Abstract
Epstein–Barr virus (EBV), a type 4 γ herpes virus, is recognized as a causative agent in nasopharyngeal carcinoma (NPC). Incidence of EBV-positive NPC have grown in recent decades along with worse outcomes compared with their EBV-negative counterparts. Latent membrane protein 1 (LMP1), encoded by EBV, induces NPC progression. The epidermal growth factor receptor (EGFR), a member of the ErbB family of receptor tyrosine kinases (RTK), is a driver of tumorigenesis, including for NPC. Little data exist on the relationship between EGFR and EBV-induced NPC. In our initial review, we found that LMP1 promoted the expression of EGFR in NPC in two main ways: the NF-κB pathway and STAT3 activation. On the other hand, EGFR also enhances EBV infection in NPC cells. Moreover, activation of EGFR signalling affects NPC cell proliferation, cell cycle progression, angiogenesis, invasion, and metastasis. Since EGFR promotes tumorigenesis and progression by downstream signalling pathways, causing poor outcomes in NPC patients, EGFR-targeted drugs could be considered a newly developed anti-tumor drug. Here, we summarize the major studies on EBV, EGFR, and LMP1-regulatory EGFR expression and nucleus location in NPC and discuss the clinical efficacy of EGFR-targeted agents in locally advanced NPC (LA NPC) and recurrent or metastatic NPC (R/M NPC) patients.
1. Epstein–Barr Virus (EBV)
1.1. Etiology of EBV
EBV was first described in 1958 by Dr. Denis Burkitt, who identified clusters of mandibular sarcomas in African children aged 2 to 14 [1]. Then, he met virologist Anthony Epstein by chance to find out if there was a viral cause of this malignant lymphoma. After years of research, Dr. Epstein provided essential insights into the association between EBV and human disease [1]. The EBV is a type 4 γ herpes virus, comprising lipoprotein capsules and 162 shell particles [2]. The genome comprises 190 kb double-stranded DNA, encoding about 100 open-reading frames [1,3]. Free circular EBV DNA usually exists in the cytoplasm of lymphocytes and can eventually be integrated into lymphocyte chromatin [2]. The latent EBV protein promotes the proliferation of host cells and boosts DNA replication of EBV in host cells by two means: infected B-cell proliferation or lytic virion production [4]. The existence of EBV culminates in disease.
1.2. EBV Causes Disease by Infecting B Cells and Epithelial Cells
It has been reported that EBV can infect both epithelial cells and B cells in vivo and shuttle back and forth between them [5]. This phenomenon is conducive to the multiplication and survival of EBV; however, the specific infection mechanisms of B lymphocytes and epithelial cells are quite different [6]. In B cells, EBV uses its envelope protein gp350 to bind to surface CD21 receptors, and gp42 to bind to HLA-II proteins to transform B cells into immortalized B lymphoblastic cell lines (LCLs). These establish a latent infection status [4,7], which depends on a group of potential genes, including six types of EBV nuclear antigens––EBNA 1, 2, 3A, 3B, and 3C and EBNA lead protein (EBNA-LP)––latent membrane proteins LMP1 and LMP2 (including LMP2A and LMP2B), EBV-encoded small RNAs (EBER1 and EBER2), and microRNAs (miRNAs) [1,2].
On the other hand, EBV infection of epithelial cells is much more complicated. The study showed that EBV-positive epithelial cells are bloodier than EBV-negative epithelial cells [8]. A recent study suggested that EphA2 and Neuropilin-1(NRP1) allow the EBV to enter epithelial cells [9,10], as does EBV gHgL, which interacts with the integrin complex αvβ6 and αvβ8 to trigger the fusion of the EBV envelope protein with the cell membrane [11].
The particles released by EBV-infected epithelial cells are rich in gp42, whereas the particles released by EBV in B cells are deficient. Interestingly, gp42 can affect the formation of the gHgL complex, thus preventing the EBV from entering epithelial cells. This bidirectional regulation promotes EBV shuttling between B cells and epithelial cells. It is worth mentioning that EBV infection cannot immortalize epithelial cells and requires direct epithelial-to-epithelial contact.
According to the diverse transcription of latent genes, there are four latencies. When EBERs are solely transcribed, it is type 0 latency, mainly occurring in dormant memory B cells [4]; when EBERS, EBNA1, and BARTS are simultaneously transcribed, it is type I latency, mainly in Burkitt’s lymphoma [12]; when EBER, EBNA1, LMPS, and BARTS are concurrently expressed, it is type II latency, commonly seen in Hodgkin’s lymphoma, gastric cancer, and NPC; and when EBERS, EBNA1, EBNA-LP, EBNA2, EBNA3A, EBNA3B, EBNA3C, and LMPs are synchronously transcribed, it is type III latency, primarily appearing in the condition of immunodeficiency [13]. Different transcriptional statuses provide an essential reference for the occurrence of various diseases.
1.3. EBV Infection can Promote the Progression of Nasopharyngeal Carcinoma
EBV infection is common worldwide, with a prevalence of 80–95%, depending on the geographical area [14]; however, people are predominantly asymptomatic [14]. Both the means of infection and the host factors may affect the outcomes of EBV infection, which mainly infects the host through lymphocytes and epithelial cells [2,3,15]. For example, infection in adolescents can lead to infectious mononucleosis, with manifestations of fever, sore throat, lymphadenectasis, and splenomegaly [16]. Burkitt’s lymphoma (BL) and classical Hodgkin’s lymphoma (HL) are related to the EBV infection of lymphocytes, whereas nasopharyngeal and gastric cancers are associated with the infection of epithelial cells [2]. In particular, NPC is a frequently reported tumor in the southeastern provinces of China, and almost 98% of NPCs are closely related to EBV infection [5].
A study of molecular mechanisms proved that the invasion and migration ability was significantly up-regulated in EBV-positive NPC cells. At the same time, the tumor formation ability of EBV-positive NPC cells was significantly higher than EBV-negative NPC cells in nude mice. In short, the malignant degree of nasopharyngeal carcinoma cells was increased in the presence of EBV in both in vivo and in vitro experiments [17]. Meanwhile, substantial scientific studies have shown that the level of free EBV DNA in the plasma of NPC patients is highly correlated with the prognosis. Patients with low EBV DNA content have shown a better prognosis, which has excellent clinical guiding significance. EBV has been used as a clinical marker in the clinical diagnosis of NPC [18].
1.4. LMP1 Protein Encoded by EBV Is Involved in the Progression of Nasopharyngeal Carcinoma
LMP1 is a membrane protein encoded by EBV, consisting of a short amino acid N-terminal, six hydrophobic alpha-helical transmembrane regions, and a large 200-amino acid cytoplasmic C-terminal tail [19]. LMP1 contains several domains, and the C-terminal which contains three functional domains, CTAR1, CTAR2, and CTAR3. Only CTAR1 uniquely induces several cellular genes, including the epidermal growth factor receptor (EGFR), TRAF1, ICAM1, and EBI3 [20]. LMP1 contributes to the development and progression of NPC by many mechanisms, including the regulation of the expression and phosphorylation of the transcription factor p53 [21], the EGFR and the STAT3 activation allowance [22], as well as necroptosis inhibition through the RIP3 promoter hypermethylation [23].
2. Epidermal Growth Factor Receptor (EGFR)
2.1. Biological Function of EGFR
EGFR (also known as ErbB1 or HER1) is a 170kDa single transmembrane glycoprotein of the receptor tyrosine kinase family (RTKs), and a member of the ErbB family (EGFR, HER2, HER3, and HER4), and is essential for epithelial cell biology [24,25]. ErbB family receptors contain an extracellular N-terminus extracellular ligand-binding domain, a hydrophobic transmembrane domain, and a conserved cytoplasmic C-terminus tyrosine kinase domain [26]. Growth factors could activate ErbB by autocrine or paracrine signalling [27]. Because the ligand binds to the extracellular domain of ErbB, it induces receptor homodimer or heterodimer formation, which phosphorylates tyrosine residues in the cytoplasmic tail, which then activates the intracellular tyrosine kinase domain [28]. Indeed, the autophosphorylation of ErbB family members is a critical step in downstream signal transduction that affects cell survival, proliferation, angiogenesis, migration, inflammatory responses, and oncogene expression [29,30].
Currently, the known downstream signalling cascades of EGFR follow five pathways: (1) Ras/Raf/MEK/mitogen-activated protein kinase (MAPK)/ERK, (2) phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR, (3) protein kinase C (PKC), (4) Src, and (5) Jak/STAT (Figure 1) [31,32,33]. In addition, the canonical receptor-dependent EGFR signalling pathway and the ligand-independent and tyrosine kinase-independent EGFR novel mechanisms were recently discovered, revealing that EGFR also plays a regulatory role in autophagy and metabolism [34]. In short, both EGFR and the EGFR downstream signalling pathways are necessary for cell fate and influence tumorigenesis.
