Abstract
This work presents a complete review of the literature on and a critical evaluation and thermodynamic optimization of the Li-Se and Na-Se binary systems. The modified quasi-chemical model in the pair approximation (MQMPA) was employed to describe the liquid solution exhibiting a high degree of short-range ordering behavior of atoms. The thermodynamic properties of the compounds Li2Se , Na2Se , NaSe and NaSe2 were also calculated by using first-principles density functional theory (DFT) calculations to assist the thermodynamic description of these two binary systems. All the available and reliable experimental data are reproduced within experimental error limits. Moreover, the phase equilibria of these two systems at low total pressure were analyzed by using the developed thermodynamic model.
1. Introduction
Mg and its alloys have great potential as absorbable medical materials from the aspect of mechanical properties and biocompatibility [1]. The density and elastic modulus of Mg and its alloys are closer to human bones than stainless steels and titanium alloys, but the strength still needs to be improved [2]. The thermomechanical properties of Mg-based alloys can be enhanced by alloying elements such as Al, Zn, Ca, Sr, Li, Na, Se, etc. [3,4,5,6,7,8]. Among various alloying elements, Li, Na, and Se can influence the mechanical properties of Mg-based alloy and play certain essential roles in the human body system.
Liu et al. [9] reported that the ductility of Mg-3.3 wt.% Li alloy could be improved by using conventional extrusion and equal-channel angular pressing process. Furui et al. [10] reported that adding 8 wt.% Li to Mg-based alloys resulted in the alloys having excellent superplastic behavior (maximum elongation achieved ~970%) after extrusion with the ECAP process. Moreover, Li is an essential trace element of the human body that greatly impacts daily physiological functioning [11]. Li also has a regulating effect on central nervous activity [12]. In their study of precipitate hardening alloys, Mendis et al. [4,5] prepared Mg-Sn-based alloys with a significantly enhanced hardening effect, using Na as a micro-alloying element. Similar results also have been reported by Sasaki et al. [6] with Na and Zn additions. Gibson et al. [7] reported that Mg-Sn-Zn-Na alloys show excellent creep resistance compared with Mg-Sn binary alloys and Mg-Sn-Zn ternary alloys. On the other hand, Na affects neuromuscular function and plays an important role in neuromuscular junction conduction. It maintains the normal water electrolyte acid–base balance and stabilizes the internal environment of the human body [13]. Thus, Na can be a candidate-alloying element for biodegradable Mg-based alloys. Persaud-Sharma et al. [8] studied the addition of Se to Mg-Zn alloys for novel bio-absorbable cardiovascular stents. They showed that the as-cast Mg-Zn-Se alloy had a better elastic modulus compared with the commercial shape-memory alloys. Moreover, its better fracture elongation can adapt to the actual complex internal environment of the human body. Se participates in the synthesis of antioxidant selenium proteins in important organs, and this also has a potential role in resisting the damage of free radicals to tissues and organs [14,15]. A large number of studies had demonstrated that Se is an effective killer of cancer cells [16], as it forms an internal environment by inhibiting the division and proliferation of cancer cells in the body [17].
Although Li, Na, and Se have been gradually applied as alloying elements for Mg-based alloys to improve the mechanical and biomedical properties, there is still a lack of systematic study to optimize the properties to meet the requirement of body implants. The microstructure of alloys can directly influence the mechanical and biomedical properties. Therefore, the evaluation of the microstructure based on the phase diagram information is essential to develop the optimum Mg-based alloy containing Li, Na, and Se.
The most efficient method for simultaneously obtaining the phase diagrams and thermodynamic properties of alloys is the Calculation of Phase Diagrams (CALPHAD) method [18]. The thermodynamic modeling of Mg-Li [19] and Mg-Na [20] binary systems was already carried out by our group. In the present work, to complete our thermodynamic description of the Mg-Li-Se and Mg-Na-Se systems, the thermodynamic modeling of the Li-Se and Na-Se binary systems were performed based on the critical evaluation of all available experimental data in the literature. In order to assist the assessments, the thermodynamic properties of several intermetallic phases were obtained by using the first principles calculations (FPC) technique [21,22]. This is also part of a large thermodynamic database development of Mg-based alloy systems containing Ag, Ca, Cu, In, Li, Na, Se, Sr, and Zn for its application to Mg-based biomaterial design [23,24,25]. The present thermodynamic calculations were carried out by using the FactSage thermodynamic software [26].
2. Critical Literature Review
2.1. The Li-Se Binary System
The phase equilibria of Li-Se binary system were investigated by Cunningham et al. [27] by using differential thermal analysis (DTA), chemical analysis, optical microscopy, and X-ray diffraction (XRD) methods. The crystal structural information of all the solid phases in the Li-Se binary system is compiled in Table 1. The existence of the intermetallic compound Li2Se with the antifluorite crystal structure () was confirmed in the referred works [27,28,29]. Moreover, the melting point of the Li2Se compound was measured to be 1302 ± 2 °C, using DTA, and 1303 ± 5 °C by Cunningham et al. [27], using optical observation, respectively. Sangster and Pelton [30] pointed out that the vapor losses in the open crucibles at this high temperature may result in an error of ±25 °C for the results obtained by Cunningham et al. [27]. The lattice parameter measured by Cunningham [27] by using XRD indicates a narrow solid solubility range of Li2Se with the excess Se when Se was added to Li containing ammonia. Bergstorm [31] reported the formation of a Li polyselenide with an average composition of Li2Se5.4 when Se was added to liquid ammonia containing Li. However, no evidence of a Li2Se5.4 compound in the Li-Se binary system was found by Cunningham et al. [27]. Only the Li2Se compound was confirmed to be stable in the Li-Se binary system by Li et al. [32] through the FPC method. Therefore, it is concluded that Li2Se is the only stable intermetallic phase, and Li2Se5.4 is unstable in the non-solvated state at ambient temperature. Moreover, in the present work, the first-principles density functional theory (DFT) method is introduced to calculate the lattice parameter of the intermetallic compound Li2Se by using different van der Waal (vdW) methods (PBE, D3, D2, optB86b, optB88, DF, and DF2). It is seen that the vdW correction can greatly improve the descriptions of the lattice parameter of Se, as the vdW force dominates the interaction between Se atomic chains. In the following, we chose DFT-D2 methods as an example to represent the DFT evaluations.
Cunningham et al. [27] measured the liquid miscibility in the Se-rich region by chemical analysis of quenched molten samples. It should be noted that an analytical error for the liquidus composition is about ±5 wt.%, as mentioned by Cunningham et al. [27]. The critical temperature for liquid immiscibility was higher than 800 °C based on the optical analysis results of quenched samples. The liquidus in the composition range from 50 to 70 at.% Se was measured by using the microscopic analysis method. The samples were equilibrated in quartz crucibles at around liquidus temperatures, followed by quenching in liquid nitrogen. Its phase assemblage was analyzed by using an optical microscope. Sangster and Pelton [30] estimated that the error in liquidus measurements by Cunningham et al. [27] would be about ±5 °C at 350 °C and ±15 °C at 700 °C, respectively.
The eutectic reaction temperatures of bcc_A2 (Li) + Li2Se ↔ liquid and hex_A8 (Se) + Li2Se ↔ liquid were reported to be 180.5 ± 0.1 °C and 220 ± 0.4 °C, respectively [27]. The terminal solid solubility has not been reported by Cunningham et al. [27]. However, it can be estimated to be very small due to the large differences in chemical nature and crystal structure between Li and Se.
The activity of Li in Li-Se liquid was measured by Cairns et al. [33], using electromotive force (emf) measurements. It should be noted that the emf values were too complicated to be analyzed due to the reaction between the molten Li-Se and LiBr-RbBr or LiF-LiCl-LiI used as the electrolyte for the emf cell. However, the Gibbs energy of formation of Li2Se compound at 360 °C was successfully derived to be −131.1 kJ/mol-atoms by Cairns et al. [33]. Later, the activity of Li in Li-Se liquid in the composition range of 0.2 to 15.9 at.% Li was measured by Chekova et al. [34], using emf technique. They employed a solid electrolyte membrane of Li2O-B2O3-LiF to prevent the reaction between the Li-Se and salt electrolyte. A constant emf value of 2.085 ± 0.002 V at 420 °C, regardless of Li content, indicated the miscibility gap of the liquid phase in the Se-rich region. The Gibbs energy of mixing of Li-Se liquid at 420 °C in the composition range of 0.2 to 15.9 at.% Li was derived by Morachevskii [35], according to the emf results from Chekova et al. [34].
