
Evidence for Horizontal Transmission and Recirculation of Shiga Toxin-Producing Escherichia coli in the Beef Production Chain in South Africa Using Whole Genome Sequencing

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sources of Isolates
2.1.1. Cattle Feedlots
2.1.2. Abattoir and Retail Outlets
2.1.3. Isolation of STEC Strains
2.1.4. Multiplex PCR to Identify Virulence
2.1.5. Validation of mPCR
2.2. Whole Genome Sequencing and Analysis
2.3. Phylogenetic Analysis of STEC Isolates
3. Results
3.1. Isolates
3.2. Multilocus Sequence Typing
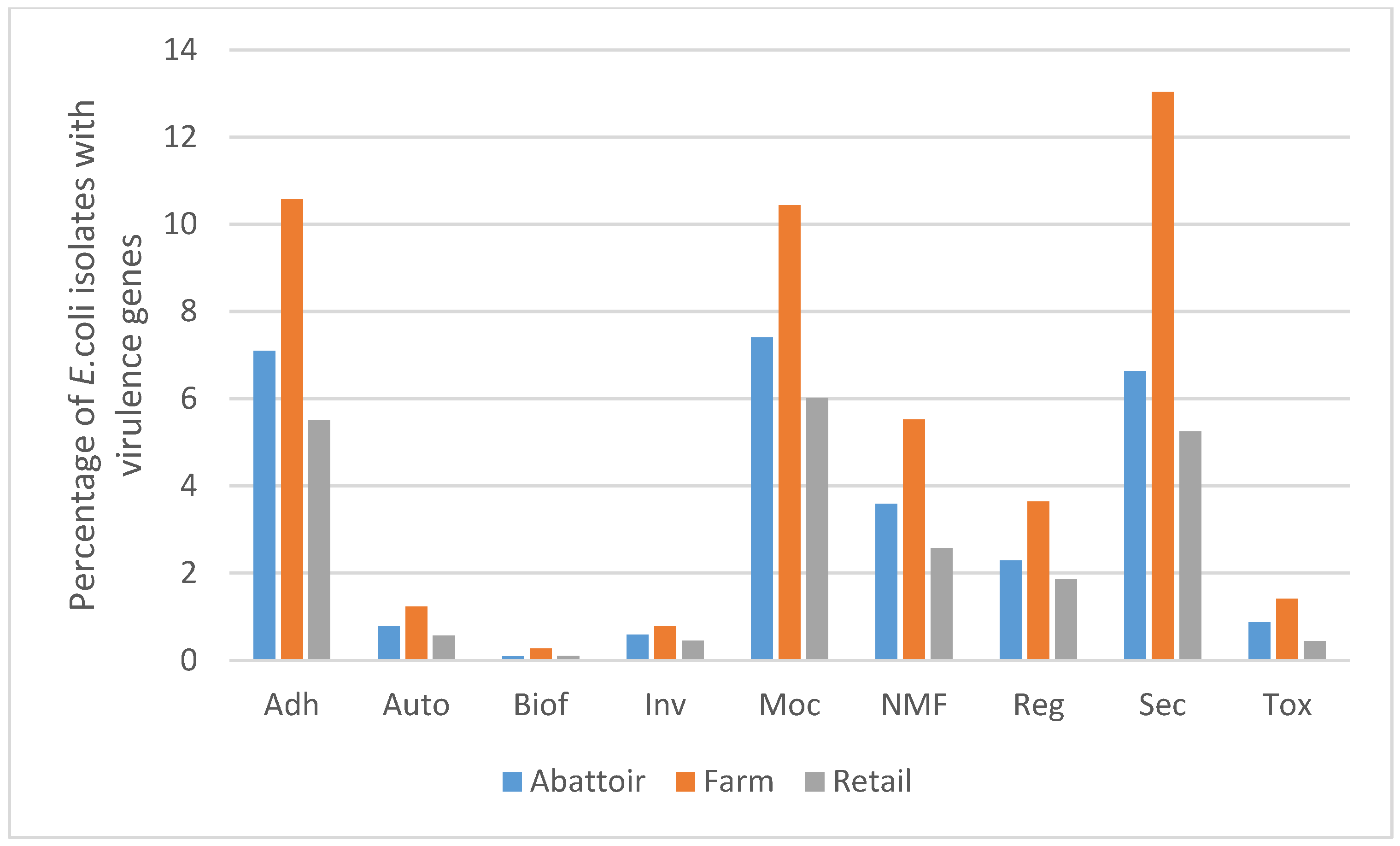
3.3. Virulence Genes
3.4. Antimicrobial Resistance Genes
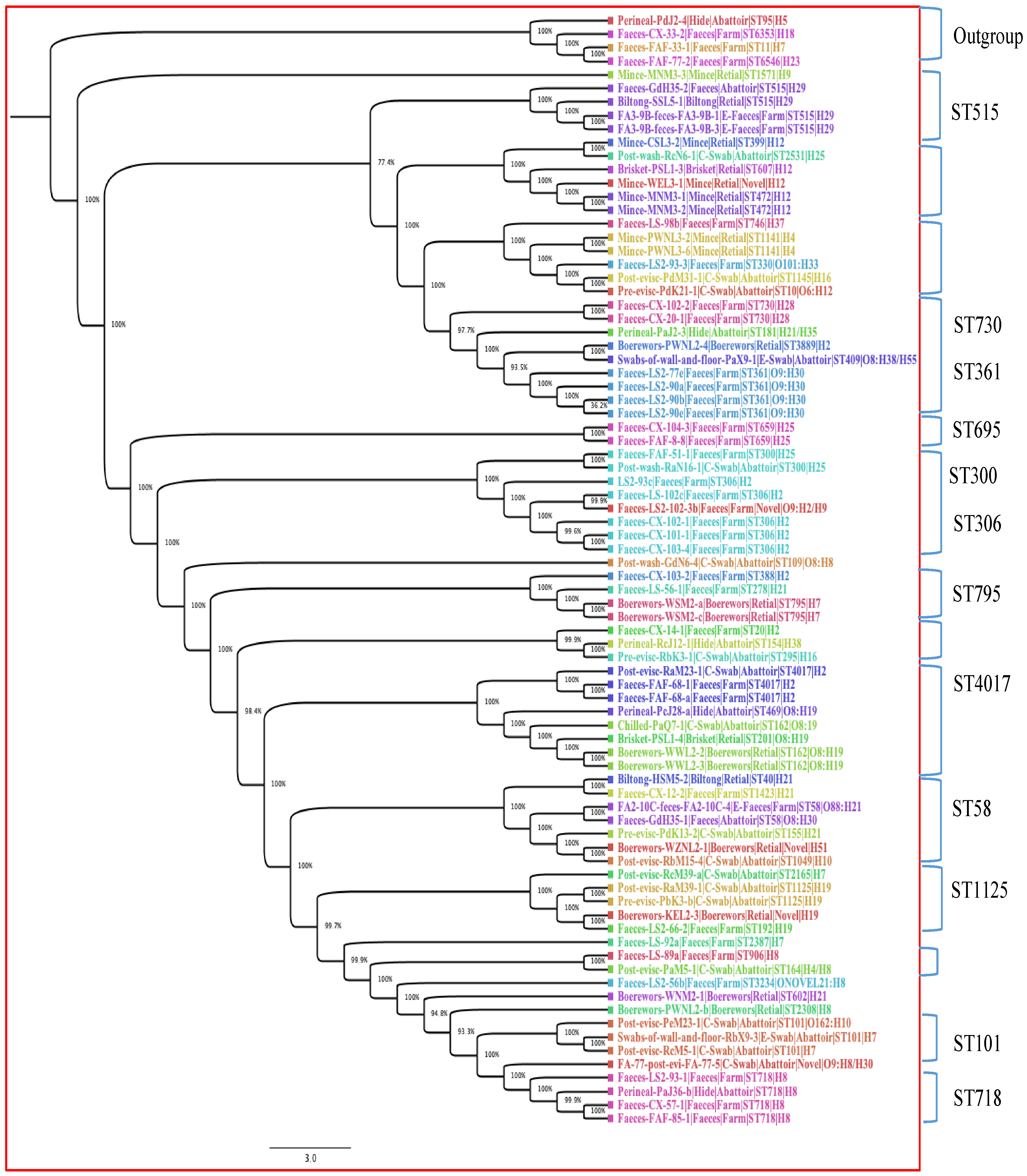
3.5. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO/WHO STEC Expert Group. Hazard identification and characterization: Criteria for categorizing Shiga toxin–producing Escherichia coli on a risk basis. J. Food Prot. 2019, 82, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.R. Shiga toxin (Stx) classification, structure, and function. Microbiol. Spectr. 2014, 2, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B. The locus of enterocyte effacement pathogenicity island of Shiga toxin-producing Escherichia coli O157:H7 and other attaching and effacing E. Coli. Jpn. J. Med. Sci. Biol. 1998, 51 (Suppl. S1), S101–S107. [Google Scholar] [CrossRef]
- Mora, A.; Herrrera, A.; López, C.; Dahbi, G.; Mamani, R.; Pita, J.M.; Alonso, M.P.; Llovo, J.; Bernárdez, M.I.; Blanco, J.E.; et al. Characteristics of the Shiga-toxin-producing enteroaggregative Escherichia coli O104: H4 German outbreak strain and of STEC strains isolated in Spain. Int. Microbiol. 2011, 14, 121–141. [Google Scholar] [CrossRef]
- Blanco, M.; Blanco, J.E.; Mora, A.; Dahbi, G.; Alonso, M.P.; González, E.A.; Bernárdez, M.I.; Blanco, J. Serotypes, Virulence Genes, and Intimin Types of Shiga Toxin (Verotoxin)-Producing Escherichia coli Isolates from Cattle in Spain and Identification of a New Intimin Variant Gene (eae-ξ). J. Clin. Microbiol. 2004, 42, 645–651. [Google Scholar] [CrossRef]
- Valilis, E.; Ramsey, A.; Sidiq, S.; DuPont, H.L. Non-O157 Shiga toxin-producing Escherichia coli—A poorly appreciated enteric pathogen: Systematic review. Int. J. Infect. Dis. 2018, 76, 82–87. [Google Scholar] [CrossRef]
