
Interlaboratory Comparison Study on Ribodepleted Total RNA High-Throughput Sequencing for Plant Virus Diagnostics and Bioinformatic Competence

 ,
,  , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Invitation and Participation of Laboratories
2.2. In-House Bioinformatic Analysis by Participating Laboratories
2.3. Virtool Analysis
3. Discussion
- (a)
- the practical performance of each laboratory to prepare the samples for HTS;
- (b)
- their ability to correctly identify the viruses using their own bioinformatic strategies;
- (c)
- the checking of the variation in sequencing quality of one commercial provider; and
- (d)
- the performance of Virtool for data analysis as an alternative standard analysis option.
4. Materials and Methods
4.1. Organisation
4.2. Preparation of Virus-Infected Plant Material
4.3. Chemicals Aliquoting and Shipment
4.4. The Sequential Steps That the Participants Were Requested to Perform
- 1.
- Preparation of pooled samples:
- 2.
- Total RNA extraction:
- 3.
- Ribosomal RNA depletion:
- 4.
- High-throughput sequencing:
- 5.
- In-house bioinformatic analysis:
- 6.
- Reporting the results:
- 7.
- Assessing Virtool:
4.5. Virtool Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coutts, R. Plant viruses: New threats to crops from changing farming practices. Nature 1986, 322, 594. [Google Scholar] [CrossRef]
- Ward, E.; Foster, S.J.; Fraaije, B.A.; Mccartney, H.A. Plant pathogen diagnostics: Immunological and nucleic acid-based approaches. Ann. Appl. Biol. 2004, 145, 1–16. [Google Scholar] [CrossRef]
- Boonham, N.; Kreuze, J.; Winter, S.; van der Vlugt, R.; Bergervoet, J.; Tomlinson, J.; Mumford, R. Methods in virus diagnostics: From ELISA to next generation sequencing. Virus Res. 2014, 186, 20–31. [Google Scholar] [CrossRef]
- Roossinck, M.J. Deep sequencing for discovery and evolutionary analysis of plant viruses. Virus Res. 2017, 239, 82–86. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Adams, I.; Fox, A. Diagnosis of plant viruses using next-generation sequencing and metagenomic analysis. In Current Research Topics in Plant Virology; Wang, A., Zhou, X., Eds.; Springer: Cham, Switzerland, 2016; ISBN 978-3-319-32917-8. [Google Scholar]
- Visser, M.; Bester, R.; Burger, J.T.; Maree, H.J. Next-generation sequencing for virus detection: Covering all the bases. Virol. J. 2016, 13, 85. [Google Scholar] [CrossRef] [Green Version]
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of HTS for routine plant virus diagnostics: State of the art and challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roenhorst, J.W.; de Krom, C.; Fox, A.; Mehle, N.; Ravnikar, M.; Werkman, A.W. Ensuring validation in diagnostic testing is fit for purpose: A view from the plant virology laboratory. EPPO Bull. 2018, 48, 105–115. [Google Scholar] [CrossRef]
- Massart, S.; Chiumenti, M.; de Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus detection by high-throughput sequencing of small RNAs: Large-scale performance testing of sequence analysis strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaafar, Y.Z.A.; Ziebell, H. Comparative study on three viral enrichment approaches based on RNA extraction for plant virus/viroid detection using high-throughput sequencing. PLoS ONE 2020, 15, e0237951. [Google Scholar] [CrossRef]
- Brandão, P.E.; Taniwaki, S.A.; Berg, M.; Hora, A.S. Complete genome of avian coronavirus vaccine strains Ma5 and BR-I. Genome Announc. 2017, 5, e00201-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaafar, Y.Z.A.; Herz, K.; Hartrick, J.; Fletcher, J.; Blouin, A.G.; MacDiarmid, R.; Ziebell, H. Investigating the pea virome in Germany—Old friends and new players in the field(s). Front. Microbiol. 2020, 11, 583242. [Google Scholar] [CrossRef] [PubMed]
- Poojari, S.; Alabi, O.J.; Fofanov, V.Y.; Naidu, R.A. Correction: A leafhopper-transmissible DNA virus with novel evolutionary lineage in the family Geminiviridae implicated in grapevine redleaf disease by next-generation sequencing. PLoS ONE 2016, 11, e0147510. [Google Scholar] [CrossRef] [PubMed]