The enthalpy of formation of Li2Se compound was first studied by Fabre [36], using aqueous solution calorimetry. A reassessed value of −147.4 ± 1.3 kJ/mol-atoms for the enthalpy of formation of Li2Se compound at 25 °C was made by Olin et al. [37] based on the data from Fabre [36]. Later, the enthalpy of formation of Li2Se compound at 25 °C was reported to be −141.97 ± 3 kJ/mol-atoms by Ader [38], using bomb calorimetry, which is less negative than the calculated one from Olin et al. [37]. The enthalpy of formation of the Li2Se compound was derived to be −139.3 kJ/mol-atoms (at 150 °C) by Morachevskii. Later, the enthalpy of formation of the Li2Se compound was calculated to be −120.3 kJ/mol-atoms by Li et al. [32], using the DFT method. As an inconsistency existed in the previous reported results [32,36,37,38], the DFT technique with different vdW methods (PBE, D3, D2, optB86b, optB88, DF, and DF2) was employed in the present work to verify the formation enthalpy of Li2Se compound. The present calculated results are compiled in Table 1 and Table 2.
The entropy of the Li2Se compound at 25 °C was estimated to be 32 ± 6.97 kJ/K·mol-atoms by Ader [38] based on the formula from Latimer’s method, and 23.3 ± 3.2 kJ/K·mol-atoms by Voronin [39], using a semi-empirical relationship involving vibrational contributions to the entropy. The Gibbs energy of formation of the Li2Se compound was determined to be −134.97 kJ/mol-atoms (at 150 °C) by Morachevskii, using the Gibbs energy equation [35].
2.2. The Na-Se Binary System
Mathewson [40] investigated the phase diagram of the Na-Se binary system in the composition range of 10 to 98 at.% Se, using thermal analysis, chemical analysis, and optical microscopy methods for the first time. He followed meticulous experimental procedures, including preparing materials, choosing protection gas (dry H2), correcting melting loss, etc. Five intermetallic compounds, namely Na2Se, NaSe, Na2Se3, NaSe2, and NaSe3, were reported. The eutectic reaction bcc_A2 (Na) + Li2Se ↔ liquid occurs nearly at pure Na composition and 97 °C (which is 0.5 °C lower than the melting temperature of pure Na). Similar eutectic reactions were also reported in the Li-Se [27] and K-Se [41] binary systems. To explain the eutectic reaction, a very limited solid solubility of Se in bcc_A2 (Na) was assumed by Sangster and Pelton [42]. Mathewson [40] reported the melting point of compound Na2Se, which had the same crystal structure as Li2Se (), to be higher than 875 °C. Sangster and Pelton [42] estimated it to be in the range of 900 to 1150 °C by considering the melting temperatures of Li2Se, Rb2Se, Li2S, and Na2S. Mathewson [40] reported the peritectic melting behavior of Na2Se, NaSe, and NaSe2 compounds. The lattice parameter of Na2Se was measured by Zintl et al. [28], using the film stress determination method. Lattice parameters of Na2Se and Na2Se4 were determined by Klemm et al. [43] by using the thermal analysis method. For the compound Na2Se2, Föppl et al. [44] measured its lattice parameter by using the X-ray diffraction method. Moreover, the lattice parameters of the intermetallic compounds Na2Se, NaSe, and NaSe2 were calculated by using DFT calculations similar to the Li-Se binary system in this work. The crystal structural information of all the solid phases in the Na-Se binary system is compiled in Table 1.

Table 1.
Structural parameters of phases and thermodynamic models used in the present work.
Table 1.
Structural parameters of phases and thermodynamic models used in the present work.
| Phase | Strukturb-ericht | Pearson Symbol | Space Group | Lattice Parameters, Å | Model | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| References | vdW Method | a | b | c | ||||||
| Liquid | - | - | - | - | - | - | 3.5093 | - | - | MQMPA a |
| bcc_A2 (Li) | A2 | cI2 | Experimental data | [30] | - | 3.4268 | - | - | CEF b | |
| Calculated data | TW c | PBE | 3.3721 | - | - | - | ||||
| TW | D3 | 3.2674 | - | - | - | |||||
| TW | D2 | 3.4569 | - | - | - | |||||
| TW | optB86b | 3.4741 | - | - | - | |||||
| TW | optB88 | 3.4534 | - | - | - | |||||
| TW | DF | 3.3920 | - | - | - | |||||
| TW | DF2 | 3.5093 | - | - | - | |||||
| hex_A8 (Se) | A8 | hP3 | Experimental data | [30] | - | 4.3659 | - | 4.9537 | CEF | |
| Calculated data | TW | PBE | 4.6347 | - | 5.0300 | - | ||||
| TW | D3 | 4.2577 | - | 5.0888 | - | |||||
| TW | D2 | 4.3682 | - | 5.0609 | - | |||||
| TW | optB86b | 4.1722 | - | 5.1180 | - | |||||
| TW | optB88 | 4.2898 | - | 5.0986 | - | |||||
| TW | DF | 4.7153 | - | 5.1036 | - | |||||
| TW | DF2 | 4.5682 | - | 5.2096 | - | |||||
| bcc_A2 (Na) | A2 | cI2 | Experimental data | [42] | - | 4.2906 | - | - | CEF | |
| Calculated data | TW | PBE | 4.1954 | - | - | - | ||||
| TW | D3 | 4.1603 | - | - | - | |||||
| TW | D2 | 3.9799 | - | - | - | |||||
| TW | optB86b | 4.1754 | - | - | - | |||||
| TW | optB88 | 4.1841 | - | - | - | |||||
| TW | DF | 4.2130 | - | - | - | |||||
| TW | DF2 | 4.1336 | - | - | - | |||||
| Li2Se | C1 | cF12 | Experimental data | [27] | - | 6.0014 | - | - | ST d | |
| [27] | - | 6.0001 | - | - | - | |||||
| [28] | - | 6.0170 | - | - | - | |||||
| [29] | - | 6.0020 | - | - | - | |||||
| Calculated data | TW | PBE | 6.0051 | - | - | - | ||||
| TW | D3 | 5.8910 | - | - | - | |||||
| Li2Se | C1 | cF12 | Calculated data | TW | D2 | 5.9999 | - | - | - | |
| TW | optB86b | 6.0012 | - | - | - | |||||
| TW | optB88 | 6.0186 | - | - | - | |||||
| TW | DF | 6.0894 | - | - | - | |||||
| TW | DF2 | 6.0890 | - | - | - | |||||
| Na2Se | C1 | cF12 | Experimental data | [28] | - | 6.8230 | - | - | ST | |
| [43] | - | 6.8130 | - | |||||||
| Calculated data | TW | PBE | 6.8535 | - | - | - | ||||
| TW | D3 | 6.7003 | - | - | - | |||||
| TW | D2 | 6.7762 | - | - | - | |||||
| TW | optB86b | 6.7693 | - | - | - | |||||
| TW | optB88 | 6.7761 | - | - | - | |||||
| TW | DF | 6.8792 | - | - | - | |||||
| TW | DF2 | 6.8636 | - | - | - | |||||
| NaSe | - | hP8 | Experimental data | [44] | - | 4.6850 | - | 10.5300 | ST | |
| Calculated data | TW | PBE | 4.7248 | - | 10.7860 | - | ||||
| TW | D3 | 4.6303 | - | 10.5777 | - | |||||
| TW | D2 | 4.6715 | - | 10.4226 | - | |||||
| TW | optB86b | 4.6352 | - | 10.6614 | - | |||||
| TW | optB88 | 4.6392 | - | 10.7487 | - | |||||
| TW | DF | 4.7253 | - | 11.0405 | - | |||||
| TW | DF2 | 4.7088 | - | 11.0724 | - | |||||
| Na2Se3 | - | - | - | - | - | - | - | - | - | ST |
| NaSe2 | - | tI48 | Experimental data | [43] | - | 10.1926 | - | 12.2177 | ST | |
| Calculated data | TW | PBE | 4.7248 | - | 10.7860 | - | ||||
| TW | D3 | 10.0389 | - | 12.2162 | - | |||||
| TW | D2 | 10.2005 | - | 11.7552 | - | |||||
| TW | optB86b | 10.1190 | - | 11.9942 | - | |||||
| TW | optB88 | 10.2042 | - | 12.0233 | - | |||||
| TW | DF | 10.5358 | - | 12.2918 | - | |||||
| TW | DF2 | 10.5920 | - | 12.1594 | - | |||||
| NaSe3 | - | - | - | - | - | - | - | - | - | ST |
a MQMPA, modified quasi-chemical model in the pair approximation; b CEF, compound energy formalism; c TW, this work; d ST, stoichiometric compound.