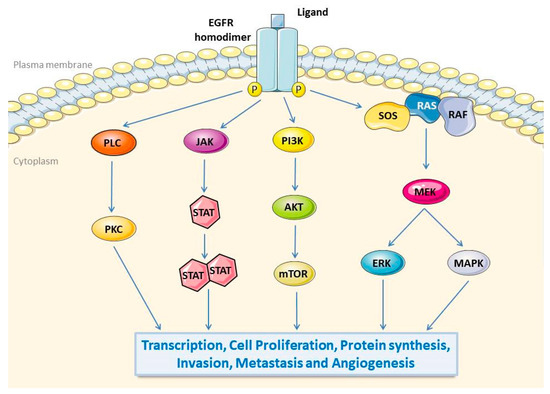
Figure 1.
EGFR signalling pathways. The binding of ligands in the extracellular domain of EGFR induces the formation of dimers and then phosphorylate tyrosine residues in the intracellular domain, leading to activated downstream signal transduction. EGFR intracellular signalling cascades include: (1) PKC pathway, (2) JAK/STAT pathway, (3) PI3K/Akt/mTOR pathway, (4) Ras/Raf/MEK/MAPK/ERK pathway. Subsequent EGFR downstream pathways affect gene transcription, proliferation, protein synthesis, invasion, metastasis, and angiogenesis. EGFR, epidermal growth factor receptor; PKC, protein kinase C; JAK, Janus kinase; STAT, signal transduction and transcriptional activator protein; PI3K, phosphoinositide-3 kinase; Akt, v-akt murine thymoma viral oncogene homolog 1; mTOR, mammalian target of rapamycin; Ras, retroviral associated DNA sequence; Raf, v-Raf 1 murine leukemia viral oncogene homologue 1; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase.
2.2. EGFR Promotes Tumorigenesis and Progression
EGFR alterations are common in many malignant tumors such as lung, breast, stomach, and colorectal cancer and glioblastoma [32]. The dominating triggers of EGFR activation in tumor tissues are EGFR gene amplification and point mutations. Transcriptional upregulation or ligand overproduction caused by autocrine/paracrine secretion has also been shown [35]. Overexpression of the EGFR can lead to a poor prognosis, drug tolerance, tumor metastasis, and a low survival rate [36,37,38]. As mentioned above, the ligand stimulates the homologous or heterologous dimerization of the receptor. It ultimately leads to the activation of the kinase, which contributes to enhanced EGFR signalling and promotes tumor development [37,39]. Because the EGFR is frequently mutated and overexpressed in tumors, it is also a promising therapeutic target for a host of tumors in clinical trials [35]. In brief, EGFR signalling is correlated with tumor proliferation, invasion, and metastasis [40].
The dimerization of ErbB allows intracellular signal transduction and tumorigenesis. The most effective of all the ErbB dimers is the ErbB-2/ErbB-3 heterodimer complex. ErbB-2/ErbB-3 dimers promote cell proliferation and transformation in vitro. They are further involved in the pathogenesis of lung cancer and breast cancer [41]. Considering the components of the heterodimer ErbB-2/ErbB-3, ErbB-2 is an excellent partner for forming a heterodimer because it is a ligand-less co-receptor; ErBb-3, a kinase-activated impaired receptor, cannot be internalized and degraded in lysosomes to prolong signalling and promote cell transformation [42].
Furthermore, ErbB-2/ErbB-3 has more vital signal transduction ability because it transmits proliferation signals not only through the Ras/Raf/MAPK pathway but also the PI3K/Akt/mTOR pathway [39]. Since a heterodimer has a weaker binding strength compared to a homodimer, it is easy to dissociate in the endosome. In addition, it cannot recruit the E3 ligase Cbl to initiate endocytosis to degradation, so the heterodimer is enhanced, which makes it more tumorigenic than the homodimer [35]. Obviously, in tumor signal transduction, ErbB heterodimers, particularly ErbB-2/ErbB-3, are more effective than homodimers.
3. The Mechanism of LMP1-Mediated EGFR Expression and Nuclear Translocation
The EBV can encode a protein mass to play a role in tumorigenesis. Existing studies have shown that only LMP1 takes part in the activation of the EGFR in NPC. Initially, Miller et al. found that EGFR expression was significantly increased when LMP1 was stably expressed in epithelial cells, which could be activated by the EGF ligands [43]. Further studies suggested that the expressions of LMP1 further led to the growth and differentiation of epithelial cells. However, regardless of its levels, LMP1 could not affect the EGFR level in B cells [43]. To clarify the relationship between LMP1 and EGFR in exosomes, researchers separated the exosomes of C666 and C666–LMP1 cells. They found that the EGFR content increased significantly after the overexpression of LMP1 [44]. More interestingly, studies found that LMP2A can inhibit the expression of LMP1 and the activation of the NF-κB pathway [45].
It is known that there are three domains in the C-terminal of LMP1, but which are involved in this process? Subsequent studies found that the CTAR1 domain at the carboxyl end of LMP1 can drive the up-regulation of the EGFR, while the CTAR2 domain cannot [46]. In-depth research found that the EGFR levels of CTAR1-solely expressed C33A cells are significantly higher than LMP1-expressed C33A cells, indicating that the presence of CTAR2 may impede the ability of CTAR1 [46]. We increased the expression of LMP1 by Tet-on in HNE2 cells then measured the EGFR expression in the nucleus by a double immunofluorescent stain using a fluorescein isothiocyanate (FITC)-conjugated anti-EGFR antibody and enhanced green fluorescent protein. The study further found that LMP1 can promote EGFR expression and facilitate the nuclear localization signal (NLS)-mediated nuclear translocation of the EGFR independent of the enhanced green fluorescent protein [47,48].
Studies have shown that LMP1 mediates the expression of the EGFR in nasopharyngeal carcinoma in several ways, including via the NF-κB pathway and STAT3 activation (Figure 2).
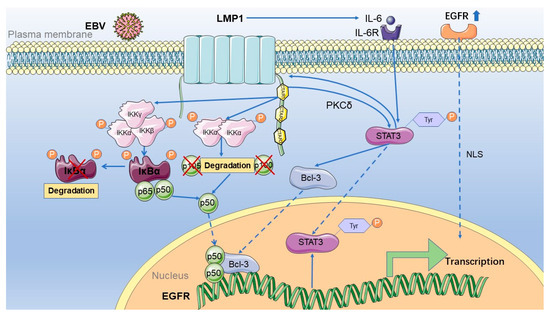
Figure 2.
The mechanism of EGFR activation by LMP1 in nasopharyngeal carcinoma. LMP1 activates the EGFR mainly in two ways: first, the expression encoded by EBV activates the NF-κB classical and non-classical pathways, and then p50 can enter the nucleus. Meanwhile, LMP1 can regulate the phosphorylation of STAT3 tyrosine 705, which PKCδ mediates. It then activates the expression of Bcl-3 and promotes it to enter the nucleus. Indeed, p50 and Bcl-3 can form a trimer or dimer, binding to the EGFR promoter to activate its transcription. Activated STAT3 can then bind to the LMP1 promoter to regulate the expression of LMP1. At the same time, IL-6 downstream of the NF-κB pathway activated by LMP1 can further activate STAT3 after binding to its receptor. Moreover, STAT3 then binds to the LMP1 promoter to promote the expression of LMP1, forming a positive autoregulatory loop. STAT3 is not only regulated by LMP1 but also regulated by the activation of EGFR. IKKα, IkappaB kinase-alpha; IKKβ, IkappaB kinase-beta; IKKγ, IkappaB kinase-gamma; IκBα, IkappaBalpha; LMP1, latent membrane protein 1; EBV, Epstein–Barr virus; Bcl-3, B-cell lymphoma 3; STAT3, signal transducer and activator of transcription 3; EGFR, epidermal growth factor receptor; NLS, nuclear localization protein; CTAR, carboxyl-terminal activating regions; IL-6, interleukin 6; IL-6R, interleukin 6 receptor.
3.1. LMP1 Activates the EGFR through the NF-κB Pathway in NPC
NF-κB is a family of transcription factors regulating various biological processes, including inflammation, apoptosis, cell cycle, and cell migration [49]. The mammalian NF-κB transcription factor family consists of five members: p65 (RelA), c-Rel, RelB, p50, and p52, all containing a Rel homology domain [50]. These five transcription factors can form dimers with each other and bind to specific sites in DNA [51]. Among them, only p50/p65 physiologically exist in the cytoplasm due to the existence of IkBα. However, when some stimuli cause IkBα to degrade, p50/p65 dimers can enter the nucleus. This pattern is the classic NF-κB pathway [52], in addition to which there is a nuclear translocation regulation (non-classical) pathway where p50 and p52 are restricted in the cytoplasm by their precursor proteins p105 and p100, respectively. After p105 and p100 are cleaved, p50 and p52 can enter the nucleus [53,54].
