Simple Summary
The mitochondrial genome of insects has been widely used in studies of molecular evolution, population genetics, phylogenetics, and species identification due to its small size (approximately 14–20 kb), relatively high evolutionary rate, low levels of recombination, and high genome copy numbers. In this study, 13 complete mitogenomes of Chiasmini, a diverse group of grassland leafhoppers, were sequenced and analyzed for the first time. The phylogenetic position of Chiasmini within leafhoppers and the phylogenetic relationships among Chiasmini genera were reconstructed. The results show that all 13 mitogenomes are composed of a circular, double-stranded molecule that consists of 37 genes with a total length ranging from 14,805 to 16,269 bp and a variable number of non-coding A + T-rich regions. The gene size, order, arrangement, base composition, codon usage, and secondary structure of tRNAs in these newly sequenced mitogenomes of 13 species are highly conserved in Chiasmini. Phylogenetic analysis of the newly sequenced genomes plus representatives of other tribes and subfamilies of Cicadellidae recover all included subfamilies of Cicadellidae and tribes of Deltocephalinae as monophyletic, except Athysanini and Opsiini in Deltocephalinae, which are paraphyletic in agreement with some other recent studies. Chiasmini form a monophyletic group consisting of seven monophyletic genera arranged as follows: ((Zahniserius + (Gurawa + (Doratura + Aconurella))) + (Leofa + (Exitianus + Nephotettix))).
Abstract
The grassland leafhopper tribe Chiasmini (Cicadellidae: Deltocephalinae) presently comprises 324 described species worldwide, with the highest species diversity occurring in the Nearctic region but a greater diversity of genera occurring in the Old World. In China, this tribe comprises 39 described species in 11 genera, but the fauna remains understudied. The complete mitogenomes of three species of this tribe have been sequenced previously. In order to better understand the phylogenetic position of Chiasmini within the subfamily Deltocephalinae and to investigate relationships among Chiasmini genera and species, we sequenced and analyzed the complete mitogenomes of 13 species belonging to seven genera from China. Comparison of the newly sequenced mitogenomes reveals a closed circular double-stranded structure containing 37 genes with a total length of 14,805 to 16,269 bp and a variable number of non-coding A + T-rich regions. The gene size, gene order, gene arrangement, base composition, codon usage, and secondary structure of tRNAs of the newly sequenced mitogenomes of these 13 species are highly conserved in Chiasmini. The ATN codon is commonly used as the start codon in protein-coding genes (PCGs), except for ND5 in Doratura sp. and ATP6 in Nephotettix nigropictus, which use the rare GTG start codon. Most protein-coding genes have TAA or TAG as the stop codon, but some genes have an incomplete T stop codon. Except for the tRNA for serine (trnS1(AGN)), the secondary structure of the other 21 tRNAs is a typical cloverleaf structure. In addition to the primary type of G–U mismatch, five other types of tRNA mismatches were observed: A–A, A–C, A–G, U–C, and U–U. Chiasmini mitochondrial genomes exhibit gene overlaps with three relatively stable regions: the overlapping sequence between trnW and trnC is AAGTCTTA, the overlapping sequence between ATP8 and ATP6 is generally ATGATTA, and the overlapping sequence between ND4 and ND4L is generally TTATCAT. The largest non-coding region is the control region, which exhibits significant length and compositional variation among species. Some Chiasmini have tandem repeat structures within their control regions. Unlike some other deltocephaline leafhoppers, the sequenced Chiasmini lack mitochondrial gene rearrangements. Phylogenetic analyses of different combinations of protein-coding and ribosomal genes using maximum likelihood and Bayesian methods under different models, using either amino acid or nucleotide sequences, are generally consistent and also agree with results of prior analyses of nuclear and partial mitochondrial gene sequence data, indicating that complete mitochondrial genomes are phylogenetically informative at different levels of divergence within Chiasmini and among leafhoppers in general. Apart from Athysanini and Opsiini, most of the deltocephaline tribes are recovered as monophyletic. The results of ML and BI analyses show that Chiasmini is a monophyletic group with seven monophyletic genera arranged as follows: ((Zahniserius + (Gurawa + (Doratura + Aconurella))) + (Leofa + (Exitianus + Nephotettix))).