The existence of liquid immiscibility in the high Se-rich region has been demonstrated by several investigators using emf measurements [45,46,47,48] and electrical resistivity [49] method. However, the critical temperature for the liquid immiscibility has not been reported. Sangster and Pelton [42] compiled the experimental results and proposed a monotectic reaction Liquid#1↔ Liquid#2 + NaSe3 at ~255 °C and the compositions of liquids at 78 and 99.95 at.% Se.
Table 2.
The calculated enthalpies of formation of compounds in the Li-Se and Na-Se binary systems in this work compared with the experimental data from References [21,22,32,35,37,38,50,51].
Table 2.
The calculated enthalpies of formation of compounds in the Li-Se and Na-Se binary systems in this work compared with the experimental data from References [21,22,32,35,37,38,50,51].
| Phase | Method | Enthalpy of Formation, (kJ/mol-atoms) | Reference |
|---|---|---|---|
| Li2Se | PBE | −120.3 | [32] |
| emf | −139.3 | [35] | |
| calorimetry | −147.4 ± 1.3 | [37] | |
| electrochemical and calorimetry | −141.97 ± 3 | [38] | |
| PBE | −123.2 | FTW a | |
| D3 | −128.4 | FTW | |
| D2 | −134.4 | FTW | |
| optB86b | −129.8 | FTW | |
| optB88 | −131.5 | FTW | |
| DF | −130.7 | FTW | |
| DF2 | −136.1 | FTW | |
| PBE | −142.1 | [21,22] | |
| CALPHAD | −143.6 | CTW b | |
| Na2Se | PBE | −109.6 | [32] |
| electrochemical and calorimetry | −114.2 | [38] | |
| emf | −131.9 | [50] | |
| calorimetry | −114.3 | [51] | |
| the NBS tables | −113.8 | [52] | |
| PBE | −106.3 | FTW | |
| D3 | −108.5 | FTW | |
| D2 | −116.7 | FTW | |
| optB86b | −108.5 | FTW | |
| optB88 | −114.1 | FTW | |
| DF | −115.1 | FTW | |
| DF2 | −119.6 | FTW | |
| PBE | −125.1 | [21,22] | |
| CALPHAD | −121.8 | CTW | |
| NaSe | PBE | −84.5 | [32] |
| emf | −100.0 | [50] | |
| calorimetry | −97.0 | [51] | |
| PBE | −82.6 | FTW | |
| D3 | −87.5 | FTW | |
| D2 | −94.7 | FTW | |
| optB86b | −85.5 | FTW | |
| NaSe | optB88 | −89.8 | FTW |
| DF | −90.7 | FTW | |
| DF2 | −93.2 | FTW | |
| PBE | −110.8 | [21,22] | |
| CALPHAD | −106.0 | CTW | |
| Na2Se3 | emf | −93.4 | [50] |
| −88.7 | CTW | ||
| NaSe2 | PBE | −58.3 | [32] |
| emf | −86.6 | [50] | |
| PBE | −56.9 | FTW | |
| D3 | −62.1 | FTW | |
| D2 | −65.4 | FTW | |
| optB86b | −58.3 | FTW | |
| optB88 | −61.0 | FTW | |
| DF | −61.6 | FTW | |
| DF2 | −62.8 | FTW | |
| PBE | −94.6 | [21,22] | |
| CALPHAD | −74.5 | CTW | |
| NaSe3 | CALPHAD | −56.3 | CTW |
a FTW, calculated by DFT in this work; b CTW, calculated by CALPHAD in this work.
There are a few thermodynamic property data in the literature for the liquid and solid phases in the Na-Se binary system. The activities of Na in the liquid solution were measured over the composition range of 54 to 99.8 at.% Se at 348 °C and 75 to 97 at.% Se at 310 °C by Tumidajski and Toguri [47], using emf measurement. Later, Morachevskii et al. [50] reported the activities of Na in liquid alloys contains 50 to 98 at.% Se at 527 °C and derived the enthalpy of mixing of the liquid solution.
The standard enthalpy of formation of the Na2Se compound was first measured to be −110.733 ± 1.967 kJ/mol-atoms by Fabre [36], using aqueous solution calorimetry. Mulder and Schmidt [51] determined the enthalpy of formation of Na2Se and NaSe to be −114.367 and −104.6 kJ/mol-atoms, respectively, using a liquid ammonia calorimeter. The enthalpies of formation of the Na2Se, NaSe, and NaSe2 compounds were calculated to be −109.6, −84.5, and −58.3 kJ/mol-atoms by Li et al. [32], using the DFT technique. As listed in Table 2, a large deviations among these reported results of the compounds’ enthalpy of formation requires a further verification. Thus, the DFT technique with different vdW methods (PBE, D3, D2, optB86b, optB88, DF, and DF2) was employed in the present work. The present calculated enthalpy of formation in the Na-Se binary system was presented in Table 2, along with the previous data [21,22,32,35,37,38,50,51,52].
3. Thermodynamic Models
The stable phases in the Li-Se and Na-Se systems considered in the present work were listed in Table 1, along with the model used to describe their thermodynamic properties. The thermodynamic parameters of the pure elements were taken from the SGTE pure substance database [53].
3.1. Liquid Phase
The liquid phase was described by the MQMPA developed by Pelton et al. [54,55]. The important advantages of the MQMPA compared with the Bragg–Williams model (BWM) for modeling the thermodynamic properties of liquid solutions, particularly in high-order systems, have been well demonstrated. The MQMPA is designed to give a better entropy expression even for highly ordered systems in order to permit ordering about any desired composition to be treated by using coordination numbers. Moreover, the energy change for the reaction is introduced as a function of composition with adjustable parameters. Please note that it is not intended as a proper theory of liquid structure, but as a mathematical formalism which has the simplicity and generality and relatively reliable interpolations and extrapolations. Thus, the MQMPA will usually provide better extrapolations into dilute solutions and more realistic entropy-of-mixing curves for higher-order systems. There are significant differences in the predicted thermodynamic properties of ternary and higher-order systems when using the binary parameters based on the MQMPA or the BWM, even if the optimizations of the binary systems look quite similar. It was beneficial to use MQMPA for the liquid phase since it takes into account the short-range-ordering of first nearest neighbors and second nearest neighbors simultaneously. In general, the MQMPA for the liquid phase can be expected to provide more accurate extrapolations regarding multicomponent systems based on parameters in the binary systems. A detailed description of the MQMPA model and its associated notations can be found elsewhere [54,55]. The same notations are used in the present study, and a brief description of the MQMPA model is given below:
For the binary A-B system, the quasi-chemical pair exchange reaction can be considered:
where pair represents the first-nearest-neighbor pair of atoms. The Gibbs energy change for the formation of one mole of pairs with the broken of A-A and B-B pairs according to Reaction (1) is . Let and be the number of moles of and , , , and be the number of moles of A-A, B-B and A-B pairs. Moreover, and are the coordination numbers of and . Then the Gibbs energy of the solution is given by the following:
where and are respectively, the standard molar Gibbs energies of the pure component and , and is the configurational entropy of mixing given by randomly distributing the A-A, B-B and A-B pairs in the one-dimensional Ising approximation. The expression for is as follows:
where , , and are the mole fractions of the A-A, B-B and A-B pairs, respectively; and YA and YB are the coordination-equivalent fractions of and :
where of ZA and ZB are the coordination number of A and B atoms in liquid solution that vary with composition, as follows:
It may be noted that there is no exact expression for the configurational entropy in three dimensions. Although Equation (3) is only an approximate expression in three dimensions, it is exact one-dimensionally (when Z = 2) [55]. As explained in Reference [55], one is forced by the approximate nature of Equation (3) to use non-exact values for the coordination numbers in order to yield good fits between the experimental data and calculated ones. The mathematical approximation of the one-dimensional Ising model of Equation (3) can be partially compensated by selecting values of ZA and ZB which are smaller than the experimental values [56]. As is known, the MQMPA model is sensitive to the ratio of coordination numbers, but less sensitive to their absolute values. From a practical standpoint for the development of large thermodynamic databases, values of ZA and ZB of the order of 6 were found to be necessary for the solutions with a small or medium degree of ordering (i.e., alloy solutions).