- Di Serio, F.; Flores, R.; Verhoeven, J.T.J.; Li, S.-F.; Pallás, V.; Randles, J.W.; Sano, T.; Vidalakis, G.; Owens, R.A. Current status of viroid taxonomy. Arch. Virol. 2014, 159, 3467–3478. [Google Scholar] [CrossRef]
- Csorba, T.; Bovi, A.; Dalmay, T.; Burgyán, J. The p122 subunit of tobacco mosaic virus replicase is a potent silencing suppressor and compromises both small interfering RNA- and microRNA-mediated pathways. J. Virol. 2007, 81, 11768–11780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, K.; Kobori, T.; Kosaka, Y.; Natsuaki, T.; Masuta, C. Characterization of silencing suppressor 2b of cucumber mosaic virus based on examination of its small RNA-binding abilities. Plant Cell Physiol. 2007, 48, 1050–1060. [Google Scholar] [CrossRef] [Green Version]
- Conti, G.; Zavallo, D.; Venturuzzi, A.L.; Rodriguez, M.C.; Crespi, M.; Asurmendi, S. TMV induces RNA decay pathways to modulate gene silencing and disease symptoms. Plant J. 2017, 89, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Gaafar, Y.; Lüddecke, P.; Heidler, C.; Hartrick, J.; Sieg-Müller, A.; Hübert, C.; Wichura, A.; Ziebell, H. First report of Southern tomato virus in German tomatoes. New Dis. Rep. 2019, 40, 1. [Google Scholar] [CrossRef] [Green Version]
- Gaafar, Y.; Richert-Pöggeler, K.R.; Hartrick, J.; Lüddecke, P.; Maaß, C.; Schuhmann, S.; Wilstermann, A.; Ziebell, H. A new tobamovirus infecting Hoya spp. New Dis. Rep. 2020, 42, 10. [Google Scholar] [CrossRef]
- EPPO. PM 7/98 (4) Specific requirements for laboratories preparing accreditation for a plant pest diagnostic activity. EPPO Bull. 2019, 49, 530–563. [Google Scholar] [CrossRef] [Green Version]
- Menzel, W.; Jelkmann, W.; Maiss, E. Detection of four apple viruses by multiplex RT-PCR assays with coamplification of plant mRNA as internal control. J. Virol. Methods 2002, 99, 81–92. [Google Scholar] [CrossRef]
- Hughes, K.J.D.; Tomlinson, J.A.; Griffin, R.L.; Boonham, N.; Inman, A.J.; Lane, C.R. Development of a one-step real-time polymerase chain reaction assay for diagnosis of Phytophthora ramorum. Phytopathology 2006, 96, 975–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fall, M.L.; Xu, D.; Lemoyne, P.; Moussa, I.E.B.; Beaulieu, C.; Carisse, O. A diverse virome of leafroll-infected grapevine unveiled by dsRNA sequencing. Viruses 2020, 12, 1142. [Google Scholar] [CrossRef] [PubMed]
- External RNA Controls Consortium. Proposed methods for testing and selecting the ERCC external RNA controls. BMC Genom. 2005, 6, 150. [Google Scholar] [CrossRef] [Green Version]
- Gaafar, Y.Z.A.; Richert-Pöggeler, K.R.; Maaß, C.; Vetten, H.-J.; Ziebell, H. Characterisation of a novel nucleorhabdovirus infecting alfalfa (Medicago sativa). Virol. J. 2019, 16, 55. [Google Scholar] [CrossRef] [Green Version]
- Green, S.K.; Tsai, W.S.; Shih, S.L.; Black, L.L.; Rezaian, A.; Rashid, M.H.; Roff, M.M.N.; Myint, Y.Y.; Hong, L.T.A. Molecular characterization of begomoviruses associated with leafcurl diseases of tomato in Bangladesh, Laos, Malaysia, Myanmar, and Vietnam. Plant Dis. 2001, 85, 1286. [Google Scholar] [CrossRef]
- Gaafar, Y.Z.A.; Richert-Pöggeler, K.R.; Sieg-Müller, A.; Lüddecke, P.; Herz, K.; Hartrick, J.; Maaß, C.; Ulrich, R.; Ziebell, H. Caraway yellows virus, a novel nepovirus from Carum carvi. Virol. J. 2019, 16, 70. [Google Scholar] [CrossRef]
- Rott, M.; Xiang, Y.; Boyes, I.; Belton, M.; Saeed, H.; Kesanakurti, P.; Hayes, S.; Lawrence, T.; Birch, C.; Bhagwat, B.; et al. Application of next generation sequencing for diagnostic testing of tree fruit viruses and viroids. Plant Dis. 2017, 101, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L. Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PLoS ONE 2014, 9, e105067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R Foundation for Statistical Computing, Vienna, Austria, 2020. Available online: https://www.R-project.org/ (accessed on 8 September 2021).