- Callaway, T.R.; Edrington, T.S.; Loneragan, G.H.; Carr, M.A.; Nisbet, D.J. Shiga toxin-producing Escherichia coli (STEC) ecology in cattle and management based options for reducing fecal shedding. Agric. Food Anal. Bacteriol. 2013, 3, 39–69. [Google Scholar]
- Munns, K.D.; Selinger, L.B.; Stanford, K.; Guan, L.; Callaway, T.R.; McAllister, T.A. Perspectives on super-shedding of Escherichia coli O157:H7 by cattle. Foodborne Pathog. Dis. 2015, 12, 89–103. [Google Scholar] [CrossRef]
- Collins, J.D. Slaughtering and processing of livestock. Agricu Mech. Autom. 2009, 2, 342. [Google Scholar] [CrossRef]
- Karmali, M.A.; Gannon, V.; Sargeant, J.M. Verocytotoxin-producing Escherichia coli (VTEC). Vet. Microbiol. 2010, 140, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.; Chinen, I.; Miliwebsky, E.; Masana, M. Risk factors for Shiga toxin-producing Escherichia coli-associated human diseases. In Enterohemorrhagic Escherichia coli and Other Shiga Toxin-Producing; Wiley: Hoboken, NJ, USA, 2015; pp. 359–380. [Google Scholar] [CrossRef]
- Fremaux, B.; Prigent-Combaret, C.; Vernozy-Rozand, C. Long-term survival of Shiga toxin-producing Escherichia coli in cattle effluents and environment: An updated review. Vet. Microbiol. 2008, 132, 1–8. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, J.M.; Doherty, A.M.; Sheridan, J.J.; Thomson-Carter, F.M.; Garvey, P.; McGuire, L.; Blair, I.; McDowell, D. The prevalence and spread of Escherichia coli O157:H7 at a commercial beef abattoir. J. Appl. Microbiol. 2003, 95, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; MacLeod, E.T.; El Bayomi, R.M.; Mohsen, R.A.; Nassar, A.H. Molecular characterization of Escherichia coli O157:H7 and non-O157 shiga toxin producing E. coli from retail meat and humans. Zagazig Vet. J. 2017, 45, 250–261. [Google Scholar] [CrossRef]
- Duffy, G.; Cummins, E.; Nally, P.; O’Brien, S.; Butler, F. A review of quantitative microbial risk assessment in the management of Escherichia coli O157:H7 on beef. Meat Sci. 2006, 74, 76–88. [Google Scholar] [CrossRef]
- Bumunang, E.W.; McAllister, T.A.; Zaheer, R.; Ortega Polo, R.; Stanford, K.; King, R.; Niu, Y.D.; Ateba, C.N. Characterization of non-O157 Escherichia coli from cattle faecal samples in the North-West Province of South Africa. Microorganisms 2019, 7, 272. [Google Scholar] [CrossRef]