Furthermore, is the model parameter to reproduce the Gibbs energy of liquid phase of the A-B binary system, which is expanded as a polynomial in terms of the pair fractions, as follows:
where is the adjustable model parameters which can be a function of the temperature. The letters s and t stand for any real number greater than or equal to 0. The equilibrium state of the system is obtained by minimizing the total Gibbs energy at a constant overall elemental composition, temperature, and pressure. The equilibrium pair distribution is calculated by setting the following:
This gives the “equilibrium constant” for the “quasi-chemical pair reaction” of Equation (1):
Moreover, the model permits ZA and ZB to vary with composition as follows:
where and are the values of when all nearest neighbors of an atom are , and when all nearest neighbors of an atom are respectively. and are defined similarly. The composition of maximum short-range-ordering is determined by the ratio of the coordination numbers . In the present study, = = 6 to keep the consistency with the previous study, but and were determined in each binary system to reproduce the phase diagram and thermodynamic properties of liquid solution.
3.2. Solid Solutions
The compound energy formalism (CEF) was introduced by Hillert to describe the Gibbs energy of solid phases with sublattices, and ideal mixing of atoms on each sublattice is assumed [57]. In this work, the solid solutions bcc_A2 and hex_A8 (Se) in the Li-Se and Na-Se binary systems were modeled with the CEF, with their sublattice stoichiometry based on the reported crystal structures. Taking the bcc_A2 solution as an example, its Gibbs energy expression, based on the CEF, is obtained by mixing Li, Na, and Se atoms on one sublattice as (Li, Na, and Se). The Gibbs energy of the bcc_A2 solution can be expressed as follows:
where , , and are the site fractions of elements Li, Na and Se on the sublattice. The values of , and represent the Gibbs free energies of the end members of the pure Li, Na and Se with bcc crystal structure. Moreover, represents the second-nearest-neighbor interaction parameters between Li↔Na, Li↔Se, or Na↔Se atoms in the sublattice, which can be expressed as follows:
The values of the temperature functions are the model parameters evaluated in the present work.
3.3. Stoichiometric Phases
The standard molar Gibbs energies of solid and liquid pure elements and all stoichiometric phases (Li2Se, Na2Se, NaSe, Na2Se3, NaSe2, and NaSe3) can be described by the following:
where is the molar enthalpy of formation of a given species from pure elements ( of element stable at 298.15 K and 1 atm is assumed to be 0 J·mol−1; reference state), is the molar entropy at 298.15 K, and is the molar heat capacity. As the experimental heat-capacity data for the intermetallics are unavailable, the heat capacities were evaluated by using the Neumann–Kopp rule [58] from its elements.
3.4. Gaseous Phase
Known gas species in this work are Li(g), Li2(g), Na(g), Na2(g), Se(g), and Se2(g) [59,60]. The standard Gibbs energy of each gas species can be expressed by Equations (15)–(17). They were taken from the FACT pure substances database, which utilized compiled values available in relevant compilations [59,60]. The ideal solution was assumed for the gaseous phase.
4. First-Principles Calculations
Due to Li, Na, and Se elements being relatively active, it is difficult to obtain accurate results, and one has to take extra precaution during an experimental procedure. Thus, it is necessary to test through the DFT technique. Based on DFT, all the first-principle calculations were performed with Vienna Ab initio Simulation Package (VASP) [61,62,63,64] and dealt with the ALKEMIE platform [65]. The Perdew–Burke–Ernzerhof functional (PBE) of generalized gradient approximation (GGA), using the projector augmented-wave (PAW) potential, was chosen to deal with the electron–ion interaction [66,67]. To provide an improved description of the van der Waals (vdW) interaction in Se, the advanced vdW corrected methods [68] and nonlocal van der Waals density functional (vdW-DF) [69] were introduced. Li (2s1), Na (3s1), and Se (4s24p4) were employed as the valence-electron configurations. The cutoff energy of 700 eV was set for both the Li-Se and Na-Se alloys. The following based on the Monkhorst–Pack scheme [70] were adopted as Brillouin Zone sampling: 14 × 14 × 14 for Li and Se, 10 × 10 × 10 for Li2Se, 8 × 8 × 8 Na2Se, 8 × 8 × 3 for NaSe, and 4 × 4 × 5 for NaSe2. The Gamma-centered MP grid was employed for some low-symmetric structures. The relaxation convergences for ions and electrons were 1 × 10−5 eV and 1 × 10−6 eV, respectively.
The formation energy, , of crystal was obtained according to , where , , and were the total energy of and chemical potentials of A and B atoms, respectively. The calculated lattice parameters and enthalpy of formation of the phases of Li2Se, Na2Se, NaSe, and NaSe2 at 0 K, using standard PBE functional and different vdW corrections, are summarized in Table 1 and Table 2, respectively.
5. Results and Discussions
5.1. Thermodynamic Modeling and Evaluation of the Li-Se Binary System
The calculated enthalpy of formation of Li2Se in the Li-Se binary system at 25 °C is shown in Figure 1 and compared with the previously reported results [32,35,37,38] and the present result with DFT. It is noted that, by including the vdW correction, the present DFT result can better reproduce the enthalpy of formation of Li2Se than the standard PBE method in the Materials Project [21,22]. The present optimized result of enthalpy of formation of Li2Se is −143.62 kJ/mol-atoms, which is in good agreement with the experimental data (−138.97 to −148.7 kJ/mol-atoms) and DFT result (−134.4 kJ/mol-atoms). The calculated entropy of formation of the Li2Se phase at 25 °C is compared in Figure 2 with the estimated data by Morachevskii [35] and Ader [38]. The optimized entropy is within the error limit of the estimated values.
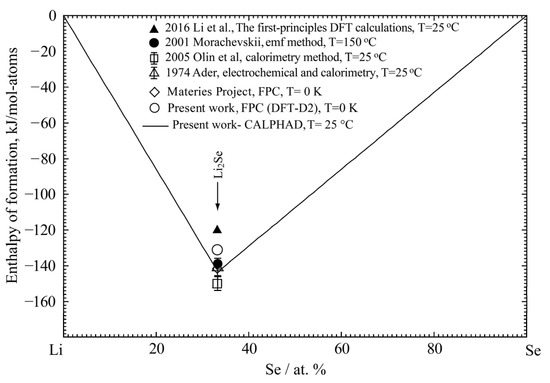
Figure 1.
The calculated enthalpy of formation of the Li-Se binary system at 25 °C compared with the previously reported data [32,35,37,38] and the present results.
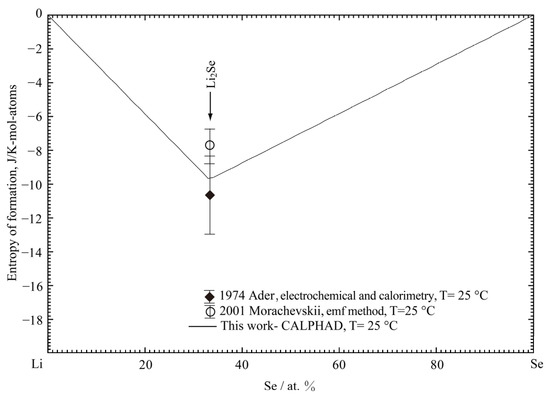
Figure 2.