1. Introduction
Insect mitochondrial genomes are typically small double-stranded circular molecules containing 37 genes, including 13 protein-coding genes (PCGs), 22 transfer RNAs (tRNAs) genes, 2 ribosomal RNAs (rRNAs) genes, and a control region (CR) or non-coding A + T-rich region [1]. The mitogenome is a suitable molecular marker for molecular evolution, phylogenetic relationships, and species identification due to its smaller size (approximately 14–20 kb), maternal inheritance, relatively high evolutionary rate, low levels of recombination, high genome copy numbers, and conservative gene components [2,3,4].
The grassland-associated leafhopper tribe Chiasmini (Cicadellidae: Deltocephalinae) presently comprises 324 described species in 21 genera distributed worldwide, with the greatest reported species diversity occurring in the Nearctic but higher genus-level diversity in the Oriental Region [5,6,7]. In China, Chiasmini comprises 39 described species in 11 genera: Aconurella Ribaut (11 species), Nephotettix Matsumura (6 spp.), Doratura Sahlberg (4 spp.), Leofa Distant (3 spp.), Gurawa Distant (4 spp.), Doraturopsis Lindberg (3 spp.), Exitianus Ball (3 spp.), Baileyus Singh-Pruthi (2 spp.), Chiasmus Mulsant & Rey (2 spp.), and Zahniserius Duan & Zhang (1 sp.) [5]. However, 13 species records are doubtful due to a lack of specimens [5,8,9,10,11,12,13,14,15]. Many species of the subfamily Deltocephalinae, including Chiasmini, are important agricultural pests, causing damage by piercing and sucking plant sap and transmitting plant pathogens [6]. Most members of the Chiasmini tribe feed on perennial grasses or sedges in native grasslands, but Nephotettix species are known to cause damage to cultivated rice, wheat, and barley, causing weakened growth, slowed development, and even the death of entire plants [15]. Exitianus species also transmit various pathogens affecting crops and forage grasses, such as maize dwarf mosaic disease and Bermuda grass white leaf disease [16,17].
Since the establishment of this tribe, various taxonomic changes have been proposed for the adjustment of various genera and species based on external body morphology, male and female genitalia, and molecular data [5,18,19,20,21,22,23]. The first phylogeny of Deltocephalinae, based on adult morphological characters and molecular data (two nuclear genes, 28S and H3), included only seven representatives of Chiasmini and did not recover the tribe as monophyletic. A subsequent more-detailed phylogenetic study of this tribe, including 21 genera and 316 species, incorporating molecular data from four genes (two mitochondrial, COII and ND1, and two nuclear genes, 28S and H3) also failed to consistently recover the tribe as monophyletic [23]. However, the recent comprehensive phylogenomic analysis of Deltocephalinae, Cao et al. [7] comprising 730 terminal taxa and >160,000 nucleotide positions obtained through anchored hybrid enrichment strongly supported the monophyly of Chiasmini and grouped New and Old World taxa into distinct lineages. Stenometopiini was recovered as the sister group of Chiasmini in the coalescent gene tree analysis, but concatenated maximum likelihood analyses suggested that the sister group of Chiasmini includes Stenometopiini plus three other grass-specialist tribes. In the most recent study, Zhang et al. [5] reconstructed the phylogenetic relationships of 20 Chinese chiasmine species from eight genera using four genes: two mitochondrial (COI and 16S) and two from the nuclear genome (28S and H3). Their results consistently supported the monophyly of Chiasmini and recovered stable relationships between the genera and species of Chiasmini, but some relationships were incongruent with those obtained in previous analyses. The phylogeny of this tribe has not yet been analyzed using full mitochondrial genome sequences. Mitochondrial genome sequences have been shown to be highly informative of relationships among other groups of deltocephaline leafhoppers, and some deltocephalines have mitochondrial gene rearrangements that provide additional phylogenetically informative characters [24]. Unfortunately, only three complete mitogenomes of Chiasmini have been sequenced and analyzed so far, so the value of such data for elucidating relationships within this tribe remains unexplored [25,26,27]. The taxonomy of the Chinese Chiasmini is well-studied [8,9,10,11,12,13,14,15], but complete mitogenome data are scarcely available. This study aimed to provide 13 new mitogenome sequences of the tribe Chiasmini, conduct comparative mitogenomic analyses, reconstruct the phylogenetic relationships between Chiasmini and other tribes in the subfamily Deltocephalinae, and estimate within the tribe using the data from published sources and newly sequenced mitogenomes.