LMP1 contains three domains, CTAR1, CTAR2, and CTAR3; however, only CTAR1 is involved in the induction of the EGFR promoter, while CTAR2 cannot mediate the EGFR promoter. What type of NF-κB is activated by LMP1? It has been reported that both of them can activate the NF-κB pathway: LMP1–CTAR2 activates the classical NF-κB signal, and LMP1–CTAR1 induces more complex NF-κB signalling, including classical and non-classical pathways [52,55]. The classical NF-κB pathway contains the IKKα, IKKβ, and IKKγ enzymes. By contrast, the non-classical NF-κB pathway contains the enzyme IKKα activated by NIK. Studies have shown that the knock-out of IKKα, IKKβ, and IKKγ does not affect LMP1-mediated EGFR up-regulation. However, the EGFR could be down-regulated after the knock-out of NIK, proving that the EGFR expression induced by LMP1 is not necessarily dependent on the classical and alternative proteasome-dependent NF-κB pathway [52].
Specifically, the two domains of the C-terminal of LMP1, CTAR1, and CTAR2, can activate different NF-κB transcription factor dimers, respectively. CTAR1 activates the p50/p50, the p50/p52, and the p52/p65 dimers, but CTAR2 only activates the p52/p65 dimer [56], which initially inhibits transcription but can acquire the transcriptional activity after combining with B-Cell Chronic Lymphocytic Leukaemia/Lymphoma-3 (Bcl-3) [57]. The level of p50 in the nucleus is significantly up-regulated by LMP1 and LMP1–CTAR1, which verifies that LMP–CTAR1 could induce the nuclear translocation of p50 [58], and that the p50 homodimer could then bind to the NF-κB binding site in the EGFR promoter [59]. The p50/Bcl-3 complex can bind to the promoter region of the EGFR. In short, LMP1 activates the NF-κB pathway to form p50 dimers, thus activating Bcl-3, and then forming the p50/Bcl-3 complex that binds to the EGFR promoter region to up-regulate EGFR expression [43,60]. In the analysis of NPC clinical samples, the expression level of p50 in the nucleus was significantly higher than in low LMP1 level samples, which supports the previous hypothesis [43]. Detections of C33A cells and NPC tissues by chromatin immunoprecipitation (ChIP) verified that p50 molecules can bind to the promoter region of the EGFR, making the molecular mechanism clear [51]. Interestingly, after knocking down p105 and p50, the expression level of Bcl-3 was significantly increased. The formation of Bcl-3 and p50 was increased, suggesting that p105 or p50 may have a negative effect on Bcl-3 [58]. In conclusion, LMP1 activates EGFR transcription by the p50/ Bcl-3 complex. Further studies are required to determine the links between other signal-transducing complexes and their contributions to NPC.
3.2. LMP1 Activates EGFR through STAT3 in NPC
The activation of STAT3 (signal transcription and signal activator 3) is related to a variety of epithelial and lymphatic system malignancies, such as breast cancer [61], multiple myeloma [62], and NPC [63]. Its transcriptional activity is regulated by phosphorylation. In particular, phosphorylation of STAT3 tyrosine 705 can induce Bcl-3 dimerization, while phosphorylation of STAT3 serine 727 can affect DNA binding and transcriptional activity [46]. However, inhibiting PKCδ by Rottlerin (PKCδ inhibitor) decreased LMP1–CTAR1-induced serine phosphorylation but not tyrosine phosphorylation, even though the EBV-encoded LMP1, can activate STAT3, which requires PKCδ [64]. The results of the two studies contradicted each other. Further research is needed to clarify this dilemma.
LMP1–CTAR1 can further upregulate the EGFR after STAT3 activation because CTAR1 promotes the tyrosine phosphorylation and activation of STAT3, which further induces the expression of Bcl-3 and the nuclear translocation of Bcl-3 and p50. Bcl-3 binds with p50 as a transcriptionally active complex to activate the EGFR expression [46]. Interestingly, activated STAT3 could bind to the L1–TR LMP1 promoter in the nucleus. Therefore, LMP1 expression was upregulated through the JAK–STAT pathway, and STATs predisposed the cell to EBV-driven tumorigenesis [65].
Meanwhile, LMP1 increases IL-6 synthesis by the NF-κB pathway. IL-6 further mediates tyrosine phosphorylation of STAT3 after binding to its receptor, after which the activated STAT3 binds to the LMP1 promoter to promote the expression of LMP1, forming a positive autoregulatory loop [66]. Moreover, STAT3 is not only regulated by LMP1 but also by the activation of the EGFR [67]. ChIP was used to detect NPC clinical samples and C33A cells and verified that Bcl-3 molecules could bind to the EGFR promoter region [58]. In short, the activation of STAT3 and Bcl-3 is critical to the transcription of the EGFR. Crosstalk between STAT3 activation and NF-κB was found in the presence of Bcl-3, but the mechanism is still not entirely clear.
3.3. Others
The ChIP analysis of CTAR1-expressing C33 cells indicated that both PIK3R1 and PIK3R3 bound to Bcl3, but the pattern remains unknown [20]. There are still various mechanisms to be investigated that are associated with LMP1 and the EGFR.
4. The Role of EGFR Pathways in Nasopharyngeal Carcinoma
4.1. The Relationship between EGFR and Nasopharyngeal Carcinoma
EGFR mutation recurs in various tumors except for NPC, and EGFR overexpression is quite common in NPC [68]. The TCGA analysis revealed that patients with a high expression of EGFR mRNA had a poorer prognosis than those with a low expression [69]. In addition, the EGFR was co-expressed with LMP1 in most NPC tissues examined by immunostaining and in situ hybridization experiments [70,71]. Several studies also showed that EGFR expression correlated with the advanced tumor node metastatic stage, clinical stage, and distant metastatic state of NPC patients by analyzing a cohort of clinical samples [72]. Consequently, it is a potential prognostic biomarker for advanced-stage patients with a poor outcome [73].
4.2. EGFR Signalling Affects the Growth of Nasopharyngeal Carcinoma Cells
In NPC cells, the abnormal expression of EGFR regulates the cell cycle and tumor growth by related genes. For example, in LMP1-positive NPC tissues of elderly individuals, the expression of the EGFR was closely related to the high enrichment of p53 in the nucleus and the expression of Bcl-2, suggesting that the up-regulation of the EGFR or Bcl-2 was associated with a poor prognosis and resistance to chemotherapy-induced apoptosis [74]. As mentioned above, LMP1 promotes EGFR binding to the promoter region of cyclin E and cyclin D1, thus accelerating the G1/S phase transition of cells [22,47]. In addition, microRNAs (miRNAs) also influence the proliferation of NPC cells through the EGFR pathway. For instance, VPS33B (vacuolar protein sorting 33B) up-regulates miR-133A-3 to suppress cell growth and induce cycle arrest by the EGFR/PI3K/Akt/c-myc/p53 pathway. Interestingly, p53 induces the expression of miR-133a-3p- to form a feedback loop. Therefore, VPS33B may be a new molecule target for developing novel NPC therapeutic methods [75].
4.3. EGFR Promotes Invasion and Metastasis of Nasopharyngeal Carcinoma Cells
EGFR signalling can promote PKM2 nuclear translocation, which stimulates the transcription of FosL1, ANTXR2, CCND1, cyclin D1, and c-Myc genes. The expression of these genes promotes cell cycle progression and the Warburg effect, which enhances the invasion and metastasis ability of NPC cells [72,76]. Moreover, the highly conserved transcription factors Forkhead box Q1 (Foxq1), regulated by miR-124, could directly bind to the EGFR promoter and increase EGFR expression, thereby inducing vasculogenic mimicry via the EGFR signalling pathway to promote NPC metastasis [77,78]. Similarly, the overexpression of LACTB (serine beta-lactamase-like protein) promotes NPC cell motility in vitro and metastasis in vivo, depending on the activation of ErbB3/EGFR–ERK signalling, which is not conducive to the survival of NPC patients [79]. On the contrary, the overexpression of PTPN12 (protein tyrosine phosphatase nonreceptor type 12) in NPC cells has decreased EGFR expression. It has enhanced caspase3 activity, preventing the proliferation and invasion of tumor cells [80]. Overall, the EGFR and EGFR signalling pathways play an essential role in the invasion and metastasis of NPC.
Recently, research showed that highly metastatic NPC cells secrete EGFR-rich extracellular vesicles (EVs) that can be absorbed by poorly metastatic NPC cells. Then, EGFR-rich EVs promote EGFR up-regulation and intracellular ROS reduction through the EGFR/PI3K/Akt pathway, thus aggravating the metastasis and progression of NPC [81]. Undoubtedly, EVs delivering EGFR or EGFR ligands promote angiogenesis, metastasis, and osteoclastogenesis in tumors, modulating the immune system and blocking these EVs’ activities to reduce drug resistance [82,83]. Therefore, the EGFR may be a favorable indicator for the progression of NPC, which would be beneficial for exploiting new anti-metastatic medicine for advanced NPC therapies. However, it remains to be studied [84].