- Karama, M.; Mainga, A.O.; Cenci-Goga, B.T.; Malahlela, M.; El-Ashram, S.; Kalake, A. Molecular profiling and antimicrobial resistance of Shiga toxin-producing Escherichia coli O26, O45, O103, O121, O145 and O157 isolates from cattle on cow-calf operations in South Africa. Sci. Rep. 2019, 9, 11930. [Google Scholar] [CrossRef]
- Onyeka, L.O.; Adesiyun, A.A.; Keddy, K.H.; Madoroba, E.; Manqele, A.; Thompson, P.N. Shiga toxin–producing Escherichia coli contamination of raw beef and beef-based ready-to-eat products at retail outlets in Pretoria, South Africa. J. Food Protect. 2020, 83, 476–484. [Google Scholar] [CrossRef]
- Onyeka, L.O.; Adesiyun, A.A.; Keddy, K.H.; Manqele, A.; Madoroba, E.; Thompson, P.N. Prevalence, risk factors and molecular characteristics of Shiga toxin-producing Escherichia coli in beef abattoirs in Gauteng, South Africa. Food Control. 2021, 123, 107746. [Google Scholar] [CrossRef]
- Onyeka, L.O.; Adesiyun, A.A.; Keddy, K.H.; Manqele, A.; Madoroba, E.; Thompson, P.N. Prevalence and patterns of fecal shedding of Shiga toxin–producing Escherichia coli by cattle at a commercial feedlot in South Africa. J. Food Safety. 2022, 42, e12961. [Google Scholar] [CrossRef]
- WOAH-OIE List of Antimicrobial Agents of Veterinary Importance 2021. Available online: https://www.woah.org/app/uploads/2021/06/a-oie-list-antimicrobials-june2021.pdf (accessed on 15 August 2024).
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updates. 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.W.; Paton, J.C. Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfb O111, and rfb O157. J. Clin. Microbiol. 1998, 36, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, R.L.; Pouseele, H.; Chen, J.C.; Strockbine, N.A.; Carleton, H.A. Implementation of whole genome sequencing (WGS) for identification and characterization of Shiga toxin-producing Escherichia coli (STEC) in the United States. Front. Microbiol. 2016, 7, 766. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Souvorov, A.; Agarwala, R.; Lipman, D.J. SKESA: Strategic k-mer extension for scrupulous assemblies. Genome biology. Genome Biol. 2018, 19, 153. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Ingle, D.J.; Valcanis, M.; Kuzevski, A.; Tauschek, M.; Inouye, M.; Stinear, T.; Levine, M.M.; Robins-Browne, R.M.; Holt, K.E. In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O: H serotype combinations within and between pathogenic lineages. Microb. Genom. 2016, 2, e000064. [Google Scholar] [CrossRef]