2. Materials and Methods
2.1. Taxon Sampling
Leafhopper samples included in this study were collected between 2010 and 2019 from various collection sites in Anhui, Guangxi, Yunnan Provinces, and the Xinjiang Autonomous Region, China (Table 1). Fresh samples were initially preserved in 95% or 100% ethanol and subsequently stored at −80 °C before the study. Prior to DNA extraction, the samples were identified at the species level based on external morphology and male genitalia using the available taxonomic literature [8,9,10,11,12,13,14,15]. The external body morphology and male genitalia of each species were examined using Nikon SMZ1500 (Nikon Corporation, Tokyo, Japan) and Motic K-700HS microscopes (MacAudi Electric Co., Ltd., Xiamen, China). All specimens studied are listed in Table 1 and are deposited at Northwest A&F University, Yangling, China (NWAFU), and Anhui Agricultural University, Hefei, China (AAU).

Table 1.
Specimens and previously published data used in this study (all from China).
2.2. DNA Extraction and Sequencing
Genomic DNA was extracted from ethanol-preserved specimens following the standard protocol of the Qiagen DNeasy Blood and Tissue kit. The concentration and quality of DNA were determined by a Nanodrop 2000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA) and 1% agarose gel electrophoresis. The mitogenomes were sequenced using high-throughput sequencing on an Illumina NovaSeq platform (United States Illumina Company, San Diego, CA, USA) for each species, generating approximately 2 GB of data per sample with a read length of 150 bp. The raw data were subjected to quality control and filtering (removing adapter contamination, trimming reads shorter than 50 bp, removing reads with an average quality score lower than 20, and removing reads with more than 3 Ns), resulting in high-quality data for subsequent analysis.
2.3. Mitogenome Sequence Assembly and Annotation
The mitogenome sequences were assembled using NOVOPlasty software under the Linux system [28]. The assembled sequences were then aligned in GENEIOUS software v. 10.2.3 (https://www.geneious.com/, accessed on 12 December 2023) [29] to process degenerate bases and detect circularity, resulting in a complete mitogenome sequence.
The assembled sequences were compared with published mitochondrial genome sequences of other chiasmine leafhopper species (Aconurella prolixa [MZ433366], Exitianus indicus [KY039128], and Nephotettix cincticeps [KP749836]) using the built-in MAFFT plugin in GENEIOUS. The sequences of 37 individual genes, including the 13 PCGs, 22 tRNAs, and 2 rRNAs, were extracted from each mitogenome. The lengths and positions of the 13 protein-coding genes were confirmed by comparing them to multiple reference sequences, and the start and stop codons were identified within the open reading frames to annotate the protein-coding genes. The annotation of RNA genes and the prediction of tRNA secondary structure were performed using the Mitos online tool (http://mitos.bioinf.uni-leipzig.de/index.py, accessed on 12 December 2023) [30]. The assembled mitochondrial genome sequence was uploaded and analyzed using the invertebrate mitochondrial codon table to determine RNA genes’ start and stop positions. The prediction of tRNA secondary structures was also conducted on the same website.
2.4. Mitogenome Sequence Analysis
The relative synonymous codon usage (RSCU) for PCGs and nucleotide composition were calculated using PhyloSuite software (version 1.2.2). The annotated sequence file was imported into PhyloSuite to extract annotations. Tandem Repeats Finder software was used to predict tandem repeats in the control region under default settings [31]. The genetic distances of the protein-coding gene sequences among samples were analyzed using MEGA software (version 7.0) with the Kimura 2-parameter model. Non-synonymous and synonymous substitution rates and sliding window analyses of protein-coding gene sequences were calculated using DnaSP software (version 3.12.03) [32] using default settings, with a 200 bp sliding window and 20 bp overlap.