4.4. Other Roles of EGFR in Nasopharyngeal Carcinoma
DLC-1 (liver cancer-1) can inhibit NPC proliferation, metastasis, and deterioration and is a candidate of NPC tumor suppressor gene. Research into the mechanisms behind the opposing roles of DLC-1 in NPC cells confirmed that the ectopic expression of DLC-1 can induce mitochondrial apoptosis. Furthermore, it also inhibits EMT and other related processes through the EGFR/Akt/NF-κB pathway [85]. This undoubtedly lays a foundation for clinical NPC application [86]. Another study suggested that curcumin inhibits the EGFR, STAT3, and growth factor receptor-bound protein 2 (GRB2) via the circRNA–miRNA–mRNA network, thereby enhancing the radio-sensitization of NPC [87]. The clinical application of anti-EGFR drugs has profound significance. However, the functions of the EGFR in NPC need further research.
5. EGFR Effects on EBV Infections
5.1. EGFR Is Overexpressed in EBV-Infected Cells
EBV infections induce many EGFR downstream pathways; however, current research has demonstrated that EGFR also gives rise to EBV infection. Studies reported that the EGFR level in EBV-positive NPC patients is significantly higher than in EBV-negative patients. In particular, EGFR and STAT3 have been detected in the nasopharyngeal tissues of EBV-positive patients, and they are up-regulated in the nucleus of epithelial cells and inflammatory cells in EBV-positive chronic nasopharyngitis patients [17,88]. In addition, LMP1 can induce STAT3 phosphorylation, EGFR expression and nuclear accumulation, EGFR and STAT3-dependent inducible nitric oxide synthase (iNOS) expression, and subsequent DNA damage. Thus, it is believed that EGFR and STAT3-dependent pathways play a crucial role in EBV-related tumors [88]. In conclusion, the EGFR may promote the neoplastic transformation of EBV-positive cells.
5.2. EGFR Enhances the Internalization and Fusion of EBV
Neuropilin 1 (NRP1) is a co-receptor of class III semaphorins and various growth factors such as EGF, VEGR, TGF-β, and FGF, which synergistically increase receptor tyrosine kinase activity [89]. After EBV contacts epithelial cells, NRP1 can directly interact with EBV gB, a conserved glycoprotein required for membrane fusion in herpesviruses, then recruit EGFR and EGF-binding receptors to up-regulate the expression of NRP1. Subsequently, EBV could activate the EGFR/Akt and EGFR/ERK pathways. Consequently, the EGFR enhances EBV infection by promoting it to enter epithelial cells via macropinocytosis and lipid raft-dependent endocytosis. Hence, the EGFR knock-out could partially inhibit EBV infections that provide an opportunity for NPC treatment [10]. Furthermore, pathogen-ErbB ligation and the ErbB receptor signalling pathway can contribute to the cellular entry of microbes. However, intracellular organisms, such as the EBV, may require ErbB signalling cascades for self-propagation [33]. In general, the EGFR is necessary for the internalization and fusion of the EBV in NPC cells and perhaps can enhance its survival in the host.
6. The Role of EGFR Targeting in Nasopharyngeal Carcinoma
Radiotherapy is the primary treatment for the early diagnosis of NPC, and palliative chemotherapy is often used for advanced NPC since EGFR overexpression promotes tumorigenesis and progression via downstream signalling pathways, causing poor outcomes in NPC patients. EGFR-targeted drugs could be considered a newly developed anti-tumor drug for NPC. Anti-EGFR therapy has been extensively applied in the treatment of NPC and has achieved a better therapeutic effect in recent years [90]. Current therapies targeting the EGFR in tumors include monoclonal humanized antibodies (mAb) that target the extracellular domain of the receptor (e.g., Cetuximab (CTX), Nimotuzumab (NTZ), and Panitumumab) and selective small-molecule inhibitors that target the tyrosine kinase domain, such as Gefitinib, Erlotinib, and Afatinib, PI3K inhibitors and antisense gene therapy [32,91].
Clinical trials have shown that EGFR-targeted therapy (CTX and NTZ) based on palliative chemotherapy for recurrent or metastatic NPC (R/M NPC) patients significantly increase the chance of progression-free survival (PFS). Moreover, toxic and non-toxic side effects are within a controllable range, suggesting the importance of anti-EGFR therapy in NPC treatment [92]. CTX and Panitumumab are the most widely used, neutralizing monoclonal antibodies, and inhibit receptor activation and downstream signalling. Furthermore, a combination of anti-EGFR antibodies is more effective than single antibodies [93]. Different subtypes of nasopharyngeal carcinoma with various genomic profiling show diverse outcomes for targeted therapy (Table 1) [94].

Table 1.
EGFR-targeted drugs in nasopharyngeal carcinoma.
6.1. EGFR Monoclonal Antibody
Studies have shown that palliative chemotherapy combined with an EGFR monoclonal antibody could significantly prolong overall survival (OS) and PFS of R/M NPC patients. Specifically, for those with locally advanced NPC (LA NPC), EGFR-targeted agents combined with induction chemotherapy have significantly higher disease-free survival (DFS) rates and fewer side effects. Meanwhile, shorter-use cycles of CTX and NTZ (meaning lower cost) in this treatment, compared to EGFR-targeted agents combined with concurrent chemoradiotherapy (CCRT), are instructive for the treatment of LA-NPC [97]. Palliative chemotherapy combined with EGFR treatment did not significantly improve OS with early R/M NPC [98]. Nevertheless, XTC-treated patients have a longer PFS than those who are NTZ-treated, and have an acceptable overall toxicity. Specifically, the toxic effects in patients treated with CTX are more common than in patients treated with NTZ [92]. The curative effect of the EGFR monoclonal antibody combined with radiotherapy, chemotherapy, and CCRT is equivalent to induction chemotherapy combined with CCRT. However, the combination with a monoclonal antibody has fewer side effects [110].
6.1.1. Cetuximab for Nasopharyngeal Carcinoma
Cetuximab, an EGFR-targeted drug that has entered clinical trials, plays an anti-tumor role by binding to the extracellular domain of the EGFR. Poly-ICLC is an immune adjuvant often used to activate mature DC cells. Studies have shown that the increase of mature DC cells caused by Poly-ICLC in combination with EGFR-targeted drugs can increase EGFR-targeted CD8+ T cells in NPC cells, thus providing a better prognosis for NPC [95]. LA NPC patients can benefit from CCRT combined with CTX treatment, and the benefit is undeniable in the T4N3 group. However, its toxic side effects are more significant [96]. In R/M NPC patients, the clinical trial demonstrated that Paclitaxel Carboplatin plus Cetuximab (PCE) could achieve a 58.3% ORR rate, with better efficacy and acceptable side effects. It is worth mentioning that patients can receive this treatment on an outpatient basis [99]. The combination of CTX has become one of the first-line treatment options for patients with advanced nasopharyngeal carcinoma.
6.1.2. Nimotuzumab for Nasopharyngeal Carcinoma
Nimotuzumab (NTZ), an EGFR-targeted drug, plays an anti-tumor role mainly by binding to the extracellular domain of EGFR and inhibiting EGF binding [111]. NTZ inhibits proliferation and promotes apoptosis and anti-vascular survival in EGFR overexpressing tumors. It is worth mentioning that the cutaneous and mucosal toxicity of NTZ is significantly lower than other EGFR-targeted agents [112]. Clinical trial results show that NTZ combined with CCRT can significantly improve a patient’s OS. However, the high cost of NTZ limits its clinical benefit [113,114]. For stage II and III NPC, NTZ combined with first-line chemoradiotherapy can significantly improve the prognosis, which is reflected in the OS improvement, DFS, and distant metastasis-free survival rate of patients [100,115]. For stage II-IVb NPC with a high EGFR expression, patients treated with CRT+NTZ/CTX exhibited better distant metastasis-free survival (DMFS) [116]. Nowadays, palliative chemotherapy is a first-line treatment for R/M-NPC patients for whom palliative care is required [100,101,115]. Therefore, NTZ combinations have become one of the first-line treatment options for advanced NPC patients.
6.2. Small Molecule EGFR Tyrosine Kinase Inhibitors (TKIs)
TKIs inhibit EGFR activation chiefly by binding to the tyrosine domain of the EGFR. Among the small-molecule inhibitors of the tyrosine kinase, experiments have shown that Lapatinib can inhibit the invasion and proliferation of nasopharyngeal carcinoma cells and promote their apoptosis [117].
In addition to inhibiting the kinase, Gefitinib also increases the formation of the inactive EGFR dimer, suggesting that Gefitinib could induce faster endocytosis and dimer degradation. Gefitinib has been shown to preferentially target cancer stem-like cells (CSC) and eliminate tumor regrowth in vivo and in vitro for NPC patients with CSC [91,102].
The mechanism of Erlotinib is to compete with adenosine triphosphate to bind the intracellular catalytic domain of EGFR and thereby inhibit EGFR phosphorylation [107]. CNE2 cells treated with Erlotinib or Cisplatin inhibit cell viability and migration ability, increase apoptosis, and enhance tumor sensitivity to radiotherapy/chemotherapy, meaning that Erlotinib and Cisplatin weaken the radiotherapy/chemotherapy resistance of tumor cells [105].