- Bessonov, K.; Laing, C.; Robertson, J.; Yong, I.; Ziebell, K.; Gannon, V.P.; Nichani, A. ECTyper: In silico Escherichia coli serotype and species prediction from raw and assembled whole-genome sequence data. Microb. Genom. 2021, 7, 000728. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Joris, M.A.; Verstraete, K.; Reu, K.D.; Zutter, L.D. Loss of vtx genes after the first subcultivation step of verocytotoxigenic Escherichia coli O157 and non-O157 during isolation from naturally contaminated fecal samples. Toxins 2011, 3, 672–677. [Google Scholar] [CrossRef]
- Mellmann, A.; Lu, S.; Karch, H.; Xu, J.G.; Harmsen, D.; Schmidt, M.A.; Bielaszewska, M. Recycling of Shiga toxin 2 genes in sorbitol-fermenting enterohemorrhagic Escherichia coli O157: NM. Appl. Environ. Microbiol. 2008, 74, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Senthakumaran, T.; Brandal, L.T.; Lindstedt, B.A.; Jørgensen, S.B.; Charnock, C.; Tunsjø, H.S. Implications of stx loss for clinical diagnostics of Shiga toxin-producing Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2361–2370. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Prager, R.; Kock, R.; Mellmann, A.; Zhang, W.; Tschäpe, H.; Tarr, P.I.; Karch, H. Shiga toxin gene loss and transfer in vitro and in vivo during enterohemorrhagic Escherichia coli O26 infection in humans. Appl. Environ. Microbiol. 2007, 73, 3144–3150. [Google Scholar] [CrossRef] [PubMed]
- Amézquita-López, B.A.; Quiñones, B.; Lee, B.G.; Chaidez, C. Virulence profiling of Shiga toxin-producing Escherichia coli recovered from domestic farm animals in Northwestern Mexico. Front. Cell Infect. Microbiol. 2014, 4, 7. [Google Scholar] [CrossRef]
- Coombes, B.K.; Wickham, M.E.; Mascarenhas, M.; Gruenheid, S.; Finlay, B.B.; Karmali, M.A. Molecular analysis as an aid to assess the public health risk of non-O157 Shiga toxin-producing Escherichia coli strains. Appl. Environ. Microbiol. 2008, 74, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Fratamico, P.M.; Bono, J.L.; Baranzoni, G.M.; Chen, C.Y. Genome sequencing and comparative genomics provides insights on the evolutionary dynamics and pathogenic potential of different H-serotypes of Shiga toxin-producing Escherichia coli O104. BMC Microbiol. 2015, 15, 83. [Google Scholar] [CrossRef]