After removing stop codons, protein-coding genes, and rRNA genes were aligned using MAFFT software (version 7.520) [33,34] with G-strategy and Q-strategy for protein-coding genes and rRNA genes, respectively. Gblocks software (version 0.91b) was used to remove gaps and ambiguous positions under default settings [35]. Concatenation of the aligned sequence files was performed using PhyloSuite software (version 1.2.2). Substitution saturation was calculated using DAMBE software (version 6.0.0) [36] by importing the concatenated sequence files and selecting the Xia method. The heterogeneity of different combination datasets was analyzed using AliGROOVE software (version 1.0.8) [37], with different sequence files being imported and selecting Ambiguity for nucleotide sequences and BLOSUM62 for amino acid sequences.
2.5. Phylogenetic Analysis
PartitionFinder software 2.1.1 [38] was used to find the best partitioning scheme and the best-fitting substitution models for the first, second, and third codon positions of protein-coding genes and amino acid sequences (Table S1). The maximum likelihood (ML) tree was constructed using IQ-TREE software (version 2.2.0) [39] by importing the partitioned model, selecting the ultrafast bootstrap (UFB) algorithm, and evaluating the support of each node based on 1000 replicates. The Bayesian (BI) tree was estimated using MrBayes software (version 3.2.2) [40] by importing the partitioned model and running 4 MCMC chains for 40 million generations, with a sampling frequency of 1000 generations. After convergence, as determined by an average standard deviation below 0.01, the first 25% of trees were discarded, and a consensus tree was calculated to evaluate the posterior probability of each node.
3. Results and Discussion
3.1. Mitogenome Analysis
In this study, a total of 13 new mitogenomes were sequenced for leafhoppers of the tribe Chiasmini from China. Comparative mitochondrial genomic analysis was performed based on both newly sequenced and three previously published mitogenomes, resulting from in a total of 16 Chiasmini species included in the subsequent phylogenetic analysis (Table 1).
3.2. Mitogenome Organization and Gene Content
All 16 Chiasmini mitochondrial genomes contain 37 genes, including 13 protein-coding genes, 22 transfer RNAs, 2 ribosomal RNAs, and a relatively long control region. The total length of the 16 Chiasmini mitochondrial genomes ranges from 14,805 to 16,269 bp, with the longest being Doratura homophyla (16,269 bp) and the shortest being Nephotettix cincticeps (14,805 bp) (Table 2). The 13 protein-coding genes range in combined length from 10,869 to 10,980 bp. The 22 tRNA genes range in combined length from 1433 to 1475 bp. The rrnL gene ranges in length from 1193 to 1216 bp. The rrnS gene ranges in length from 737 to 801 bp. The control region (CR) is by far the most variable, ranging in length from 399 to 1828 bp, and is the main factor affecting variation in mitochondrial genome size. Each newly sequenced mitogenome contains a typical set of 37 mitochondrial genes, 13 PCGs, 22 tRNAs, 2 rRNAs, and 1 control region (Figure 1 and Figure 2). We did not detect any gene rearrangements within Chiasmini; mitochondrial gene rearrangements are apparently rare among leafhoppers but have been reported in some other tribes of Deltocephalinae [24].
Table 2.
Length and base compositions of Chiasmini mitochondrial genomes.
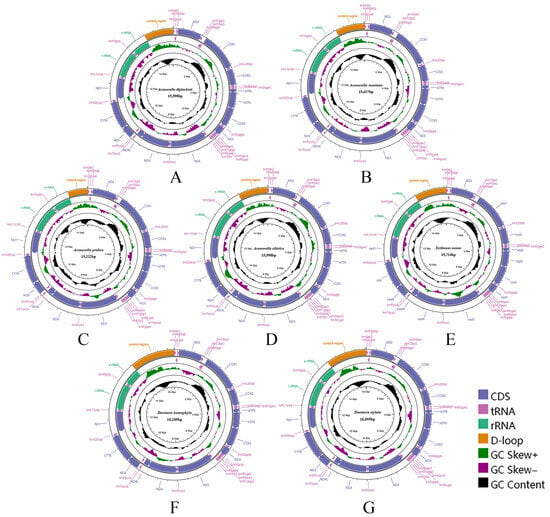
Figure 1.
Circular maps of the complete mitochondrial genomes of Chiasmini species. (A) Aconurella diplachnis; (B) Aconurella montana; (C) Aconurella prolixa; (D) Aconurella sibirica; (E) Exitianus nanus; (F) Doratura homophyla; (G) Doratura stylata.