- Ginestet, C. ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. Ser. B Stat. Methodol. 2011, 174, 245–246. [Google Scholar] [CrossRef]
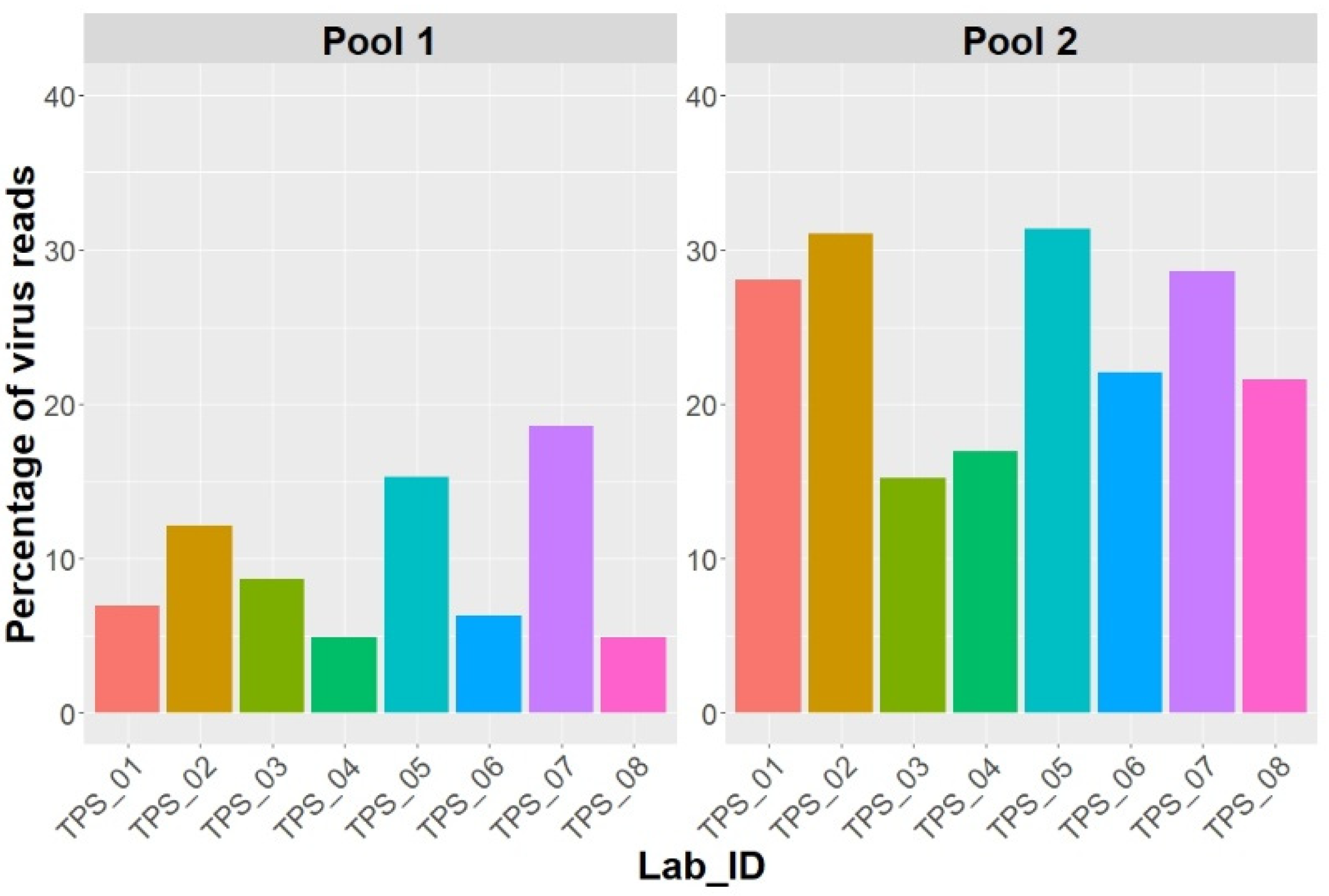
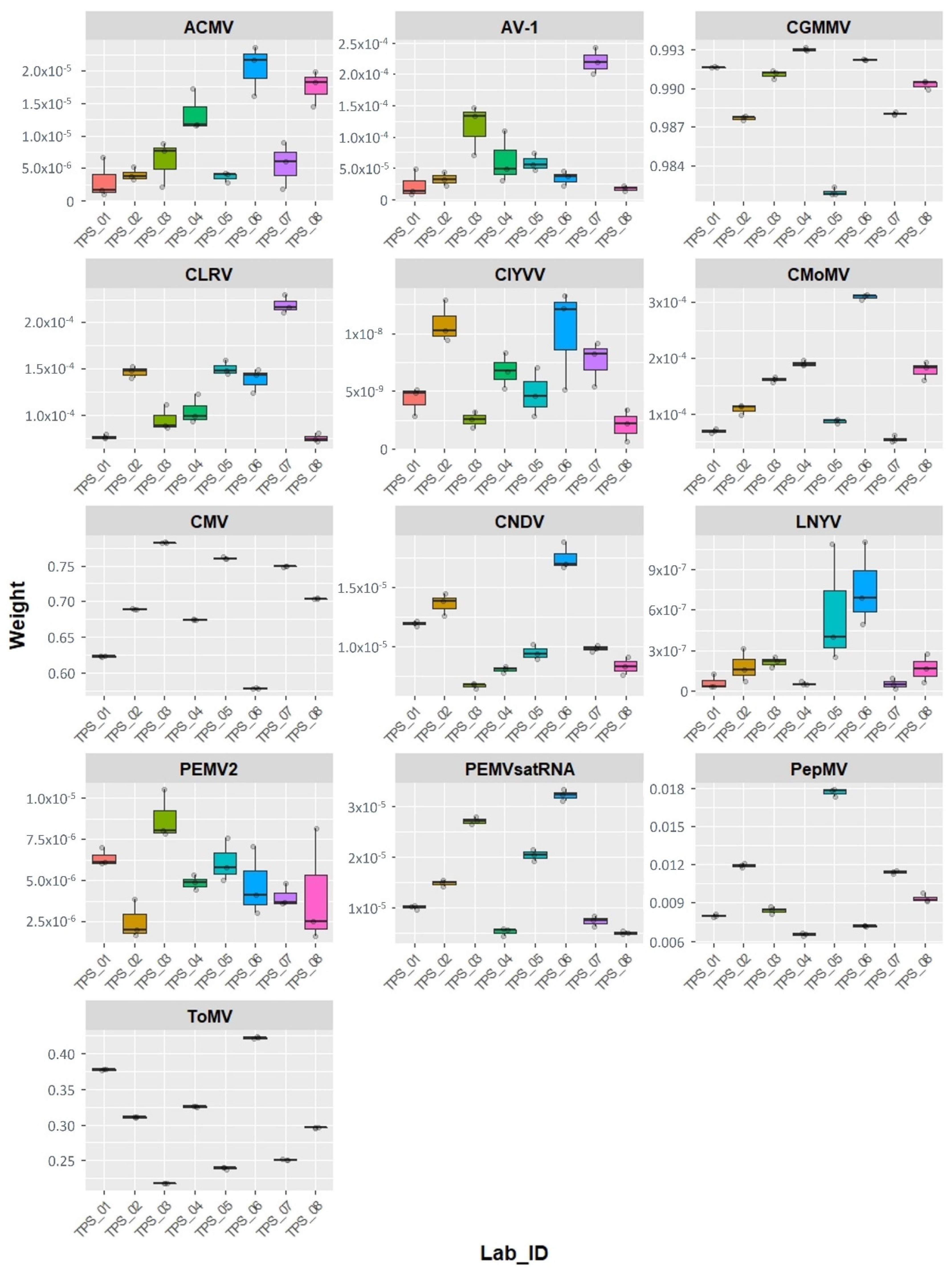
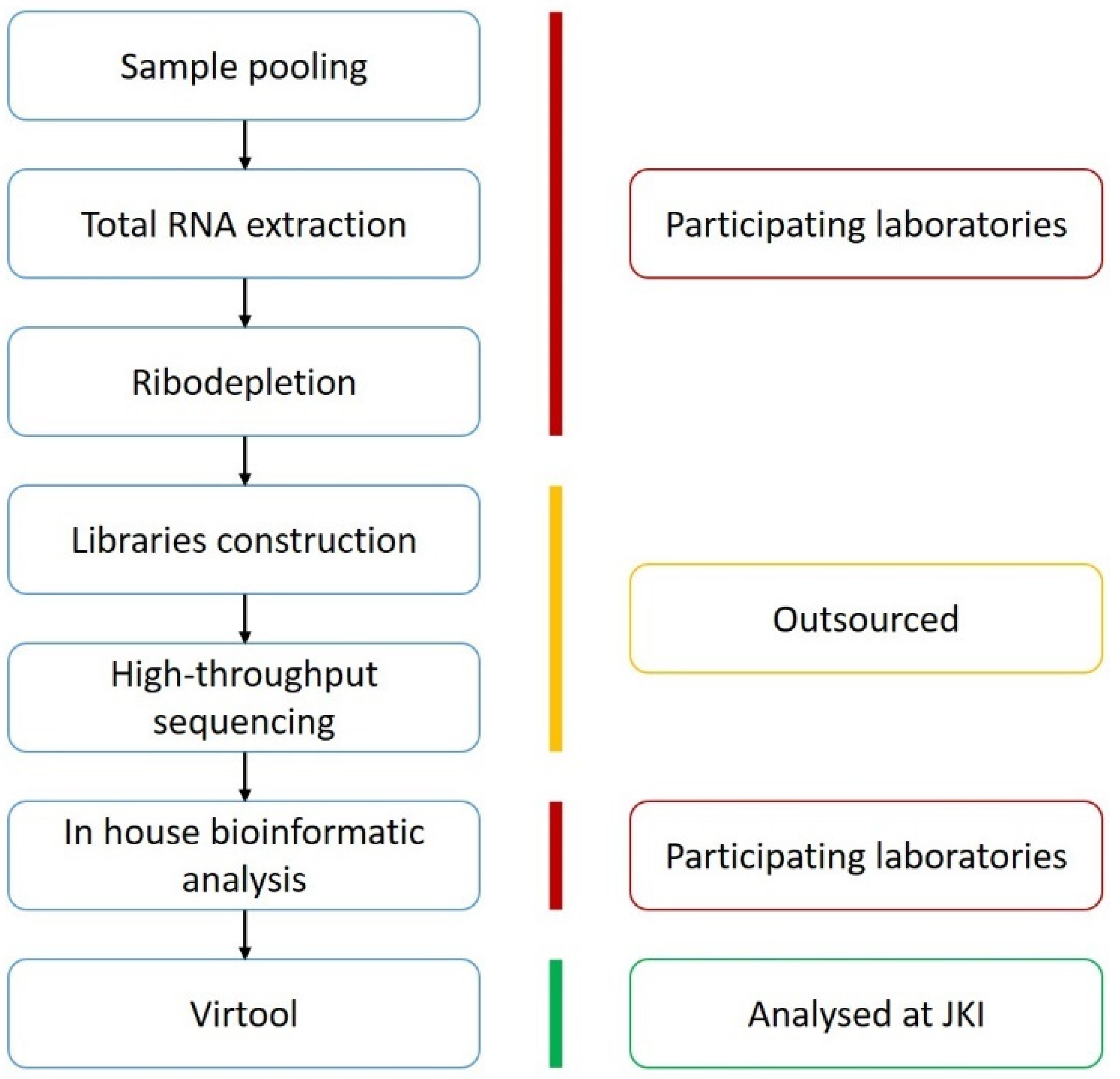

{kind=link}
{kind=link}
{kind=link}
| Lab ID | Pool 1 | Pool 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| totRNA Quantity (ng/μL) | Ribodepleted totRNA Quantity (ng/μL) | Number of Raw Reads | Q ≥ 30 | totRNA Quantity (ng/μL) | Ribodepleted totRNA Quantity (ng/μL) | Number of Raw Reads | Q ≥ 30 | |
| TPS_01 | 357.5 | 57.2 | 54,074,222 | 92.3 | 281.1 | 43.7 | 48,768,068 | 92.8 |
| TPS_02 | 318.6 | 11.8 | 19,541,014 | 91.9 | 146 | 13.3 | 15,829,278 | 91.1 |
| TPS_03 | 28.5 | 3.8 | 16,692,936 | 93.1 | 73 | 9.1 | 20,928,004 | 93.1 |
| TPS_04 | 73.8 | 4.2 | 20,388,244 | 91.6 | 237.6 | 16.9 | 22,487,248 | 92.1 |
| TPS_05 a | 73 | 5.7 | 75,831,846 | 83.2 | 86.8 | 4.2 | 79,993,710 | 84.1 |
| TPS_06 | 245 | 21.8 | 39,932,780 | 93.5 | 249 | 25.5 | 35,501,376 | 94.1 |
| TPS_07 | Nr b | 405 c | 12,948,820 | 93.3 | Nr b | 104 c | 9,871,160 | 92.6 |
| TPS_08 | 20 | 7.7 | 17,740,834 | 91.9 | 225 | 24 | 19,539,256 | 91.2 |
| Lab ID | Bioinformatic Analysis | Complete Analysis Date |
|---|---|---|
