherichia coli. J. Antimicrob. Chemother. 2013, 68, 2477–2481. [Google Scholar] [CrossRef] [PubMed]
- Alumasa, J.N.; Manzanillo, P.S.; Peterson, N.D.; Lundrigan, T.; Baughn, A.D.; Cox, J.S.; Keiler, K.C. Ribosome rescue inhibitors kill actively growing and nonreplicating persister Mycobacterium tuberculosis cells. ACS Infect. Dis. 2017, 3, 634–644. [Google Scholar] [CrossRef]
- Aron, Z.D.; Mehrani, A.; Hoffer, E.D.; Connolly, K.L.; Srinivas, P.; Torhan, M.C.; Alumasa, J.N.; Cabrera, M.; Hosangadi, D.; Barbor, J.S.; et al. Trans-translation inhibitors bind to a novel site on the ribosome and clear Neisseria gonorrhoeae in vivo. Nat. Commun. 2021, 12, 1779. [Google Scholar] [CrossRef]
- Senges, C.H.R.; Stepanek, J.J.; Wenzel, M.; Raatschen, N.; Ay, Ü.; Märtens, Y.; Prochnow, P.; Hernández, M.V.; Yayci, A.; Schubert, B.; et al. Comparison of proteomic responses as global approach to antibiotic mechanism of action elucidation. Antimicrob. Agents Chemother. 2020, 65, e01373-20. [Google Scholar] [CrossRef]
- Macé, K.; Demay, F.; Guyomar, C.; Georgeault, S.; Giudice, E.; Goude, R.; Trautwetter, A.; Ermel, G.; Blanco, C.; Gillet, R. A genetic tool to quantify trans-translation activity in vivo. J. Mol. Biol. 2017, 429, 3617–3625. [Google Scholar] [CrossRef] [PubMed]
- Brunel, R.; Descours, G.; Durieux, I.; Doublet, P.; Jarraud, S.; Charpentier, X. KKL-35 exhibits potent antibiotic activity against Legionella species independently of trans-translation inhibition. Antimicrob. Agents Chemother. 2018, 62, e01459-17. [Google Scholar] [CrossRef]
- Guyomar, C.; Thépaut, M.; Nonin-Lecomte, S.; Méreau, A.; Goude, R.; Gillet, R. Reassembling green fluorescent protein for in vitro evaluation of trans-translation. Nucleic Acids Res. 2020, 48, e22. [Google Scholar] [CrossRef]
- Tresse, C.; Radigue, R.; Gomes Von Borowski, R.; Thepaut, M.; Le, H.H.; Demay, F.; Georgeault, S.; Dhalluin, A.; Trautwetter, A.; Ermel, G.; et al. Synthesis and evaluation of 1,3,4-oxadiazole derivatives for development as broad-spectrum antibiotics. Bioorganic Med. Chem. 2019, 27, 115097. [Google Scholar] [CrossRef]
- Cole, S.T. Microbiology. Pyrazinamide—Old TB drug finds new target. Science 2011, 333, 1583–1584. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, X.; Jiang, X.; Yuan, H.; Lee, J.S.; Barry, C.E.; Wang, H.; Zhang, W.; Zhang, Y. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y.; Bi, J.; Cai, Q.; Liao, X.; Li, W.; Guo, C.; Zhang, Q.; Lin, T.; Zhao, Y.; et al. Structural basis for targeting the ribosomal protein S1 of Mycobacterium tuberculosis by pyrazinamide. Mol. Microbiol. 2015, 95, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Dillon, N.A.; Peterson, N.D.; Feaga, H.A.; Keiler, K.C.; Baughn, A.D. Anti-tubercular activity of pyrazinamide is independent of trans-translation and RpsA. Sci. Rep. 2017, 7, 6135. [Google Scholar] [CrossRef] [PubMed]
- Vallejos-Sánchez, K.; Lopez, J.M.; Antiparra, R.; Toscano, E.; Saavedra, H.; Kirwan, D.E.; Amzel, L.M.; Gilman, R.H.; Maruenda, H.; Sheen, P.; et al. Mycobacterium tuberculosis ribosomal protein S1 (RpsA) and variants with truncated C-terminal end show absence of interaction with pyrazinoic acid. Sci. Rep. 2020, 10, 8356. [Google Scholar] [CrossRef] [PubMed]
- Reverdatto, S.; Burz, D.S.; Shekhtman, A. Peptide aptamers: Development and applications. Curr. Top. Med. Chem. 2015, 15, 1082–1101. [Google Scholar] [CrossRef]
- Liu, P.; Huang, D.; Hu, X.; Tang, Y.; Ma, X.; Yan, R.; Han, Q.; Guo, J.; Zhang, Y.; Sun, Q.; et al. Targeting inhibition of SmpB by peptide aptamer attenuates the virulence to protect zebrafish against Aeromonas veronii infection. Front. Microbiol. 2017, 8, 1766. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Zhou, Z.; Huang, H.; Ren, Y.; Zhang, Y.; Li, G.; Zhou, Z.; Wang, L. Complete genome sequence of Aeromonas veronii Strain B565. J. Bacteriol. 2011, 193, 3389–3390. [Google Scholar] [CrossRef]
- Liu, P.; Chen, Y.; Wang, D.; Tang, Y.; Tang, H.; Song, H.; Sun, Q.; Zhang, Y.; Liu, Z. Genetic selection of peptide aptamers that interact and inhibit both small protein B and alternative ribosome-rescue factor A of Aeromonas veronii C4. Front. Microbiol. 2016, 7, 1228. [Google Scholar] [CrossRef]
- Thépaut, M.; Campos-Silva, R.; Renard, E.; Barloy-Hubler, F.; Ennifar, E.; Boujard, D.; Gillet, R. Safe and easy in vitro evaluation of tmRNA-SmpB-mediated trans-translation from ESKAPE pathogenic bacteria. RNA 2021, 27, 1390–1399. [Google Scholar] [CrossRef]
- Wilson, B.A.P.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos-Silva, R.; D’Urso, G.; Delalande, O.; Giudice, E.; Macedo, A.J.; Gillet, R. Trans-Translation Is an Appealing Target for the Development of New Antimicrobial Compounds. Microorganisms 2022, 10, 3. https://doi.org/10.3390/microorganisms10010003
Campos-Silva R, D’Urso G, Delalande O, Giudice E, Macedo AJ, Gillet R. Trans-Translation Is an Appealing Target for the Development of New Antimicrobial Compounds. Microorganisms. 2022; 10(1):3. https://doi.org/10.3390/microorganisms10010003
Chicago/Turabian StyleCampos-Silva, Rodrigo, Gaetano D’Urso, Olivier Delalande, Emmanuel Giudice, Alexandre José Macedo, and Reynald Gillet. 2022. "Trans-Translation Is an Appealing Target for the Development of New Antimicrobial Compounds" Microorganisms 10, no. 1: 3. https://doi.org/10.3390/microorganisms10010003
APA StyleCampos-Silva, R., D’Urso, G., Delalande, O., Giudice, E., Macedo, A. J., & Gillet, R. (2022). Trans-Translation Is an Appealing Target for the Development of New Antimicrobial Compounds. Microorganisms, 10(1), 3. https://doi.org/10.3390/microorganisms10010003