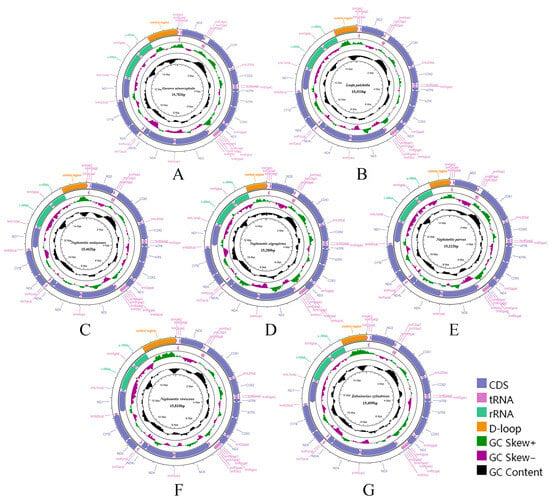
Figure 2.
Circular maps of the complete mitochondrial genomes of Chiasmini species. (A) Gurawa minorcephala; (B) Leofa pulchella; (C) Nephotettix malayanus; (D) Nephotettix nigropictus; (E) Nephotettix parvus; (F) Nephotettix virescens; (G) Zahniserius cylindricus.
3.3. Nucleotide Composition
The AT content of the 16 Chiasmini mitochondrial genomes ranges from 73.6% to 77.4%, and each component exhibits a relatively high AT content. Protein-coding genes (PCGs) have the lowest AT content, followed by tRNAs, rrnS, and rrnL. The highest AT content is found in the control region (CR), with a mean value exceeding 81%. This indicates a clear AT bias in the complete mitogenome in Chiasmini species (Table 2 and Figure 3).
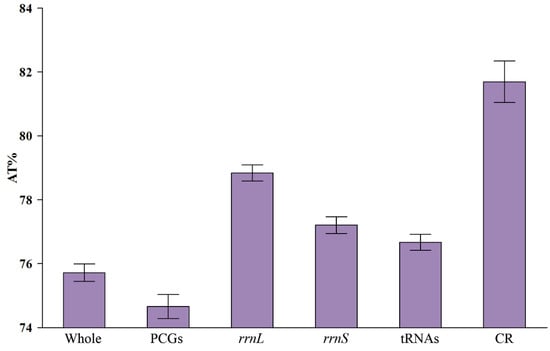
Figure 3.
AT% in different regions of Chiasmini mitochondrial genomes.
3.4. Protein-Coding Genes and Codon Usage
The protein-coding genes (PCGs) of the Chiasmini species have a total of six initiation codons: ATG (88 times), ATA (47 times), ATT (45 times), TTG (16 times), ATC (9 times), and GTG (3 times). Regarding the stop codons, the majority are TAA or TAG, but some are T. Specifically, TAA is used 167 times, TAG is used 24 times, and T is used 17 times (Table 3). Statistical results of the relative synonymous codon usage (RSCU) show that the frequently used amino acids in the Chiasmini mitochondrial genome are Ile, Leu2, Phe, and Met (Figure 4).
Table 3.
The use of start and stop codons of mitochondrial PCGs in Chiasmini.
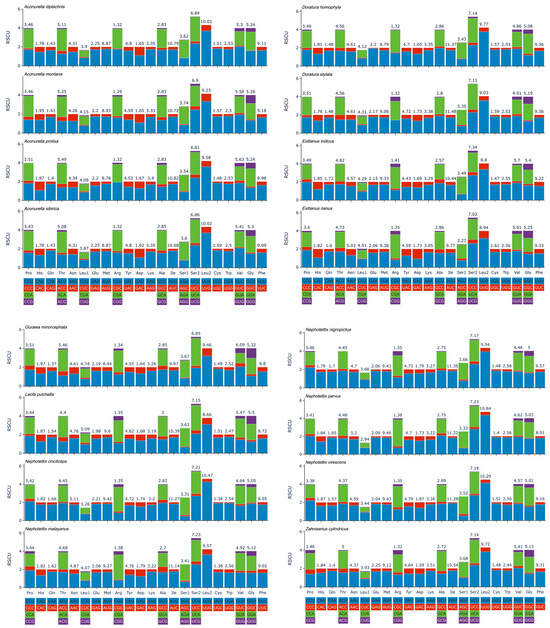
Figure 4.