Afatinib inhibits EGFR and ErbB-2 tyrosine kinase activity and suppresses NPC cell proliferation by arresting the cell cycle [108]. The combination of Afatinib and Gemcitabine (GEM) have shown significant anti-tumor efficacy in NPC xenograft models [108,109]. In addition, Erlotinib and Afatinib could enhance the sensitivity of tumors to chemoradiotherapy by inhibiting DNA damage repair [105,108]. However, no clinical trials have shown that TKIs can significantly improve the prognosis of NPC patients [107,118]. Therefore, the use of TKIs is limited to NPC patients.
7. Conclusions
The EBV is a type 4 γ herpes virus, consisting of lipoprotein capsules and 162 shell particles [2,15]. Incidence of EBV-positive NPC have grown in recent decades, with worse outcomes than for their EBV-negative counterparts. The malignant degree of nasopharyngeal carcinoma cells increased in the presence of the EBV in both in vivo and in vitro experiments [17]. Now, it is being used as a clinical marker in the clinical diagnosis of NPC [18]. LMP1 is a membrane protein encoded by the EBV, consisting of a short amino acid N-terminal, six hydrophobic alpha-helical transmembrane regions, and a large 200-amino acid cytoplasmic C-terminal tail [19].
The EGFR is necessary for cell development and homeostasis, the overexpression of which is common in many malignant tumors, and is a driver of tumorigenesis in various cancers, including NPC [35]. As mentioned above, the EGFR forms homodimers or heterodimers by binding to the ligands, phosphorylating and activating the tyrosine kinase domain in the cytoplasm. Afterwards, the EGFR activates downstream signal transduction, such as the PI3K/Akt/mTOR, JAK/STAT, and Ras/Raf/MAPK pathways. Ultimately, these EGFR pathways affect cell survival, proliferation, oncogene transcription, and other cancer-associated reactions.
Subsequent studies found that the CTAR1 domain at the carboxyl end of LMP1 can drive EGFR up-regulation, while the CTAR2 domain cannot [46,58]. In-depth research found that EGFR levels of CTAR1-solely-expressed C33A cells are remarkably higher than LMP1-expressed C33A cells, indicating that the presence of CTAR2 may impede the ability of CTAR1 [46]. Specifically, LMP1 up-regulates EGFR expression and promotes the nuclear translocation of the EGFR through the NF-κB pathway and the activation of STAT3. On the one hand, LMP1 activates the NF-κB pathway to form p50 dimers, activating Bcl-3, and then forms the p50/Bcl-3 complex, which binds to the EGFR promoter region to up-regulate EGFR expression [43,60]. On the other hand, LMP1–CTAR1 can further upregulate the EGFR after STAT3 activation. This occurs when CTAR1 promotes the tyrosine phosphorylation and activation of STAT3, which further induces the expression of Bcl-3, which then induces the nuclear translocation of Bcl-3 and p50. Bcl-3 binds with p50 as a transcriptionally active complex to activate EGFR expression [46,51]. Nevertheless, it is unknown whether EBV can regulate the EGFR through other encoded proteins.
The EGFR is overexpressed in NPC. Several studies confirmed that its expression is strongly related to NPC cell proliferation, invasion, metastasis, and pathogenesis. The latest research showed that the EGFR enhances the internalization and membrane fusion of the EBV in NPC cells. However, the findings of EGFR-assisted EBV infection are rarely known [10]. Moreover, the entry and invasion of pathogens depend on ErbB family receptors. Thus, other receptors in the ErbB family may drive EBV infection and pathogen-induced cellular transformation [33]. Further study on the relationship between ErbB receptors and EBV infections is needed.
The overexpression of the EGFR is common in NPC. Meanwhile, it promotes tumorigenesis and progression via the EGFR signalling pathway, thus reducing NPC patient survival rates. EGFR-targeting drugs could be considered as NPC anti-tumor drugs, and recently, therapies targeting the EGFR in NPC have included monoclonal humanized antibodies (CTX, NTZ and Panitumumab), selective small molecule inhibitors (Gefitinib, Erlotinib, and Afatinib), PI3K inhibitors, and antisense gene therapy [32,91]. Many clinical studies have shown that monoclonal antibodies against the EGFR can significantly prolong the OS and PFS of middle and advanced NPC patients. However, no clinical studies have shown that selective TKIs can improve the prognosis of patients with middle and advanced NPC, which may require further investigation or clinical trials.
Author Contributions
Y.T., S.L., X.P., Y.Z. contributed to writing and editing the review. X.P., Y.Z. contributed to designing the figures and correcting the manuscript and mainly contributed to the final form of the manuscript and its improvement. All authors contributed to the idea and formation of the manuscript and approved the submitted version. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China [82073097 (SL), 81874139 (SL), 81672991 (SL), 82072594 (YT), and 81672787 (YT)].
Acknowledgments
We thank the contributions of all of Yongguang Tao’s lab members.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
- Fitzsimmons, L.; Kelly, G.L. EBV and Apoptosis: The Viral Master Regulator of Cell Fate? Viruses 2017, 9, 339. [Google Scholar] [CrossRef]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390 Pt 1, 151–209. [Google Scholar]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Genet. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein–Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Young, L.; Yap, L.-F.; Murray, P.G. Epstein–Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Kanda, T. EBV-Encoded Latent Genes. Adv. Exp. Med. Biol. 2018, 1045, 377–394. [Google Scholar]
- Edwards, R.H.; DeKroon, R.; Raab-Traub, N. Alterations in cellular expression in EBV infected epithelial cell lines and tumors. PLoS Pathog. 2019, 15, e1008071. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein–Barr virus entry. Nat. Microbiol. 2018, 3, 164–171. [Google Scholar] [CrossRef]
- Wang, H.-B.; Zhang, H.; Zhang, J.-P.; Li, Y.; Zhao, B.; Feng, G.-K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.-L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 6240. [Google Scholar] [CrossRef]
- Chen, J.; Longnecker, R. Epithelial cell infection by Epstein–Barr virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Ferressini Gerpe, N.M.; Vistarop, A.G.; Moyano, A.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Distinctive EBV infection characteristics in children from a developing country. Int. J. Infect. Dis. 2020, 93, 139–145. [Google Scholar] [CrossRef]
- Dalton, T.; Doubrovina, E.; Pankov, D.; Reynolds, I.R.C.; Scholze, H.; Selvakumar, A.; Vizconde, T.; Savalia, B.; Dyomin, V.; Weigel, C.; et al. Epigenetic reprogramming sensitizes immunologically silent EBV+ lymphomas to virus-directed immunotherapy. Blood 2020, 135, 1870–1881. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Verma, D.; Burton, A.; Swaminathan, S. Cellular RNA Helicase DHX9 Interacts with the Essential Epstein-Barr Virus (EBV) Protein SM and Restricts EBV Lytic Replication. J. Virol. 2019, 93, e01244-18. [Google Scholar] [CrossRef]
- Elgui de Oliveira, D.; Müller-Coan, B.G.; Pagano, J.S. Viral Carcinogenesis beyond Malignant Transformation: EBV in the Pro-gression of Human Cancers. Trends Microbiol. 2016, 24, 649–664. [Google Scholar] [CrossRef]
- Cohen, J.I. Primary Immunodeficiencies Associated with EBV Disease. Curr. Top. Microbiol. Immunol. 2015, 390, 241–265. [Google Scholar]
- Wu, H.-C.; Lin, Y.-J.; Lee, J.-J.; Liu, Y.-J.; Liang, S.-T.; Peng, Y.; Chiu, Y.-W.; Wu, C.-W.; Lin, C.-T. Functional Analysis of EBV in Nasopharyngeal Carcinoma Cells. Lab. Investig. 2003, 83, 797–812. [Google Scholar] [CrossRef]