- Friesema, I.H.; Keijzer-Veen, M.G.; Koppejan, M.; Schipper, H.S.; van Griethuysen, A.J.; Heck, M.E.; van Pelt, W. Hemolytic uremic syndrome associated with Escherichia coli O8: H19 and Shiga toxin 2f gene. Emerg. Infect. Dis. 2015, 21, 168. [Google Scholar] [CrossRef]
- Fan, R.; Shao, K.; Yang, X.; Bai, X.; Fu, S.; Sun, H.; Xu, Y.; Wang, H.; Li, Q.; Hu, B.; et al. High prevalence of non-O157 Shiga toxin-producing Escherichia coli in beef cattle detected by combining four selective agars. BMC Microbiol. 2019, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Karama, M.; Cenci-Goga, B.T.; Malahlela, M.; Smith, A.M.; Keddy, K.H.; El-Ashram, S.; Kabiru, L.M.; Kalake, A. Virulence characteristics and antimicrobial resistance profiles of shiga toxin-producing Escherichia coli isolates from humans in South Africa: 2006–2013. Toxins 2019, 11, 424. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.A.; Kudva, I.T. Antibiotic-resistant Shiga toxin-producing Escherichia coli: An overview of prevalence and intervention strategies. Zoonoses Public. Health. 2019, 66, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Iweriebor, B.C.; Iwu, C.J.; Obi, L.C.; Nwodo, U.U.; Okoh, A.I. Multiple antibiotic resistances among Shiga toxin producing Escherichia coli O157 in feces of dairy cattle farms in Eastern Cape of South Africa. BMC Microbiol. 2015, 15, 213. [Google Scholar] [CrossRef]
- Piddock, L.J. Multidrug-resistance efflux pumps? not just for resistance. Nat. Rev. Microbiol. 2006, 4, 629–636. [Google Scholar] [CrossRef]
- Nikaido, H. Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 77, 1. [Google Scholar]
- Nishino, K.; Yamasaki, S.; Nakashima, R.; Zwama, M.; Hayashi-Nishino, M. Function and inhibitory mechanisms of multidrug efflux pumps. Front. Microbiol. 2021, 12, 737288. [Google Scholar] [CrossRef]
- WHO Ten Threaths to Global Health in 2019. Available online: https://www.who.int/news-room/spotlight/ten-threats-to-global-health-in-2019 (accessed on 15 August 2024).
- Grace, D.; Lindahl, J.F.; Nguyen-Viet, H.; Kakkar, M. Antimicrobial use in developing countries. In Proceedings of the World Veterinary Association (WVA)/World Medical Association (WMA) Global Conference on One Health, Madrid, Spain, 21–22 May 2015; ILRI:: Nairobi, Kenya, 2015. Available online: https://cgspace.cgiar.org/handle/10568/67030 (accessed on 8 August 2024).