Relative synonymous codon usage (RSCU) of the PCGs of species of Chiasmini.
Based on the sliding window analysis of 13 PCGs in the Chiasmini 16 species, the nucleotide diversity of ATP8 (Pi = 0.345) is the highest. At the same time, CYTB (Pi = 0.199), ND1 (Pi = 0.198), COX3 (Pi = 0.197), and COX1 (Pi = 0.169) show lower nucleotide diversity (Figure 5). According to a genetic distance analysis of the 13 PCGs, the ATP8 (0.45) gene has the highest genetic distance and fastest evolution rate, and the COX1 (0.19) gene has the lowest genetic distance and slowest evolution rate. The ratio of synonymous (Ks) to non-synonymous (Ka) substitution rates (Ka/Ks, ω) among the 13 PCGs ranges from 0.09 to 0.696 (0 < ω < 1). All ω values are less than 1, indicating that the 13 PCGs are under purifying selection at the gene level. COX1 has the lowest ω value (0.09), while ATP8 has the highest ω value (0.70), showing a significant difference (Figure 6).
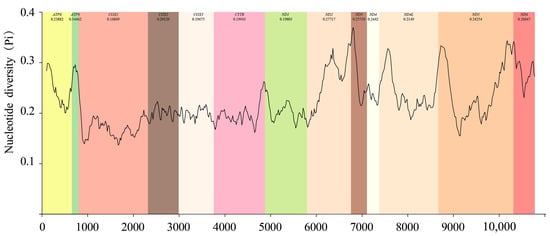
Figure 5.
Sliding-window analyses based on 13 aligned PCGs among species of Chiasmini.
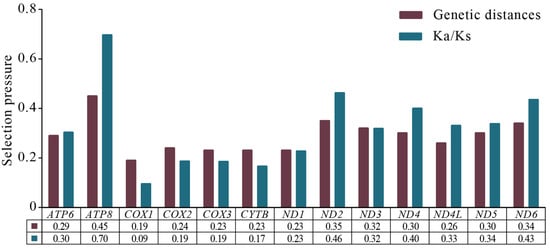
Figure 6.
Genetic distances and ratios of non-synonymous (Ka) to synonymous (Ks) substitution rates of 13 aligned PCGs among species of Chiasmini. The average value for each PCG is shown below the gene name.
3.5. RNA Genes
All 22 tRNA genes of the Chiasmini mitochondrial genome range in length from 60 to 70 bp. The dihydrouridine (DHU) arm of trnS1 (AGN) is missing and forms a circular structure (Figure 7), while the remaining 21 tRNAs are predicted to have a typical cloverleaf structure (Figures S1–S14). In addition to A–U and G–C pairing, the predicted secondary structure of tRNA includes five mismatched types: A–A, A–C, A–G, U–C, and U–U (Table 4). The rrnL length ranges from 1193 to 1216 bp, and the rrnS length ranges from 737 to 801 bp in the rRNA (Table 4).
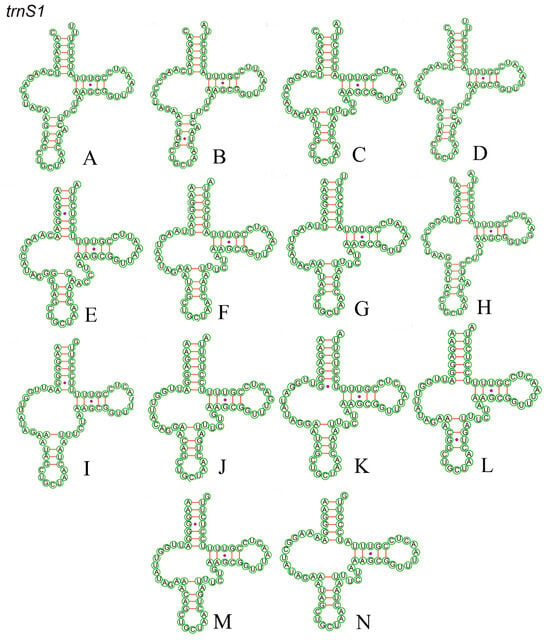
Figure 7.