The calculated entropy of formation of the Li-Se binary system at 25 °C compared with the experimental data [35,38].
There is no information reported for the short-range-ordering behavior of Li and Se atoms in the liquid solution. The proposed composition with maximum short-range-ordering of liquid was estimated according to the composition with most negative of enthalpy of formation of solids, as shown in Figure 2. Thus, in order to reproduce such short-range ordering behavior, binary coordination numbers of and were set for the MQMPA in the present study, as listed in Table 3.
Table 3.
Optimized model parameters of the liquid phase with MQMPA in the Li-Se and Na-Se binary systems.
Very limited thermodynamic property data are available for the liquid Li-Se solution. Morachevskii [35] estimated the Gibbs energy of mixing in the Se-rich region from the emf data at 420 °C by Chekova et al. [34] in the composition range from 0.2 to 15.9 at.% Li. According to the phase diagram data, the composition range of the experiment by Chekova et al. [34] is in the liquid miscibility gap. Therefore, the partial Gibbs energy of Li in liquid solution should be constant. Based on this assumption and the experimental activity coefficient of Li in nearly pure Se [34], Morachevskii [35] estimated the integral Gibbs energy of liquid solution in the Se-rich region at 420 °C. The calculated Gibbs energy of mixing of the liquid phase of the Li-Se binary system at 420 °C is shown in Figure 3 in comparison with the estimated data by Morachevskii [35]. As shown in Figure 3, the present calculated results are in good agreement with the estimated values.
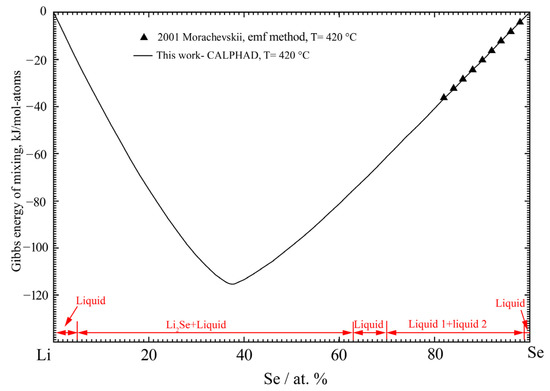
Figure 3.
The calculated Gibbs energy of mixing for the liquid phase of the Li-Se binary system at 420 °C compared with the reported results by Morachevskii [35].
The model parameters for solid Li2Se and liquid Li-Se solution were optimized to reproduce the phase diagram data in Figure 4 and all the available thermodynamic data in Figure 1, Figure 2 and Figure 3. Using the optimized model parameters, the thermodynamic properties and phase equilibria can be back calculated. Figure 5 shows the calculated entropy of mixing of liquid solution at 1400 °C in the present study. Extremely negative entropy of mixing of liquid solution (~−1.4 J/(K∙mol-atoms)) is calculated near 33.3 at.% Se. The entropy of mixing shows the typical M-shaped curve, which indicates that the liquid phase has the maximum short-range-order, with the composition being around 33.3 at.% Se.
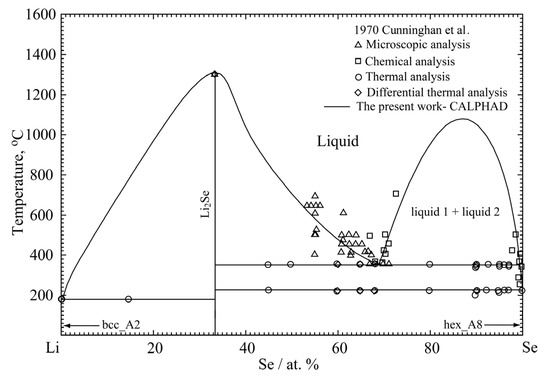
Figure 4.
The calculated phase diagram of the Li-Se binary system compared with the experimental data [27].
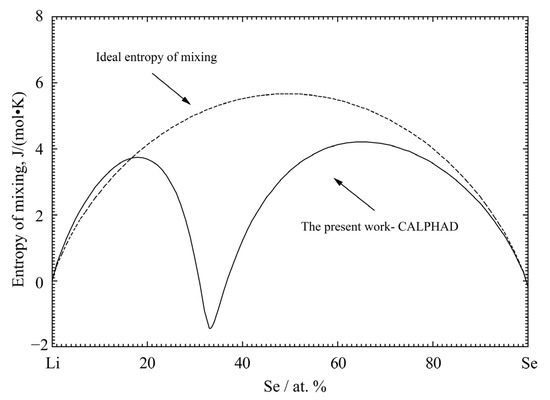
Figure 5.
The calculated entropy of mixing for the liquid phase of the Li−Se binary system at 1400 °C.
The calculated phase diagram of the Li-Se binary system is shown in Figure 4, along with the experimental data from Cunningham et al. [27]. As can be seen, the phase equilibria of the Li-Se binary system are well reproduced. The melting point of the Li2Se compound was calculated to be 1302 °C in the present optimization; thus, it is in good agreement with the experimental data [27]. According to the present optimization results, the critical temperature of liquid immiscibility in the Se-rich region was calculated to be 1068 °C with a liquid composition of 86.9 at.% Se. The optimized model parameters for the Li-Se binary system are listed in Table 3 and Table 4. The calculated invariant reactions and the experimental results from Cunningham et al. [27] are summarized in Table 5.
Table 4.
Optimized thermodynamic parameters of solid solutions and compounds in the Li-Se and Na-Se binary systems.
Table 5.
Calculated invariant reactions in the Li-Se system compared with experimental data [27].
Although the Gibbs energies of liquid and solid phases in alloy systems are not very sensitive to the external pressure, the Gibbs energy of the gas phase depends highly upon the external pressure. The standard vapor pressure of Se is very high at high temperatures, so there is a high possibility of vaporization of alloy in the Se-rich region at high temperatures with decreasing total pressure of the system. The phase diagram of the Li-Se binary system under different pressures is calculated in Figure 6. In Figure 6, colored curves represent the isobars of Li-Se ideal mixed gas at different pressure. For example, the red curve represents the Li-Se ideal mixed gas isobar at the pressure of 10−3 atm. As can be seen from Figure 6, the evaporation temperature of the liquid phase decreases with decreasing pressure, meaning that the evaporation of liquid phase can be significantly influenced by the pressure. Moreover, the sublimation of intermediate compound can be expected with the decreasing of pressure. This high vapor pressure of the system can easily induce difficulty in the phase diagram measurement at high temperatures. For example, if experiments were carried out with an open crucible under gas-purging condition, the significant Se would be evaporated, and it would be hard to maintain the exact initial composition. Therefore, special care would be necessary to handle the materials and perform the experiments of this binary alloy system.
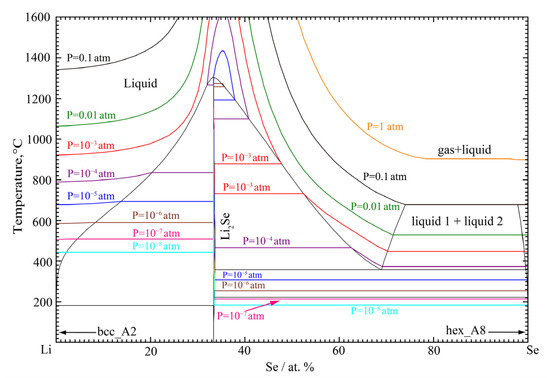
Figure 6.
The calculated phase diagram of the Li-Se binary system under different pressures.
5.2. Thermodynamic Modeling and Evaluation of the Na-Se Binary System
Figure 7 shows the calculated enthalpies of formation of all solid phases at 25 °C compared with the experimental data from References [32,38,50,51,52] and the present DFT results. The results obtained with FPC are more negative than the experimental values. This is because the vdW correction can better reproduce the enthalpy of formation of compounds in the Na-Se binary system. As shown in Figure 7, a reasonable agreement was obtained in the present optimization.