- Nilsson, J.S.; Forslund, O.; Andersson, F.C.; Lindstedt, M.; Greiff, L. Intralesional EBV-DNA load as marker of prognosis for naso-pharyngeal cancer. Sci. Rep. 2019, 9, 15432. [Google Scholar] [CrossRef] [PubMed]
- Dawson, C.W.; Port, R.J.; Young, L.S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin. Cancer Biol. 2012, 22, 144–153. [Google Scholar] [CrossRef]
- Edwards, R.H.; Marquitz, A.R.; Raab-Traub, N. Changes in Expression Induced by Epstein-Barr Virus LMP1-CTAR1: Potential Role of bcl3. mBio 2015, 6, e00441-15. [Google Scholar] [CrossRef]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortaliza-tion. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y. Epstein-Barr Virus encoded LMP1 regulates cyclin D1 promoter activity by nuclear EGFR and STAT3 in CNE1 cells. J. Exp. Clin. Cancer Res. 2013, 32, 90. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Zhou, M.; Shang, L.; Du, Q.; Li, Y.; Xie, L.; Liu, X.; Tang, M.; Luo, X.; Fan, J.; et al. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 2019, 9, 2424–2438. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Receptor Tyrosine Kinases: Legacy of the First Two Decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, S.; Koyama, S.; Nishikawa, H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat. Rev. Cancer 2021, 21, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, P.; Zhang, C.; Ma, Z.-L. Epidermal growth factor receptor (EGFR): A rising star in the era of precision medicine of lung cancer. Oncotarget 2017, 8, 50209–50220. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.I.; Nikonova, A.S.; Sun, D.; Golemis, E.A. Proliferative signaling by ERBB proteins and RAF/MEK/ERK effectors in pol-ycystic kidney disease. Cell. Signal. 2020, 67, 109497. [Google Scholar] [CrossRef]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncology 2018, 20, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Mirghani, H.; Amen, F.; Moreau, F.; Guigay, J.; Hartl, D.; Guily, J.L.S. Oropharyngeal cancers: Relationship between epidermal growth factor receptor alterations and human papillomavirus status. Eur. J. Cancer 2014, 50, 1100–1111. [Google Scholar] [CrossRef]
- Chen, J.; He, W.; Hu, X.; Shen, Y.; Cao, J.; Wei, Z.; Luan, Y.; He, L.; Jiang, F.; Tao, Y. A role for ErbB signaling in the induction of reactive astrogliosis. Cell Discov. 2017, 3, 17044. [Google Scholar] [CrossRef]
- Jotte, R.M.; Spigel, D.R. Advances in molecular-based personalized non-small-cell lung cancer therapy: Targeting epidermal growth factor receptor and mechanisms of resistance. Cancer Med. 2015, 4, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-targeted therapies in the post-genomic era. Cancer Metast. Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Moyes, D.; Tavassoli, M.; Naglik, J.R. The Role of ErbB Receptors in Infection. Trends Microbiol. 2017, 25, 942–952. [Google Scholar] [CrossRef]
- Tan, X.; Lambert, P.F.; Rapraeger, A.C.; Anderson, R.A. Stress-Induced EGFR Trafficking: Mechanisms, Functions, and Therapeutic Implications. Trends Cell Biol. 2016, 26, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Chung, I.; Akita, R.W.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [PubMed]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Waterman, H.; Alroy, I.; Strano, S.; Seger, R.; Yarden, Y. The C-terminus of the kinase-defective neuregulin receptor ErbB-3 confers mitogenic superiority and dictates endocytic routing. EMBO J. 1999, 18, 3348–3358. [Google Scholar] [CrossRef]
- Miller, W.E.; Earp, H.S.; Raab-Traub, N. The Epstein-Barr virus latent membrane protein 1 induces expression of the epidermal growth factor receptor. J. Virol. 1995, 69, 4390–4398. [Google Scholar] [CrossRef]
- Meckes, D.G.; Shair, K.H.Y.; Marquitz, A.R.; Kung, C.-P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Dawson, C.W.; Takada, K.; Curnow, J.; Moody, C.A.; Sixbey, J.W.; Young, L.S. Epstein-Barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-kappaB transcription factor pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 15730–15735. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Raab-Traub, N. Epstein-Barr Virus Latent Membrane Protein 1 Induces Expression of the Epidermal Growth Factor Receptor through Effects on Bcl-3 and STAT. J. Virol. 2008, 82, 5486–5493. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Song, X.; Deng, X.; Xie, D.; Lee, L.M.; Liu, Y.; Li, W.; Li, L.; Deng, L.; Wu, Q.; et al. Nuclear accumulation of epidermal growth factor receptor and acceleration of G1/S stage by Epstein–Barr-encoded oncoprotein latent membrane protein. Exp. Cell Res. 2005, 303, 240–251. [Google Scholar] [CrossRef]
- Tao, Y.; Song, X.; Tan, Y.; Lin, X.; Zhao, Y.; Zeng, L.; Tang, M.; Li, W.; Wu, Q.; Cao, Y. Nuclear translocation of EGF receptor regulated by Epstein-Barr virus encoded latent membrane protein. Sci. China Ser. C Life Sci. 2004, 47, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Thornburg, N.J.; Pathmanathan, R.; Raab-Traub, N. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer Res. 2003, 63, 8293–8301. [Google Scholar] [PubMed]
- Kung, C.P.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 modulates distinctive NF-kappaB pathways through C-terminus-activating region 1 to regulate epidermal growth factor receptor expression. J. Virol. 2010, 84, 6605–6614. [Google Scholar] [CrossRef] [PubMed]
- Salmerón, A.; Janzen, J.; Soneji, Y.; Bump, N.; Kamens, J.; Allen, H.; Ley, S.C. Direct phosphorylation of NF-kappaB1 p105 by the IkappaB kinase complex on serine 927 is essential for signal-induced p105 proteolysis. J. Biol. Chem. 2001, 276, 22215–22222. [Google Scholar] [CrossRef] [PubMed]
- Heissmeyer, V.; Krappmann, D.; Wulczyn, F.G.; Scheidereit, C. NF-kappaB p105 is a target of IkappaB kinases and controls signal induction of Bcl-3-p50 complexes. EMBO J. 1999, 18, 4766–4778. [Google Scholar] [CrossRef] [PubMed]
- Luftig, M.; Yasui, T.; Soni, V.; Kang, M.S.; Jacobson, N.; Cahir-McFarland, E.; Seed, B.; Kieff, E. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2004, 101, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Paine, E.; Scheinman, R.I.; Baldwin, A.S., Jr.; Raab-Traub, N. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-kappa B/Rel family proteins. J. Virol. 1995, 69, 4572–4576. [Google Scholar] [CrossRef]
- Westerheide, S.D.; Mayo, M.W.; Anest, V.; Hanson, J.L.; Baldwin, A.S. The Putative Oncoprotein Bcl-3 Induces Cyclin D1 To Stimulate G1 Transition. Mol. Cell. Biol. 2001, 21, 8428–8436. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, N.J.; Raab-Traub, N. Induction of epidermal growth factor receptor expression by Epstein-Barr virus latent mem-brane protein 1 C-terminal-activating region 1 is mediated by NF-kappaB p50 homodimer/Bcl-3 complexes. J. Virol. 2007, 81, 12954–12961. [Google Scholar] [CrossRef] [PubMed]
- Baldassarre, F.; Mallardo, M.; Mezza, E.; Scala, G.; Quinto, I. Regulation of NF-kappa B through the nuclear processing of p105 (NF-kappa B1) in Epstein-Barr virus-immortalized B cell lines. J. Biol. Chem. 1995, 270, 31244–31248. [Google Scholar] [CrossRef]
- Chung, G.T.; Lou, W.P.; Chow, C.; To, K.F.; Choy, K.W.; Leung, A.W.; Tong, C.Y.; Yuen, J.W.; Ko, C.W.; Yip, T.T.; et al. Constitutive activation of distinct NF-κB signals in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 311–322. [Google Scholar] [CrossRef]
- Siersbæk, R.D.; Scabia, V.; Nagarajan, S.; Chernukhin, I.; Papachristou, E.K.; Broome, R.; Johnston, S.J.; Joosten, S.E.; Green, A.R.; Kumar, S.; et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor α Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020, 38, 412–423.e9. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chung, T.-H.; Chng, P.Y.; Toh, S.H.M.; Chng, W.J. IL6R-STAT3-ADAR1 (P150) interplay promotes oncogenicity in multiple myeloma with 1q21 amplification. Haematology 2019, 105, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.R.; Jin, Y.T.; Tsai, S.T.; Shiau, A.L.; Wu, C.L.; Su, W.C. Constitutive activation of STAT3 and STAT5 is present in the majority of nasopharyngeal carcinoma and correlates with better prognosis. Br. J. Cancer 2003, 89, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Meckes, D.G., Jr.; Raab-Traub, N. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCdelta. J. Virol. 2011, 85, 4399–4408. [Google Scholar] [CrossRef]