Putative secondary structures of the trnS1 in 14 Chiasmini mitogenomes that do not possess a DHU-arm. Dashes (−) indicate Watson−Crick base pairing and dots (•) indicate G−U pairing. (A) Aconurella diplachnis; (B) Aconurella montana; (C) Aconurella prolixa; (D) Aconurella sibirica; (E) Exitianus nanus; (F) Doratura stylata; (G) Doratura homophyla; (H) Gurawa minorcephala; (I) Leofa pulchella; (J) Nephotettix malayanus; (K) Nephotettix nigropictus; (L) Nephotettix parvus; (M) Nephotettix virescens; (N) Zahniserius cylindricus.
Table 4.
Comparison of transfer RNA and mitochondrial RNA in Chiasmini mitochondrial genomes.
3.6. Gene Overlap and Non-Coding Regions
The gene arrangement of the Chiasmini mitochondrial genome is relatively compact, with overlapping regions ranging from 6 bp (in Nephotettix nigropictus) to 12 bp (in Leofa pulchella). There are three stable gene overlaps: the overlapping sequence of trnW and trnC is AAGTCTTA, the overlapping sequence of ATP8 and ATP6 is generally ATGATTA, and the overlapping sequence of ND4 and ND4L is generally TTATCAT. The Chiasmini mitochondrial genome also contains intergenic spacer regions, ranging from 11 bp (in Aconurella diplachnis) to 19 bp (in Nephotettix nigropictus). Except for a 548 bp-long spacer between trnM and ND2 in Exitianus indicus, the spacer lengths are generally small, usually less than 30 bp.
3.7. Phylogenetic Relationships
This study aimed to reconstruct the phylogenetic relationships among the cicadellid subfamilies, tribes in the subfamily Deltocephalinae, and the genera and species of Chiasmini. Therefore, we selected all available Deltocephalinae species and no more than two representative species from each other subfamily instead of the entire 153 cicadellid mitogenomes currently available from NCBI. In this phylogenetic analysis, we selected 65 Membracoidea species (including 62 leafhoppers and 3 treehoppers) as the ingroup and 2 Cicadoidea and 2 Cercopoidea species as the outgroups (Table 5).
Table 5.
Summary of mitochondrial genome information used in this study.
Mitogenome data of the 65 species representing 14 subfamilies of Cicadellidae were concatenated into five datasets: 13 PCG sequences (P123); 13 PCG and 2 rRNA sequences (P123R); 13 PCG sequences with the third codon position removed (P12); 13 PCG sequences with the third codon position removed, as well as 2 rRNA sequences (P12R); and amino acid sequences (AA). Maximum likelihood (ML) and Bayesian inference (BI) methods were used for phylogenetic analysis, and tests of base substitution saturation indicated that the selected datasets were suitable for phylogenetic analysis. There were no significant differences in sequence divergence among the different datasets, and removing the third codon position in the nucleotide datasets reduced heterogeneity between species. Based on 10 phylogenetic trees reconstructed herein using five datasets and two analysis methods (Figure 8 and Figure 9; Figures S15–S22), the included representatives of each subfamily for which more than one representative was included (Coelidiinae, Deltocephalinae, Eurymelinae, Evacanthinae, Hylicinae, Iassinae, Ledrinae, Megophthalminae, Mileewinae, and Typhlocybinae) form monophyletic groups (bootstrap values, BS = 100; posterior probabilities, PP = 1.0). As in other recent molecular phylogenetic analyses, branches pertaining to relationships among leafhopper subfamilies received only low to moderate support and were unstable among analyses, with the exception of Megophthalminae consistently grouping with the treehopper lineage (Membracidae + Aetalionidae) with maximum support. Ledrinae is the earliest diverging cicadellid subfamily in both AA-ML and AA-BI trees (Figure 8), while in the remaining eight phylogenetic trees, Typhlocybinae is sister to the remaining leafhoppers (Figure 9; Figures S15–S22). In the P12R-BI, AA-ML, and AA-BI trees, (Hylicinae + (Iassinae + Coelidiinae)) formed a clade, sister to Deltocephalinae, but the support for this node was generally low (P12R-BI, PP = 0.39; AA-ML, BS = 62; AA-BI, PP = 0.71). In the remaining seven phylogenetic trees, Hylicinae formed a clade sister to Deltocephalinae, with support in P123-BI and P123R-BI analyses of 0.93 and 0.86 (PP), respectively.