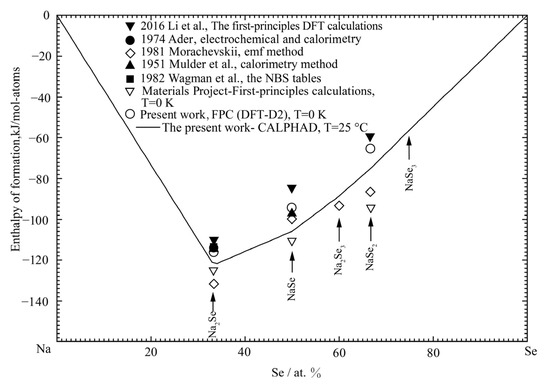
Figure 7.
The calculated enthalpy of formation of the Na-Se binary system at 25 °C compared with the previously reported data [32,38,50,51,52] and the present DFT results.
Figure 8 presents the calculated entropies of formation of all solid phases at 25 °C compared with the experimental results [38,71]. The optimized entropies are within the error limit of the experimental value. It should be noted that the enthalpies of formation of solids are extremely negative, so its negative entropies of formation would be reasonable. It is similar to the Li-Se binary liquid solution, as discussed above. The proposed composition with maximum short-range-ordering of liquid was estimated according to the composition with the most negative of enthalpy of formation of solids. Thus, according to the enthalpy of formation of solids, as shown in Figure 7, the binary coordination numbers of and of the Na-Se binary system were set for the MQMPA in the present study, as listed in Table 3.
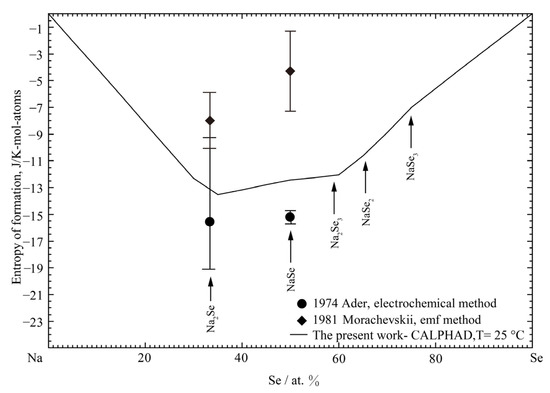
Figure 8.
The calculated entropy of formation of the Na-Se binary system at 25 °C compared with the experimental data [38,71].
Only a few thermodynamic data of the liquid solution were available in the Na-Se binary system. Similar to the Li-Se system, Morachevskii [35] estimated the Gibbs energy of mixing in the Se-rich region from the emf data at 523 °C in the composition range from 20 to 50 at.% Se. According to the phase diagram data, the composition range of the experiment by Morachevskii [35] is in the liquid region. Therefore, the partial Gibbs energy of Na in liquid solution should be constant. The calculated Gibbs energy of the mixing of liquid phase at 523 °C compared with the experimental data from Morachevskii [35] is shown in Figure 9. As shown in Figure 9, the present calculated results are in good agreement with the estimated values. Figure 10 shows the calculated entropy of the liquid phase at 1400 °C.
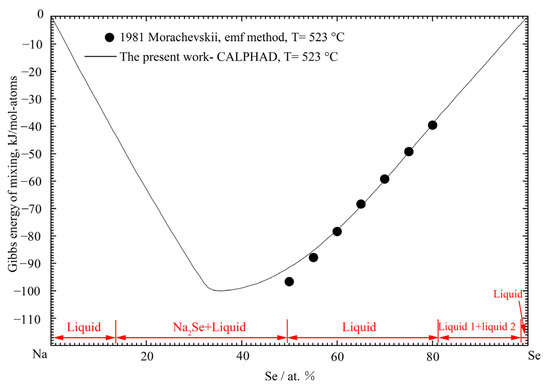
Figure 9.
The calculated Gibbs energy of mixing for the liquid phase of the Na-Se binary system at 523 °C compared with the previously calculated result by Romanchenko [71].
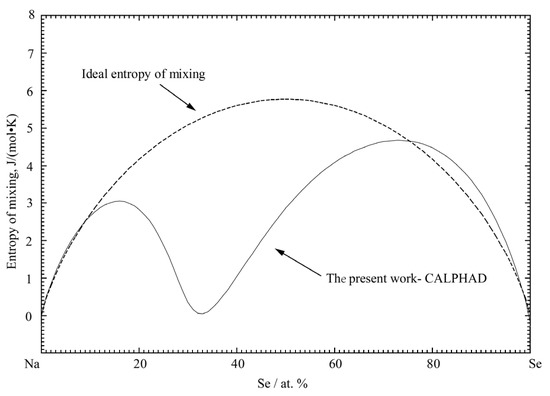
Figure 10.
The calculated entropy of mixing for the liquid phase of the Na-Se binary system at 1400 °C.
The calculated phase diagram of the Na-Se binary system is shown in Figure 11, along with the experimental data from References [40,49,50]. The present calculated results are consistent with most of the experimental results shown in Figure 11. The Na-rich part of the calculated curve is slightly different from the experimental data measured by Mathewson [40]. As the high vapor pressure and oxidation of Na-Se alloys occur in the experimental processes, the accuracy of the experimental results is questionable. Therefore, the difference between the present calculated results and the experimental results is acceptable. The melting point of the Na2Se compound was estimated to be in the range of 900 to 1150 °C by Sangster and Pelton [42], taking the melting temperatures of Li2Se, Rb2Se, Li2S, and Na2S as references. Therefore, according to the present optimization of the Na-Se system, the melting point of the Na2Se compound was fixed to be 1087 °C. Moreover, the critical temperature of liquid immiscibility in the Se-rich region was calculated to be 752 °C with the liquid composition of 91.9 at.% Se. All the optimized parameters of the stable phases are listed in Table 3 and Table 4. The calculated invariant reactions and the experimental results from Sangster et al. [42] are all listed in Table 6.
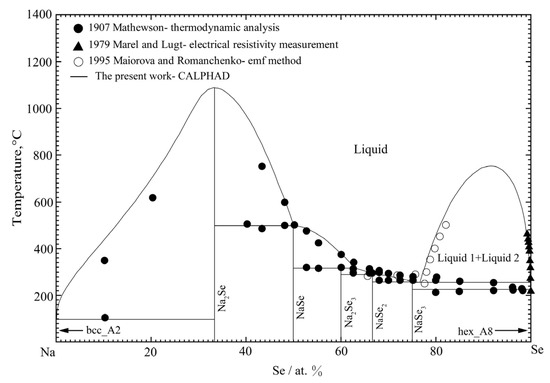
Figure 11.
The calculated phase diagram of the Na-Se binary system compared with the experimental data [40,49,50].
Table 6.
Calculated invariant reactions in the Na-Se system compared with the experimental data [42].
The phase diagram of the Na-Se binary system under different pressures is calculated in Figure 12. In Figure 12, colored curves represent the isobars of Na-Se ideal mixed gas at different pressure. For example, the red curve represents the Na-Se ideal mixed gas isobar at the pressure of 10−3 atm. It can be seen that the evaporation temperature of the liquid phase decreases with the decreasing external pressure. This is similar to the Li-Se binary system in Figure 6. It also means that, as the external pressure decreases, the evaporation of the liquid phase can be significant, and even the sublimation of intermediate compounds can be expected. This high vapor pressure of the Na-Se system can easily induce difficulty in the phase diagram measurement at high temperatures. To sum up, the thermodynamic properties and phase equilibria of the Na-Se binary system were reproduced well in the present work.
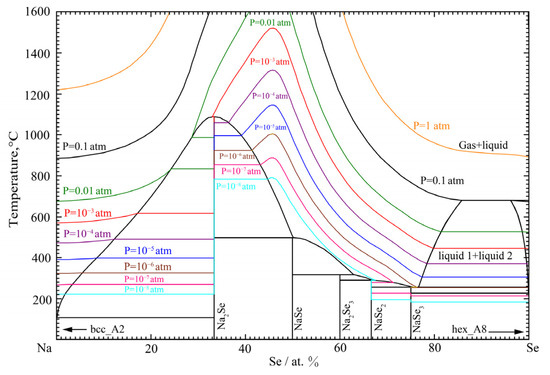
Figure 12.
The calculated phase diagram of the Na-Se binary system under different pressures.