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sciences. 2015, 112, 5649–5654. [Google Scholar] [CrossRef]
- Riley, L.W. Pandemic lineages of extraintestinal pathogenic Escherichia coli. Clin. Microbiol. Infect. 2014, 20, 380–390. [Google Scholar] [CrossRef]
- Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L.H.; Karch, H.; Reeves, P.R.; Maiden, M.C.J.; Ochman, H.; et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006, 60, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.C.; Stanford, K.; McAllister, T.A.; Thomas, J.; Reuter, T. Further development of sample preparation and detection methods for O157 and the top 6 non-O157 STEC serogroups in cattle faeces. J. Microbiol. Methods 2014, 105, 22–30. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Lab id | Sample Type * | Source | Season ** | Location | Serogenotype *** | STEC | Adherence | Haemolysin |
|---|---|---|---|---|---|---|---|---|
| GdH35(2) | Faeces | abattoir | W | Gauteng east | H29 | stx2a, stx2b | eaeH | hlyE |
| PaJ36(b) | perineum HS | abattoir | W | Gauteng north | H8 | stx1a, stx1b, stx2a, stx2d1A | eaeH | hlyA, hlyB, hlyC, hlyD, hlyE |
| PcJ28(a) | perineum HS | abattoir | W | Gauteng north | O8:H19 | stx1a, stx1b, stx2a, stx2b | eaeH, ehaA | hlyA, hlyB, hlyC, hlyD, hlyE |
| RaM39(1) | post-evisc CS | abattoir | W | Gauteng west | H19 | stx2a, stx2b | eaeH | hlyA, hlyB, hlyC, hlyD, hlyE |
| RcM39(a) | post-evisc CS | abattoir | W | Gauteng west | H7 | stx1a, stx1b, stx2a, | eaeH, ehaA | hlyA, hlyB, hlyD, hlyE |
| RaM23(1) | post-evisc CS | abattoir | S | Gauteng west | H2 | stx1a, stx1b, stx2a, | ehaA- | hlyA, hlyC, hlyD, hlyE |
| RaN16(1) | post-evisc CS | abattoir | S | Gauteng west | H25 | stx1a, stx1b | eae, eaeH, tir, nleBI, nleB2 | hlyA, hlyB, hlyD, hlyE |
| PbK3(b) | pre-evisc CS | abattoir | S | Gauteng north | H19 | stx2a, stx2b | eaeH | hlyA, hlyB, hlyC, hlyD, hlyE |
| LS 92a | faeces | farm | S | Gauteng north | H7 | stx2a, stx2dB | eaeH | hlyE |
| CX 104(3) | faeces | farm | S | Gauteng north | H25 | stx2a, stx2dB | eae, eaeH, ehaA, tir, nleBI | hlyA, hlyB, hlyD, hlyE |
| LS2 66(2) | faeces | farm | S | Gauteng north | H19 | stx2a | eaeH | hlyE |
| CX 57(1) | faeces | farm | S | Gauteng north | H8 | stx1a, stx1b, stx2b, stx2d1A | eaeH, ehaA | hlyA, hlyB, hlyC, hlyD, hlyE |
| LS2 102-3b | faeces | farm | S | Gauteng north | O9:H2/H9 | stx1a, stx1b | eae, tir, nleB2-2 | hlyD, hlyE |
| LS2 93c | faeces | farm | S | Gauteng north | H2 | stx1a, stx1b | eae, eaeH, tir, nleB2-2 | hlyA, hlyB, hlyC, hlyD, hlyE |
| CX 101(1) | faeces | farm | S | Gauteng north | H2 | stx1a, stx1b | eae, eaeH, tir | hlyA, hlyB, hlyD, hlyE |
| CX 102(1) | faeces | farm | S | Gauteng north | H2 | stx1a, stx1b | eae, eaeH, tir, nleBI | hlyA, hlyD, hlyE |
| CX 103(4) | faeces | farm | S | Gauteng north | H2 | stx1a, stx1b | eae, eaeH, tir | hlyB, hlyD, hlyE |
| LS 102c | faeces | farm | S | Gauteng north | H2 | stx1a, stx1b | eae, eaeH, tir | hlyB, hlyD, hlyE |
| CX 8(4) | faeces | farm | S | Gauteng north | 0 | stx1a | eaeH, eaeX | 0 |
| FAF 33(1) | faeces | farm-abattoir | S | Gauteng north | H7 | stx2b | eae, eaeH, tir, nleB2 | hlyD, hlyE |
| FAF 8(8) | faeces | farm-abattoir | S | Gauteng north | H25 | stx2a, stx2dB | eae, eaeH, ehaA, tir | 0 |
| FAF 85(1) | faeces | farm-abattoir | S | Gauteng north | H8 | stx1a, stx1b, stx2b, stx2d1A | eaeH | hlyA, hlyB, hlyC, hlyD, hlyE |
| FAF 68(1) | faeces | farm-abattoir | S | Gauteng north | H2 | stx1a, stx1b, stx2a, stx2dB | eaeH, saa | hlyA, hlyB, hlyC, hlyD, hlyE |