| TPS_01 | Geneious (Biomatters Limited): The raw reads were normalised using the BBNorm tool (version 38.84), quality trimmed, and size-filtered using Geneious. De novo assembly using the Geneious assembler (medium sensitivity/fast), followed by BLASTn search against virus/viroid sequences in the nt database on the NCBI non-redundant database (December 2018), was performed. | 15 December 2018 |
| TPS_02 | SGA, IDBA_UD, SSPACE, Bowtie2, bbmap, Biopython, and BLASTn. | 18 March 2019 |
| TPS_03 | CLC Genomic Workbench 11 custom-built pipeline (databases: NCBI viral RefSeq, November 2018; Pfam [v31]), DIAMOND (database: NCBI nr, June 2018). | 31 December 2018 |
| TPS_04 | Cutadapt v1.16, Prinseq v0.20.4, PEAR v0.9.8, SortmeRNA v2.0, VirusDetect v1.5 with databank v229, CLC Genomic Workbench 10, and BLASTn/BLASTx. | 13 November 2018 |
| TPS_05 | CLC Genomic Workbench 11, BLASTn, DIAMOND, and Krona (database September-2018). | 8 December 2018 |
| TPS_06 | FastQC, Rcorrector, trim_galore, Bowtie2, Trinity, Samtools, and R packages: Iranges, dplyr, and BLASTn. | 8 March 2019 |
| TPS_07 | VirusDetect v1.7 (Linux version) and BLASTn/BLASTx (database December 2018) | 17 August 2019 |
| TPS_08 | Geneious (Biomatters Limited): The raw reads were normalised using the BBNorm tool (version 38.84), quality trimmed, and size-filtered using Geneious. De novo assembly using the Geneious assembler (medium sensitivity/fast), followed by BLASTn search against virus/viroid sequences in the nt database on the NCBI non-redundant database (October 2018), was performed. | 30 November 2018 |