6. Conclusions
A critical evaluation and thermodynamic optimization of the Li-Se and Na-Se binary systems were presented in the present work. The MQMPA model was employed to describe the liquid solution exhibiting a high degree of short-range-ordering behavior. The thermodynamic properties of the compounds Li2Se , Na2Se , NaSe , and NaSe2 were calculated by using DFT to assist the thermodynamic medeling of these two binary systems. All the available and reliable experimental data were reproduced within experimental error limits. The phase equilibria of these two systems at low total pressure were analyzed by using the developed thermodynamic models. Finally, a self-consistent thermodynamic database of Li-Se and Na-Se binary systems was constructed as a part of the work of a comprehensive research program to develop the thermodynamic database of novel medical absorbable materials [72,73,74,75].
Author Contributions
Conceptualization, J.W.; data curation, J.M. and C.J.; investigation, J.M. and C.J.; formal analysis, J.M. and D.H.; writing—original draft, J.M.; software, C.J.; validation, Z.C. and J.W.; writing—review and editing, J.W., B.S., and S.C.; supervision, J.W. and S.C.; funding acquisition, J.M. and J.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Anhui Province, 2108085QE213; the Nature Science Foundation of the Universities of Anhui Province, KJ2019A0776; and the Technology Project of Shenzhen City, JCYJ20170815153210359.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Atrens, A.; Liu, M.; Abidin, N. Corrosion mechanism applicable to biodegradable magnesium implants. Mater. Sci. Eng. B 2011, 176, 1609–1636. [Google Scholar] [CrossRef]
- Staiger, M.P.; Pietak, A.M.; Jerawala, H.; George, D. Magnesium and its alloys as orthopedic biomaterials: A review. Biomaterials 2006, 27, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Dolly, M.; Sravanthr, R.; Li, Y.C.; Kumar, M.J.; Harishankar, N.; Hodgson, P.D.; Wen, C.; Pande, G. Zirconium, calcium, and strontium contents in magnesium based biodegradable alloys modulate the efficiency of implant-induced osseointegration. Int. J. Nanomed. 2013, 8, 2887–2902. [Google Scholar]
- Mendis, C.L.; Bettles, C.J.; Gibson, M.A.; Hutchinson, C.R. An enhanced age hardening response in Mg-Sn based alloys containing Zn. Mater. Sci. Eng. A 2006, 435-436, 163–171. [Google Scholar] [CrossRef]
- Mendis, C.L.; Bettles, C.J.; Gibson, M.A.; Gorsse, S.; Hutchinson, R. Refinement of precipitate distributions in an age-hardenable Mg–Sn alloy through microalloying. Phil. Mag. Lett. 2006, 86, 443–456. [Google Scholar] [CrossRef]
- Sasaki, T.T.; Oh-ishi, K.; Ohkubo, T.; Hono, K. Enhanced age hardening response by the addition of Zn in Mg-Sn alloys. Scr. Mater. 2006, 55, 251–254. [Google Scholar] [CrossRef]
- Gibson, M.A.; Fang, X.; Bettles, C.J.; Hutchinson, C.R. The effect of precipitate state on the creep resistance of Mg-Sn alloys. Scr. Mater. 2010, 63, 899–902. [Google Scholar] [CrossRef]
- Persaud-Sharma, D.; Budiansky, N.; McGoron, A.J. Mechanical properties and tensile failure analysis of novel bio-absorbable Mg-Zn-Cu and Mg-Zn-Se alloys for endovascular applications. Metals 2013, 3, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, Y.D.; Wu, S.D.; Peng, R.L.; Huang, C.X.; Jiang, C.B.; Li, S.X. Textures and mechanical behavior of Mg-3.3%Li alloy after ECAP. Scr. Mater. 2004, 51, 1057–1061. [Google Scholar] [CrossRef]
- Mitsuaki Furui, C.X.; Tetsuo, A.; Makoto, I.; Hiroshi, A.; Langdon, T.G. Improving the superplastic properties of a two-phase Mg–8%Li alloy through processing by ECAP. Mater. Sci. Eng. A 2005, 410-411, 439–442. [Google Scholar] [CrossRef]
- Schrauzer, G.N. Lithium: Occurrence, Dietary intakes, nutritional essentiality. J. Am. Coll. Nutr. 2013, 21, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Young, W. Review of lithium effects on brain and blood. Cell Transplant. 2009, 18, 951–975. [Google Scholar] [CrossRef] [PubMed]
- Alderman, M.H. Summary of the international workshop of dietary sodium and human health in China and the United States. Am. J. Hypertens. 2009, 22, 925–928. [Google Scholar] [CrossRef][Green Version]
- Kipp, A.P.; Strohm, D.; Brigelius-Flohé, R.; Schomburg, L.; Bechthold, A.; Leschik-Bonnet, E.; Heseker, H.; German Nutrition Society (DGE). Revised reference values for selenium intake. J. Trace Elem. Med. Biol. 2015, 32, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Fairweather-Tait, S.J.; Bao, Y.P.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in human health and disease. Antioxid. Redox Signal. 2017, 14, 1337–1383. [Google Scholar] [CrossRef] [PubMed]
- Rederstorff, M.; Krol, A.; Lescure, A. Understanding the importance of selenium and selenoproteins in muscle function. Cell. Mol. Life Sci. 2006, 63, 52–59. [Google Scholar] [CrossRef]
- Flores-Mateo, G.; Ana, A.N.; Pastor-Barriuso, R.; Guallar, E. Selenium and coronary heart disease: A meta-analysis. Am. Soc. Nutr. 2006, 84, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.; Miodownik, A.P. Calphad (Calculation of Phase Diagrams) A Comprehensive Guide; Pergamon: Oxford, UK, 1992. [Google Scholar]
- Spencer, P.J. Mg-Li system. In Thermodynamic Database; CRCT, Ed.; CRCT: Montreal, QC, Canada, 2006. [Google Scholar]
- Chartrand, P. Mg-Na system. In Thermodynamic Database; CRCT, Ed.; CRCT: Montreal, QC, Canada, 2003. [Google Scholar]
- Jomard, G.; Petit, T.; Magaud, L.; Pasturel, A. First principles calculations to describe zirconia pseudopolymorphs. MRS Proc. 1997, 492, 4044–4052. [Google Scholar] [CrossRef]
- Jain, A.; Hautier, G.; Ong, S.; Moore, C.; Fischer, C.; Persson, K.; Ceder, G. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 2011, 84, 045115. [Google Scholar] [CrossRef]
- Wang, J.; Han, D.; Zhang, Z.; Jung, I.-H.; Rao, W.-F. Experimental measurement and thermodynamic evaluation of the Mg + Cu + Sr ternary system. J. Chem. Thermodyn. 2021, 163, 106582. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Li, S.; Meng, L.; Cao, Z.; Rao, W.-F. Experimental investigation and thermodynamic modeling of the Mg-Cu-Ca ternary system. Calphad 2021, 75, 102325. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Jung, I.-H.; Sheng, L. Experimental investigation and thermodynamic modeling of the Mg–Sn–Sr ternary system. Calphad 2021, 72, 102237. [Google Scholar] [CrossRef]
- Bale, C.W.; Chartrand, P.; Degterov, S.A.; Eriksson, G.; Hack, K.; Mahfoud, R.B.; Melancon, J.; Pelton, A.D.; Petersen, S. FactSage thermochemical software and databases. Calphad 2002, 26, 189–228. [Google Scholar] [CrossRef]
- Cunningham, P.; Johnson, S.; Cairns, E. Phase equilibria in lithium-chalcogen systems: I. lithium-selenium. J. Electrochem. Soc. 1971, 118, 1941. [Google Scholar] [CrossRef]
- Zintl, E.; Harder, A.; Dauth, B. Gitterstruktur der oxyde, sulfide, selenide und telluride des lithiums, natriums und kaliums. Z. Für Elektrochem. Und Angew. Phys. Chem. 2010, 40, 588–593. [Google Scholar] [CrossRef]