| FAF 68(a) | faeces | farm-abattoir | S | Gauteng north | H2 | stx1a, stx1b, stx2a, | eaeH | hlyA, hlyB, hlyC, hlyD, hlyE |
| FAF 51(1) | faeces | farm-abattoir | S | Gauteng north | H25 | stx1a, stx1b | eae, eaeH, tir, nleB2 | hlyA, hlyB, hlyD, hlyE |
| FA3 9B(1) | faeces | farm-env | S | Gauteng north | H29 | stx2b, stx2dB | eaeH | hlyE |
| FA3 9B(3) | faeces | farm-env | S | Gauteng north | H29 | stx2b, stx2d1A, stx2dB | eaeH | hlyE |
| SSL5(1) | biltong | retail | S | Gauteng north | H29 | stx2d1 | eaeH | hlyE |
| WWL2(2) | boerewors | retail | S | Gauteng north | O8:H19 | stx2a, stx2dB | eaeH | hlyA, hlyB, hlyC, hlyD, hlyE |
| WWL2(3) | boerewors | retail | S | Gauteng north | O8:H19 | stx2a, stx2dB | eaeH, saa | hlyA, hlyC, hlyD, hlyE |
| KEL2(3) | boerewors | retail | A | Gauteng north | H19 | stx2a | eaeH | hlyE |
| PSL1(3) | brisket | retail | A | Gauteng north | H12 | stx2a, stx2dB | - | hlyE |
| PSL1(4) | brisket | retail | A | Gauteng north | O8:H19 | stx1a, stx1b, stx2a, stx2dB | eaeH, saa | hlyA, hlyB, hlyC, hlyD, hlyE |
| Clonal Complex | Sample | Source | Location * | ST † | Serogenotype | stx-Subtypes |
|---|---|---|---|---|---|---|
| 5 matching loci | Post-evisceration-RbM15-4 | abattoir | GW | ST1049 | H10 | - |
| Pre-evisceration-PdK13-2 | abattoir | GN | ST155 | H21 | - | |
| Faeces-GdH35-1 | abattoir | GE | ST58 | O8:H30 | - | |
| 5 matching loci | Brisket-PSL1-4 | retail | GN | ST201 | O8:H19 | stx1a, stx1b, stx2a, stx2dB |
| Chilled-PaQ7-1 | abattoir | GN | ST162 | O8:H19 | - | |
| Perineal-PcJ28-a | abattoir | GN | ST469 | O8:H19 | stx1a, stx1b, stx2a, stx2b | |
| 6 matching loci | Post-evisceration-PdM31-1 | abattoir | GN | ST1145 | H16 | - |
| Pre-evisceration-PdK21-1 | abattoir | GN | ST10 | O6:H12 | - | |
| Mince-PWNL3-2 | retail | GN | ST1141 | H4 | - |
| Antimicrobial Class | Resistance Genes | No. of Isolates Positive for AMR Genes | Antimicrobial Compounds |
|---|---|---|---|
| Aminoglycosides | aph (3″)-Ib | 8 | Aminoglycoside |
| aph (6)-Id | 8 | Aminoglycoside | |
| aadA | 4 | Aminoglycoside | |
| aadA2 | 2 | Aminoglycoside | |
| aadA3 | 3 | Aminoglycoside | |
| aadA4 | 1 | Aminoglycoside | |
| kdpE | 83 | Aminoglycoside | |
| Amphenicols | cmlA6 | 1 | Amphenicols |
| floR/chloramphenicol | 3 | Amphenicols | |
| Beta-lactam/beta-lactamase-inhibitor | ampC | 83 | Cephalosporin, penam |
| TEM-1 | 2 | Penam, monobactam, penem, cephalosporin | |
| TEM-150 | 1 | Penam, monobactam, penem, cephalosporin | |
| Fluoroquinolones | patA | 84 | Fluoroquinolone |
| Glycopeptides | bacA | 84 | Peptide |
| eptA/PmrC | 85 | Peptide | |
| pmrF | 85 | Peptide | |
| ugd/pmrE | 73 | Peptide | |
| yojI | 84 | Peptide | |
| Macrolide | mphB | 4 | Macrolide |
| Multidrug (MDR) efflux pumps | CRP | 85 | Penam, macrolide, fluoroquinolone |
| acrA | 85 | Tetracycline, glycylcycline, rifamycin, phenicol, penam, cephalosporin, fluoroquinolone, disinfecting agents and antiseptics | |
| emrE | 63 | Macrolide | |
| mdfA | 84 | Disinfecting agents and antiseptics, tetracycline | |
| H-NS | 83 | Macrolide, tetracycline, penam, cephalosporin, fluoroquinolone, cephamycin | |
| acrB | 82 | Tetracycline, glycylcycline, rifamycin, phenicol, penam, cephalosporin, fluoroquinolone, disinfecting agents and antiseptics | |
| acrD | 82 | Aminoglycoside | |
| acrE | 82 | Penam, cephamycin, fluoroquinolone, cephalosporin | |
| acrF | 79 | Penam, cephamycin, fluoroquinolone, cephalosporin | |
| acrS | 80 | Tetracycline, cephamycin, glycylcycline, rifamycin, phenicol, penam, cephalosporin, fluoroquinolone, disinfecting agents and antiseptics | |