| Pool | Pool 1 | Pool 2 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Virus | ACMV | AV-1 | CGMMV | CLRV | ClYVV | CMoMV | LNYV | PEMV2 | PEMVsatRNA | PepMV | Detection % | AaNV a | CMV | CNDV | PhCMoV | ToMV | Detection Percentage | |
| Lab ID | TPS_01 | + | + | + | + | + | + | + | + | + | + | 100 | + | + | + | + | + | 100 |
| TPS_02 | + | + | + | + | − | + | + | − | − | + | 70 | − | + | − | + | + | 60 | |
| TPS_03 | + | + | + | + | + | + | + | + | + | + | 100 | − | + | + | + | + | 80 | |
| TPS_04 | + | + | + | + | + | + | + | + | − | + | 90 | − | + | + | + | + | 80 | |
| TPS_05 | + | + | + | + | + | + | + | + | + | + | 100 | − | + | + | + | + | 80 | |
| TPS_06 | + | + | + | + | + | + | + | + | − | + | 90 | + b | + | + | + | + | 100 | |
| TPS_07 | + | + | + | + | + | + | + | + | − | − | 80 | − | + | − | + c | + | 60 | |
| TPS_08 | + | + | + | + | + | + | + | + | + | + | 100 | + | + | + | + | + | 100 | |
| Pool | Virus | Accession Number | Closest nt Sequence |
|---|---|---|---|
| Pool 1 | ACMV | MW848516 and MW848517 | 97.3% and 95.4% X17095 and X17096 |
| AV-1 | MW848534 | 90.4% KJ830761 | |
| CGMMV | MW848531 | 99.7% MH271420 | |
| CLRV | MW848518 and MW848519 | 85.1% and 82.9% KC937022 and FR851462 | |