- Chen, H.; Lee, J.M.; Zong, Y.; Borowitz, M.; Ng, M.H.; Ambinder, R.F.; Hayward, S.D. Linkage between STAT Regulation and Epstein-Barr Virus Gene Expression in Tumors. J. Virol. 2001, 75, 2929–2937. [Google Scholar] [CrossRef]
- Chen, H.; Hutt-Fletcher, L.; Cao, L.; Hayward, S.D. A Positive Autoregulatory Loop of LMP1 Expression and STAT Activation in Epithelial Cells Latently Infected with Epstein-Barr Virus. J. Virol. 2003, 77, 4139–4148. [Google Scholar] [CrossRef]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef]
- Tu, C.; Zeng, Z.; Qi, P.; Li, X.; Guo, C.; Xiong, F.; Xiang, B.; Zhou, M.; Liao, Q.; Yu, J.; et al. Identification of genomic alterations in naso-pharyngeal carcinoma and nasopharyngeal carcinoma-derived Epstein-Barr virus by whole-genome sequencing. Carcinogenesis 2018, 39, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, S.K.; Wang, Y.; Fan, Z.X.; Li, C.R.; Feng, M.; Xu, P.; Wang, W.D.; Lang, J.Y. p53, MDM2, eIF4E and EGFR expression in na-sopharyngeal carcinoma and their correlation with clinicopathological characteristics and prognosis: A retrospective study. Oncol. Lett. 2015, 9, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-H.; Gao, M.; Chen, C.-L.; Yeh, P.-Y.; Cheng, A.-L. Inhibitors of Epidermoid Growth Factor Receptor Suppress Cell Growth and Enhance Chemosensitivity of Nasopharyngeal Cancer Cells in vitro. Oncology 2005, 68, 538–547. [Google Scholar] [CrossRef]
- Sheen, T.-S.; Huang, Y.-T.; Chang, Y.-L.; Ko, J.-Y.; Wu, C.-S.; Yu, Y.-C.; Tsai, C.-H.; Hsu, M.-M. Epstein-Barr Virus-encoded Latent Membrane Protein 1 Co-expresses with Epidermal Growth Factor Receptor in Nasopharyngeal Carcinoma. Jpn. J. Cancer Res. 1999, 90, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Youhong, T.; Tan, Y.; He, Y.; Ban, Y.; Cai, J.; Li, X.; Xiong, W.; Zeng, Z.; Li, G.; et al. EGFR-PKM2 signaling promotes the metastatic potential of nasopharyngeal carcinoma through induction of FOSL1 and ANTXR. Carcinogenesis 2020, 41, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Chua, D.T.; Nicholls, J.M.; Sham, J.S.; Au, G.K. Prognostic value of epidermal growth factor receptor expression in patients with advanced stage nasopharyngeal carcinoma treated with induction chemotherapy and radiotherapy. Int. J. Radiat. Oncol. 2004, 59, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bourouba, M.; Benyelles-Boufennara, A.; Terki, N.; Baraka-Kerboua, E.; Bouzid, K.; Touil-Boukoffa, C. Epidermal growth factor receptor (EGFR) abundance correlates with p53 and Bcl-2 accumulation and patient age in a small cohort of North African nasopharyngeal carcinoma patients. Eur. Cytokine Netw. 2011, 22, 38–44. [Google Scholar] [CrossRef]
- Liang, Z.; Liu, Z.; Cheng, C.; Wang, H.; Deng, X.; Liu, J.; Liu, C.; Li, Y.; Fang, W. VPS33B interacts with NESG1 to modulate EGFR/PI3K/AKT/c-Myc/P53/miR-133a-3p signaling and induce 5-fluorouracil sensitivity in nasopharyngeal carcinoma. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3343–3347. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, J.; Wang, F.; Liu, X.; Lu, J.; Yu, X.; Ma, X.; Peng, X.; Li, X. Foxq1 promotes metastasis of nasopharyngeal carcinoma by inducing vasculogenic mimicry via the EGFR signaling pathway. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- A Elian, F.; Are, U.; Ghosh, S.; Nuin, P.; Footz, T.; McMullen, T.P.; Brindley, D.N.; A Walter, M. FOXQ1 is Differentially Expressed Across Breast Cancer Subtypes with Low Expression Associated with Poor Overall Survival. Breast Cancer Targets Ther. 2021, 13, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.-X.; Wang, M.-D.; Xie, P.; Yang, J.-P.; Sun, R.; Zheng, L.-S.; Mei, Y.; Meng, D.-F.; Peng, X.-S.; Lang, Y.-H.; et al. LACTB promotes metastasis of nasopharyngeal carcinoma via activation of ERBB3/EGFR-ERK signaling resulting in unfavorable patient survival. Cancer Lett. 2021, 498, 165–177. [Google Scholar] [CrossRef]
- Lin, Q.; Wang, H.; Lin, X.; Zhang, W.; Huang, S.; Zheng, Y. PTPN12 Affects Nasopharyngeal Carcinoma Cell Proliferation and Mi-gration Through Regulating EGFR. Cancer Biother. Radiopharm. 2018, 33, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, X.; Sun, R.; Ou, J.; Huang, J.; Yang, N.; Xu, T.; Li, J.; He, X.; Li, C.; et al. EGFR-rich extracellular vesicles derived from highly metastatic nasopharyngeal carcinoma cells accelerate tumour metastasis through PI3K/AKT pathway-suppressed ROS. J. Extracell. Vesicles 2020, 10, e12003. [Google Scholar] [CrossRef] [PubMed]
- Frawley, T.; Piskareva, O. Extracellular Vesicle Dissemination of Epidermal Growth Factor Receptor and Ligands and Its Role in Cancer Progression. Cancers 2020, 12, 3200. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Bonner, S.E.; Martin-Fernandez, M.L.; Huber, V. Mechanisms of Action of EGFR Tyrosine Kinase Receptor Incorporated in Extracellular Vesicles. Cells 2020, 9, 2505. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Bonner, S.E.; Iyer, R.S.; Martin-Fernandez, M.L.; Huber, V. Cooperation and Interplay between EGFR Signalling and Extracellular Vesicle Biogenesis in Cancer. Cells 2020, 9, 2639. [Google Scholar] [CrossRef]
- Huang, W.; Liu, J.; Feng, X.; Chen, H.; Zeng, L.; Huang, G.; Liu, W.; Wang, L.; Jia, W.; Chen, J.; et al. DLC-1 induces mitochondrial apoptosis and epithelial mesenchymal transition arrest in nasopharyngeal carcinoma by targeting EGFR/Akt/NF-κB pathway. Med. Oncol. 2015, 32, 115. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Li, C.; Liu, W.; Chen, H.; Zhou, W.; Wang, L.; Zhu, B.; Yao, K.; Jiang, X.; Ren, C. DLC-1, a candidate tumor suppressor gene, inhibits the proliferation, migration and tumorigenicity of human nasopharyngeal carcinoma cells. Int. J. Oncol. 2013, 42, 1973–1984. [Google Scholar] [CrossRef][Green Version]
- Yang, J.; Zhu, D.; Liu, S.; Shao, M.; Liu, Y.; Li, A.; Lv, Y.; Huang, M.; Lou, D.; Fan, Q. Curcumin enhances radiosensitization of nasopha-ryngeal carcinoma by regulating circRNA network. Mol. Carcinog. 2020, 59, 202–214. [Google Scholar] [CrossRef]
- Ma, N.; Kawanishi, M.; Hiraku, Y.; Murata, M.; Huang, G.-W.; Huang, Y.; Luo, D.-Z.; Mo, W.-G.; Fukui, Y.; Kawanishi, S. Reactive nitrogen species-dependent DNA damage in EBV-associated nasopharyngeal carcinoma: The relation to STAT3 activation and EGFR expression. Int. J. Cancer 2008, 122, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Zachary, I.C. How neuropilin-1 regulates receptor tyrosine kinase signalling: The knowns and known unknowns. Biochem. Soc. Trans. 2011, 39, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Huang, W.; Zhang, Z.; Lin, X.; Lin, H.; Peng, L.; Chen, T. Highly Uniform Synthesis of Selenium Nanoparticles with EGFR Targeting and Tumor Microenvironment-Responsive Ability for Simultaneous Diagnosis and Therapy of Nasopharyngeal Carcinoma. ACS Appl. Mater. Interfaces 2019, 11, 11177–11193. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, Y.; Zhang, X.; Fu, S.; Lin, Z.; Fang, W.; Yang, Y.; Huang, Y.; Zhao, H.; Hong, S.; et al. Anti-epidermal growth factor receptor monoclonal antibody plus palliative chemotherapy as a first-line treatment for recurrent or metastatic nasopharyngeal car-cinoma. Cancer Med. 2020, 9, 1721–1732. [Google Scholar] [CrossRef]
- Campoli, M.; Ferris, R.L.; Ferrone, S.; Wang, X. Immunotherapy of Malignant Disease with Tumor Antigen-Specific Monoclonal Antibodies. Clin. Cancer Res. 2009, 16, 11–20. [Google Scholar] [CrossRef]