| baeR | 85 | Aminocoumarin, aminoglycoside | |
| baeS | 85 | Aminocoumarin, aminoglycoside | |
| cpxA | 84 | Aminocoumarin, aminoglycoside | |
| emrA | 85 | Fluoroquinolone | |
| Antimicrobial Class | Resistance Genes | Number of Resistant Isolates | Antimicrobial Compounds |
| Multidrug (MDR) efflux pumps | emrB | 85 | Fluoroquinolone |
| emrK | 83 | Tetracycline | |
| emrR | 85 | Fluoroquinolone | |
| emrY | 79 | Tetracycline | |
| evgA | 84 | Penam, tetracycline, macrolide, fluoroquinolone | |
| evgS | 78 | Penam, tetracycline, macrolide, fluoroquinolone | |
| gadW | 83 | Penam, macrolide, fluoroquinolone | |
| gadX | 81 | Penam, macrolide, fluoroquinolone | |
| marA | 85 | Tetracycline, glycylcycline, rifamycin, phenicol, penam, cephalosporin, cephamycin, penem, monobactam, carbapem, fluoroquinolone, disinfecting agents and antiseptics | |
| mdtA | 84 | Aminocoumarin | |
| mdtB | 84 | Aminocoumarin | |
| mdtC | 84 | Aminocoumarin | |
| mdtE | 82 | Fluoroquinolone, macrolide, penam | |
| mdtF | 81 | Fluoroquinolone, macrolide, penam | |
| mdtH | 84 | Fluoroquinolone | |
| mdtK | 66 | Fluoroquinolone | |
| mdtM | 83 | Nucleoside, lincosamide, fluoroquinolone, phenicol, disinfectant agents and antiseptics | |
| mdtN | 85 | Nucleoside, disinfectant and antiseptics | |
| mdtO | 82 | Nucleoside, disinfectant and antiseptics | |
| mdtP | 83 | Nucleoside, disinfectant and antiseptics | |
| msbA | 85 | Nitroimidazole | |
| tolC | 85 | Peptide, aminoglycoside, tetracycline, aminocoumarin, penem, phenicol, fluoroquinolone, carbapem, macrolide, disinfecting agents and antiseptics, cephalosporin, glycylcycline, rifamycin, cephamycin | |
| vgaC | 6 | Streptogramin, pleuromutilin, streptogramin A, lincosamide | |
| Phosphonics | FosA7 | 4 | Phosphonic acid |
| mdtG | 84 | Phosphonic acid | |
| Sulfonamides | sul1 | 5 | Sulphonamide |
| sul2 | 7 | Sulphonamide | |
| sul3 | 1 | Sulphonamide | |
| Tetracyclines | tet(A) | 2 | Tetracycline |
| tet(C) | 6 | Tetracycline | |
| tet(D) | 1 | Tetracycline | |
| Trimethoprim-derivatives | dfrA12 | 4 | Diaminopyrimidine |
| dfrA15 | 3 | Diaminopyrimidine |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onyeka, L.O.; Adesiyun, A.A.; Ismail, A.; Allam, M.; Keddy, K.H.; Thompson, P.N. Evidence for Horizontal Transmission and Recirculation of Shiga Toxin-Producing Escherichia coli in the Beef Production Chain in South Africa Using Whole Genome Sequencing. Pathogens 2024, 13, 732. https://doi.org/10.3390/pathogens13090732
Onyeka LO, Adesiyun AA, Ismail A, Allam M, Keddy KH, Thompson PN. Evidence for Horizontal Transmission and Recirculation of Shiga Toxin-Producing Escherichia coli in the Beef Production Chain in South Africa Using Whole Genome Sequencing. Pathogens. 2024; 13(9):732. https://doi.org/10.3390/pathogens13090732
Chicago/Turabian StyleOnyeka, Libby Obumneke, Abiodun A. Adesiyun, Arshad Ismail, Mushal Allam, Karen H. Keddy, and Peter N. Thompson. 2024. "Evidence for Horizontal Transmission and Recirculation of Shiga Toxin-Producing Escherichia coli in the Beef Production Chain in South Africa Using Whole Genome Sequencing" Pathogens 13, no. 9: 732. https://doi.org/10.3390/pathogens13090732
APA StyleOnyeka, L. O., Adesiyun, A. A., Ismail, A., Allam, M., Keddy, K. H., & Thompson, P. N. (2024). Evidence for Horizontal Transmission and Recirculation of Shiga Toxin-Producing Escherichia coli in the Beef Production Chain in South Africa Using Whole Genome Sequencing. Pathogens, 13(9), 732. https://doi.org/10.3390/pathogens13090732