| ClYVV | MW848532 | 88.1% MN399730 | |
| CMoMV | MW848525 | 97.4% FJ188472 | |
| LNYV | MW848533 | 80.8% AJ867584 | |
| PEMV2 | MW848526 | 98.4% MN399707 | |
| PEMVsatRNA | MW848527 | 95.9% U03564 | |
| PepMV | MW848530 | 99.7% AJ606361 | |
| Pool 2 | AaNV | MW848524 | 99.9% MG948563 |
| CMV | MW848520, MW848521, and MW848522 | 99.9%, 99.9%, and 99.7% D00356, D00355, and D10538 | |
| CNDV | MW848523 | 76.9% NC_038320 | |
| PhCMoV | MW848528 | 97.5% KY706238 | |
| ToMV | MW848529 | 99.9% KY912162 |
| Number | Virus | Acronym | Genus | Family | Genome | Confirmation | Primers/antibodies | Pool | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | African cassava mosaic virus | ACMV | Begomovirus | Geminiviridae | ssDNA | Bipartite | PCR | [28] | 1 |
| 2 | Alfalfa-associated nucleorhabdovirus a | AaNV | Betanucleorhabdovirus | Rhabdoviridae | (−ve) ssRNA | Monopartite | ELISA | JKI-1607 | 2 |
| 3 | Asparagus virus 1 | AV-1 | Potyvirus | Potyviridae | (+ve) ssRNA | Monopartite | ELISA | JKI-44 | 1 |
| 4 | Carrot mottle mimic virus | CMoMV | Umbravirus | Tombusviridae | (+ve) ssRNA | Monopartite | RT-PCR | JKI-881 TACCCTAACATGTACGCCGC and JKI-882 GCGTTCAGATATTGCCGCTG | 1 |
| 5 | Carrot necrotic dieback virus | CNDV | Sequivirus | Secoviridae | (+ve) ssRNA | Monopartite | ELISA | JKI-45 | 2 |