- Ali, S.M.; Yao, M.; Yao, J.; Wang, J.; Cheng, Y.; Schrock, A.B.; Chirn, G.-W.; Chen, H.; Mu, S.; Gay, L.; et al. Comprehensive genomic profiling of different subtypes of nasopharyngeal carcinoma reveals similarities and differences to guide targeted therapy. Cancer 2017, 123, 3628–3637. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.S.Y.; Wong, B.; Gangodu, N.R.; Lee, A.Z.E.; Kian Fong Liou, A.; Loh, K.S.; Li, H.; Yann Lim, M.; Salazar, A.M.; Lim, C.M. Enhancing the immune stimulatory effects of cetuximab therapy through TLR3 signalling in Epstein-Barr virus (EBV) positive nasopha-ryngeal carcinoma. Oncoimmunology 2018, 7, 1500109. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.R.; Zhu, H.F.; Xu, J.; Jiang, X.S.; Yin, L.; Jiang, N.; Zong, D.; Wang, F.J.; Huang, S.F.; Bian, X.H.; et al. Effectiveness of Cetuximab in Combination with Concurrent Chemoradiotherapy in Locoregionally Advanced Nasopharyngeal Carcinoma: A 1:2 Pro-pensity Score-matched Analysis. J. Cancer 2018, 9, 1642–1651. [Google Scholar] [CrossRef]
- Peng, H.; Tang, L.L.; Liu, X.; Chen, L.; Li, W.F.; Mao, Y.P.; Zhang, Y.; Liu, L.Z.; Tian, L.; Guo, Y.; et al. Anti-EGFR targeted therapy delivered before versus during radiotherapy in locoregionally advanced nasopharyngeal carcinoma: A big-data, intelligence plat-form-based analysis. BMC Cancer 2018, 18, 323. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-S.; Liang, Y.-J.; Li, X.-Y.; Liu, S.-L.; Chen, Q.-Y.; Tang, L.-Q.; Mai, H.-Q. Palliative chemotherapy with or without anti-EGFR therapy for de novo metastatic nasopharyngeal carcinoma: A propensity score-matching study. Drug Des. Dev. Ther. 2019, 13, 3207–3216. [Google Scholar] [CrossRef]
- Ueda, Y.; Enokida, T.; Okano, S.; Fujisawa, T.; Ito, K.; Tahara, M. Combination Treatment with Paclitaxel, Carboplatin, and Ce-tuximab (PCE) as First-Line Treatment in Patients with Recurrent and/or Metastatic Nasopharyngeal Carcinoma. Front. Oncol. 2020, 10, 571304. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Mei, Q.; Li, J.-B.; You, R.; Liu, Y.-P.; Sun, R.; Hu, G.-Y.; Chen, M.-Y.; Hua, Y.-J. The long-term survival of patients with III-IVb stage nasopharyngeal carcinoma treated with IMRT with or without Nimotuzumab: A propensity score-matched analysis. BMC Cancer 2019, 19, 1122. [Google Scholar]
- Zhu, Y.; Yang, S.; Zhou, S.; Yang, J.; Qin, Y.; Gui, L.; Shi, Y.; He, X. Nimotuzumab plus platinum-based chemotherapy versus plati-num-based chemotherapy alone in patients with recurrent or metastatic nasopharyngeal carcinoma. Ther. Adv. Med. Oncol. 2020, 12, 1758835920953738. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, G.; Miao, X.-B.; Deng, X.-B.; Wu, Y.; Liu, Y.; Jin, Z.-R.; Li, X.-Q.; Liu, Q.-Z.; Sun, D.-X.; et al. Cancer stem-like cell properties are regulated by EGFR/AKT/β-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. 2013, 280, 2027–2041. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Wu, L.; Wang, Z.; Wang, X.; Yü, Y.; Zhao, Q.; Luo, F. MiRNA-125a-5p: A regulator and predictor of gefitinib’s effect on nasopharyngeal carcinoma. Cancer Cell Int. 2014, 14, 24. [Google Scholar] [CrossRef]
- Yugui, F.; Wang, H.; Sun, D.; Zhang, X. Nasopharyngeal cancer combination chemoradiation therapy based on folic acid modified, gefitinib and yttrium 90 co-loaded, core-shell structured lipid-polymer hybrid nanoparticles. Biomed. Pharmacother. 2019, 114, 108820. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, F.; Zhang, J.; Zou, Q.; Fan, Q.; Zhang, F. Erlotinib enhanced chemoradiotherapy sensitivity via inhibiting DNA damage repair in nasopharyngeal carcinoma CNE2 cells. Ann. Palliat. Med. 2020, 9, 2559–2567. [Google Scholar] [CrossRef]
- Lan, M.-Y.; Hsu, Y.-B.; Chen, J.-P.; Lu, Y.-J. Polyethylene Glycol-Coated Graphene Oxide Loaded with Erlotinib as an Effective Therapeutic Agent for Treating Nasopharyngeal Cancer Cells. Int. J. Nanomed. 2020, 15, 7569–7582. [Google Scholar] [CrossRef]
- You, B.; Le Tourneau, C.; Chen, E.X.; Wang, L.; Jarvi, A.; Bharadwaj, R.R.; Kamel-Reid, S.; Perez-Ordonez, B.; Mann, V.; Siu, L.L. A Phase II Trial of Erlotinib as Maintenance Treatment After Gemcitabine Plus Platinum-based Chemotherapy in Patients with Recurrent and/or Metastatic Nasopharyngeal Carcinoma. Am. J. Clin. Oncol. 2012, 35, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Liang, X.; Min, X.; Zhang, Y.; Wang, G.; Peng, Z.; Peng, F.; Li, M.; Chen, L.; Chen, Y. Simultaneous Inhibition of EGFR and HER2 via Afatinib Augments the Radiosensitivity of Nasopharyngeal Carcinoma Cells. J. Cancer 2019, 10, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xue, C.; Tian, Y.; Zhang, J.; Zhao, Y.; Zhan, J.; Fang, W. In vitro and in vivo efficacy of afatinib as a single agent or in combination with gemcitabine for the treatment of nasopharyngeal carcinoma. Drug Des. Dev. Ther. 2016, 10, 1299–1306. [Google Scholar] [CrossRef]
- Lin, M.; You, R.; Liu, Y.-P.; Zhang, Y.-N.; Zhang, H.-J.; Zou, X.; Yang, Q.; Li, C.-F.; Hua, Y.-J.; Yu, T.; et al. Beneficial effects of anti-EGFR agents, Cetuximab or Nimotuzumab, in combination with concurrent chemoradiotherapy in advanced nasopharyngeal carcinoma. Oral Oncol. 2018, 80, 1–8. [Google Scholar] [CrossRef]
- Talavera, A.; Friemann, R.; Gómez-Puerta, S.; Martinez-Fleites, C.; Garrido, G.; Rabasa, A.; Requena, A.L.; Pupo, A.; Johansen, R.F.; Sanchez, O.; et al. Nimotuzumab, an Antitumor Antibody that Targets the Epidermal Growth Factor Receptor, Blocks Ligand Binding while Permitting the Active Receptor Conformation. Cancer Res. 2009, 69, 5851–5859. [Google Scholar] [CrossRef]
- Si, X.; Wu, S.; Wang, H.; Zhang, X.; Wang, M.; Zeng, X.; Zhang, L. Nimotuzumab combined with chemotherapy as first-line treatment for advanced lung squamous cell carcinoma. Thorac. Cancer 2018, 9, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Fei, Z.; Xu, T.; Li, M.; Chen, T.; Li, L.; Qiu, X.; Chen, C. Effectiveness and cost-effectiveness analysis of nimotuzumab for the radio-therapy of locoregionally advanced nasopharyngeal carcinoma. Radiat. Oncol. 2020, 15, 230. [Google Scholar] [CrossRef]
- Yao, J.J.; Zhang, L.L.; Gao, T.S.; Peng, Y.L.; Lawrence, W.R.; Zhang, W.J.; Zhang, F.; Zhou, G.Q.; Wang, S.Y.; Sun, Y. Comparing treatment outcomes of concurrent chemoradiotherapy with or without nimotuzumab in patients with locoregionally advanced naso-pharyngeal carcinoma. Cancer Biol. Ther. 2018, 19, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- You-Ping, L.; Hua, Y.-J.; Liu, Y.-P.; Yang, Q.; Zhang, Y.-N.; Li, J.-B.; Li, C.-F.; Zou, X.; Jing-Yu, C.; Cao, J.-Y.; et al. Concurrent Chemoradiotherapy with or without Anti-EGFR-Targeted Treatment for Stage II-IVb Nasopharyngeal Carcinoma: Retrospective Analysis with a Large Cohort and Long Follow-up. Theranostics 2017, 7, 2314–2324. [Google Scholar]
- Mao, L.; Tan, J.; Wang, F.; Luo, Y.; Liu, W.; Zeng, F.; Yu, B.; Huang, H.; Lu, J.; Peng, X.; et al. Retrospective study comparing anti-EGFR monoclonal antibody plus cisplatin-based chemoradiotherapy versus chemoradiotherapy alone for stage II-IVb nasopha-ryngeal carcinoma and prognostic value of EGFR and VEGF expression. Clin. Otolaryngol. 2019, 44, 572–580. [Google Scholar] [CrossRef]
- Lui, V.W.Y.; Lau, C.P.Y.; Ho, K.; Ng, M.H.L.; Cheng, S.H.; Tsao, S.-W.; Tsang, C.M.; Lei, K.I.K.; Chan, A.T.; Mok, T.S.K. Anti-invasion, anti-proliferation and anoikis-sensitization activities of lapatinib in nasopharyngeal carcinoma cells. Investig. New Drugs 2010, 29, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Chua, D.T.; Wei, W.I.; Wong, M.P.; Sham, J.S.; Nicholls, J.; Au, G.K. Phase II study of gefitinib for the treatment of recurrent and met-astatic nasopharyngeal carcinoma. Head Neck 2008, 30, 863–867. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).