- Johnston, W.; Heikes, R. A study of the system LixMn(1-X)Se. J. Am. Chem. Soc. 1958, 80, 5904–5907. [Google Scholar] [CrossRef]
- Sangster, J.; Pelton, A. The Li-Se (lithium-selenium) system. J. Phase Equilibria 1997, 18, 181–184. [Google Scholar] [CrossRef]
- Bergstrom, F. The polysulfides and polyselenides of lithium, sodium and potassium. J. Am. Chem. Soc. 1926, 48, 146–151. [Google Scholar] [CrossRef]
- Li, Q.; Liu, H.; Yao, Z.; Cheng, J.; Li, T.; Li, Y.; Wolverton, C.; Wu, J.; Dravid, V. Electrochemistry of selenium with sodium and lithium: Kinetics and reaction mechanism. ACS Nano 2016, 10, 8788. [Google Scholar] [CrossRef]
- Cairns, E.; Kucera, G.; Cunningham, P. Thermodynamic studies of the lithium-selenium system by an EMF method. J. Electrochem. Soc. 1973, 120, 595–602. [Google Scholar] [CrossRef]
- Chekoev, N.G.; Morachevskij, A.G. Propriétés thermodynamiques des masses fondues du systèmes lithium-selenium. Gen. Chem. Phys. Chem. 1978, 51, 2348–2350. [Google Scholar]
- Morachevskii, A. Thermodynamic analysis of alloying in the system lithium-selenium. Russ. J. Appl. Chem. 2001, 74, 569–571. [Google Scholar] [CrossRef]
- Fabre, C. Thermal studies on the selenides. Ann. Chim. Phys. 1887, 10, 472–550. [Google Scholar]
- Olin, Å.; Noläng, B.; Osadchii, E.; Öhman, L.-O.; Rosén, E. Chemical Thermodynamics of Selenium; Elsevier: Amsterdam, Holland, 2005. [Google Scholar]
- Ader, M. Cheminform abstract: Enthalpy of formation of lithium monoselenide. J. Chem. Thermodyn. 1974, 6, 587–597. [Google Scholar] [CrossRef]
- Voronin, G. Estimation of the standard entropies of chemical compounds. Zhur. Fiz. Khim. 1970, 44, 3013–3017. [Google Scholar]
- Mathewson, C. The selenides of sodium. J. Am. Chem. Soc. 1907, 29, 867–880. [Google Scholar] [CrossRef][Green Version]
- Sangster, J.; Pelton, A. The K-Se (potassium-selenium) system. J. Phase Equilibria 1997, 18, 177–180. [Google Scholar] [CrossRef]
- Sangster, J.; Pelton, A. The Na-Se (sodium-selenium) system. J. Phase Equilibria 1997, 18, 185–189. [Google Scholar] [CrossRef]
- Klemm, W.; Sodomann, H.; Langmesser, P. Beiträge zur kenntnis der alkalimetallchalkogenide. Z. Anorg. Allg. Chem. 2004, 241, 281–304. [Google Scholar] [CrossRef]
- Föppl, H.; Busmann, E.; Frorath, F.K. Die kristallstrukturen von α-Na2S2 und K2S2, β-Na2S2 und Na2Se2. Z. Anorg. Allg. Chem. 2004, 314, 12–20. [Google Scholar] [CrossRef]
- Morachevskii, A.; Bykova, M.; Rozova, T. Thermodynamic Properties of Liquid Alloys in the Sodium-Indium System; Liniya: Leningrad, Russia, 1970; pp. 1611–1612. [Google Scholar]
- Maiorova, E.; Morachevskii, A. Thermodynamic properties of dilute sodium solutions in liquid selenium. Soviet Electrochem. 1978, 14, 1266–1267. [Google Scholar]
- Tumidajski, P.J.; Toguri, J. Thermodynamic properties of the Na-Se and Rb-Se systems. Can. Metall. Q. 1991, 30, 169–171. [Google Scholar] [CrossRef]
- Maiorova, E.A.; Romanchenko, N.M.; Morachevskii, A.G. Investigation of polysulfide melts by means of a sodium microelectrode. Russ. J. Appl. Chem. 1981, 17, 429–432. [Google Scholar]
- Van der Marel, C.; van der Lugt, W. Electrical resistivity and phase separation in dilute liquid alloys of sodium in selenium. Z. Naturforsch. A 1979, 34, 832–835. [Google Scholar] [CrossRef]
- Morachevskii, A.; Romanchenko, N.; Maiorova, E. Thermodynamic analysis of components’ interaction in the sodium-selenium system. Russ. J. Appl. Chem. 1995, 68, 327–330. [Google Scholar]
- Mulder, H.D.; Schmidt, F.C. Heats of solution and reaction in liquid ammonia. VIII. J. Am. Chem. Soc. 1951, 73, 5575–5577. [Google Scholar] [CrossRef]
- Wagman, D.; Evans, W.; Parker, V.; Schumm, R.; Har-low, I.; Bailey, S.; Churney, K.; Nuttall, R. The NBS tables of chemical thermodynamic properties. J. Phys. Chem. Ref. Data 1982, 11, 2–61. [Google Scholar]
- Dinsdale, A.T. SGTE data for pure elements. Calphad 1991, 15, 317–425. [Google Scholar] [CrossRef]
- Pelton, A.D.; Chartrand, P. The modified quasi-chemical model: Part II. Multicomponent solutions. Metall. Mater. Trans. A 2001, 32, 1355–1360. [Google Scholar] [CrossRef]
- Pelton, A.D.; Degterov, S.A.; Eriksson, G.; Robelin, C.; Dessureault, Y. The modified quasichemical model I—Binary solutions. Metall. Mater. Trans. B 2000, 31, 651–659. [Google Scholar] [CrossRef]
- Kang, Y.B.; Pelton, A.D.; Chartrand, P.; Fuerst, C.D. Critical evaluation and thermodynamic optimization of the Al-Ce, Al-Y, Al-Sc and Mg-Sc binary systems. Calphad 2008, 32, 413–422. [Google Scholar] [CrossRef]
- Hillert, M. The compound energy formalism. J. Alloy. Compd. 2001, 320, 161–176. [Google Scholar] [CrossRef]
- Kopp, H. Investigations of the specific heat of solid bodies. Phil. Trans. R. Soc. 1865, 155, 71–202. [Google Scholar] [CrossRef]
- Gurvich, L.V.; Veyts, I.V.; Alcock, C.B. Thermodynamic Properties of Individual Substances, 4th ed.; Hemisphere Publishing Corporation: New York, NY, USA, 1996. [Google Scholar]
- Chase, M.W. NIST-JANAF Thermochemical Tables; AIP: Woodbury, MN, USA, 1998. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. B 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Lushtak, Y.; Burke, D.; Li, Y.; Lyndaker, A.; Liu, X.; Ying, L. Vacuum system design, construction, and operation for the cornell high energy synchrotron source upgrade. Vacuum 2021, 186, 110064. [Google Scholar] [CrossRef]
- Perdew, J.P.; Yue, W. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Klimes, J.; Bowler, D.R.; Michaelides, A. Chemical accuracy for the van der Waals density functional. J. Phys. Condens. Matter. 2010, 22, 022201. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Romanchenko, N.; Maiorova, E.; Morachevskii, A. Determination of the Limits of Phase-Separation in the Sodium-Selenium System by Electromotive-Force Measurements; Plenum Publishing Corporation: New York, NY, USA, 1981; pp. 615–616. [Google Scholar]
- Wang, J.; Chen, P.; Meng, L.; Jin, L. Investigation on the mechanical and corrosion properties of ZnMnSr alloys for biodegradable orthopedic implants. Adv. Eng. Mater. 2022, 2101581. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Zhang, Y.; Jin, L.; Sheng, L. Experimental measurement on the phase equilibria of the Mg–Ag–Cu ternary system at 350 and 400 °C. J. Magnes. Alloy. 2022, 10, 449–457. [Google Scholar] [CrossRef]
- Wen, C.L.; Qian, J.M.; Luo, L.L.; Zeng, J.H.; Sa, B.S.; Zhan, X.; Wang, J.; Sheng, L.Y.; Zheng, Y.F. Effect of nitrogen on the structure evolution and biological properties of mesoporous bioactive glass nanospheres: Experiments and simulations. J. Non-Cryst. Solids 2022, 578, 121329. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z.; Jung, I.H.; Rao, W.F. Critical evaluation and thermodynamic modeling of the Ag-X (X=Mn, Y, Sr) binary systems. Intermetallics 2021, 136, 107260. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).