| 6 | Cherry leaf roll virus (strain carrot) | CLRV | Nepovirus | Secoviridae | (+ve) ssRNA | Bipartite | ELISA | [29] | 1 |
| 7 | Clover yellow vein virus | ClYVV | Potyvirus | Potyviridae | (+ve) ssRNA | Monopartite | ELISA | JKI-98 | 1 |
| 8 | Cucumber green mottle mosaic virus | CGMMV | Tobamovirus | Virgaviridae | (+ve) ssRNA | Monopartite | ELISA | JKI-1773 | 1 |
| 9 | Cucumber mosaic virus | CMV | Cucumovirus | Bromoviridae | (+ve) ssRNA | Tripartite | ELISA | JKI-1745 | 2 |
| 10 | Lettuce necrotic yellows virus | LNYV | Cytorhabdovirus | Rhabdoviridae | (−ve) ssRNA | Monopartite | ELISA | JKI-2073 | 1 |
| 11 | Pea enation mosaic virus 2 b | PEMV2 | Umbravirus | Tombusviridae | (+ve) ssRNA | Monopartite | RT-PCR | [12] | 1 |
| 12 | Pepino mosaic virus | PepMV | Potexvirus | Alphaflexiviridae | (+ve) ssRNA | Monopartite | ELISA | JKI-1452 | 1 |
| 13 | Physostegia chlorotic mottle virus | PhCMoV | Alphanucleorhabdovirus | Rhabdoviridae | (−ve) ssRNA | Monopartite | ELISA | JKI-2051 | 2 |
| 14 | Tomato mosaic virus | ToMV | Tobamovirus | Virgaviridae | (+ve) ssRNA | Monopartite | ELISA | JKI-68 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaafar, Y.Z.A.; Westenberg, M.; Botermans, M.; László, K.; De Jonghe, K.; Foucart, Y.; Ferretti, L.; Kutnjak, D.; Pecman, A.; Mehle, N.; et al. Interlaboratory Comparison Study on Ribodepleted Total RNA High-Throughput Sequencing for Plant Virus Diagnostics and Bioinformatic Competence. Pathogens 2021, 10, 1174. https://doi.org/10.3390/pathogens10091174
Gaafar YZA, Westenberg M, Botermans M, László K, De Jonghe K, Foucart Y, Ferretti L, Kutnjak D, Pecman A, Mehle N, et al. Interlaboratory Comparison Study on Ribodepleted Total RNA High-Throughput Sequencing for Plant Virus Diagnostics and Bioinformatic Competence. Pathogens. 2021; 10(9):1174. https://doi.org/10.3390/pathogens10091174
Chicago/Turabian StyleGaafar, Yahya Z. A., Marcel Westenberg, Marleen Botermans, Krizbai László, Kris De Jonghe, Yoika Foucart, Luca Ferretti, Denis Kutnjak, Anja Pecman, Nataša Mehle, and et al. 2021. "Interlaboratory Comparison Study on Ribodepleted Total RNA High-Throughput Sequencing for Plant Virus Diagnostics and Bioinformatic Competence" Pathogens 10, no. 9: 1174. https://doi.org/10.3390/pathogens10091174
APA StyleGaafar, Y. Z. A., Westenberg, M., Botermans, M., László, K., De Jonghe, K., Foucart, Y., Ferretti, L., Kutnjak, D., Pecman, A., Mehle, N., Kreuze, J., Muller, G., Vakirlis, N., Beris, D., Varveri, C., & Ziebell, H. (2021). Interlaboratory Comparison Study on Ribodepleted Total RNA High-Throughput Sequencing for Plant Virus Diagnostics and Bioinformatic Competence. Pathogens, 10(9), 1174. https://doi.org/10.3390/pathogens10091174