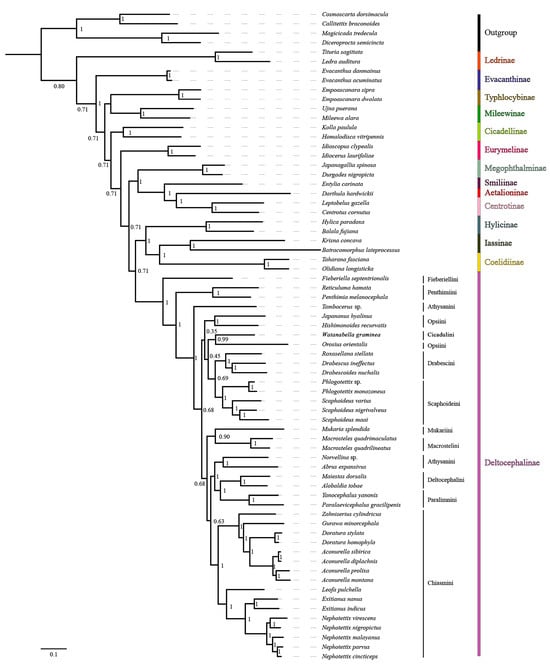
Figure 8.
The phylogenetic relationships using the Bayesian inference (BI) analysis method based on the AA datasets. Numbers on each node correspond to the posterior probability (PP) values.
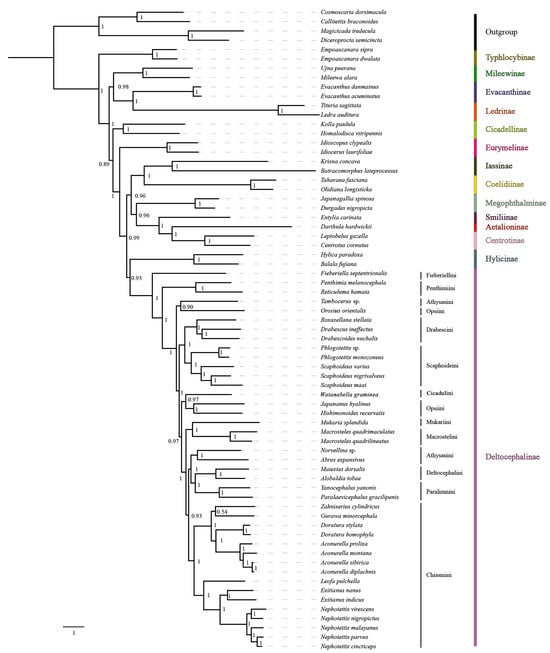
Figure 9.
The phylogenetic relationships using the Bayesian inference (BI) analysis method based on the PCG datasets. Numbers on each node correspond to the posterior probability (PP) values.
Within the Deltocephalinae, the phylogenies based on mitogenome sequence data are consistent with the recent anchored-hybrid-based phylogeny of Cao et al. [7], except in a few parts of the tree that received less than maximum branch support. As in the Cao et al. [7] analysis, Fieberiellini, Penthimiini, and Tambocerus represent early diverging lineages, and the tribe Athysanini is polyphyletic. Orosius orientalis of tribe Opsiini is separate from other opsiines (Hishimonoides recurvatis + Japananus hyalinus), indicating the non-monophyly of Opsiini, but the branches separating the included opsiines received low support (0.35–0.45) in our Bayesian analysis of amino acid sequences.
The seven included genera within Chiasmini consistently form a strongly supported clade, with the phylogenetic relationships supporting a stable sister-group relationship between Exitianus and Nephotettix, consistent with their morphological similarity, with Leofa being the closest relative. The four species of Aconurella, which are difficult to distinguish based on morphological characters, are also grouped into a larger clade, which shows a close sister-group relationship with Zahniserius, Doratura, and Gurawa, presenting a topology of ((Zahniserius + (Gurawa + (Doratura + Aconurella))) + (Leofa + (Exitianus + Nephotettix))). The phylogenetic results obtained from the two different analytical methods do not vary significantly, and relationships within this clade are well-resolved under a homogenous substitution model; therefore, we chose to perform subsequent phylogenetic analysis under the homogenous model only. All analyses yielded results largely consistent with previous phylogenetic studies of this tribe by Zahniser and Dietrich [23], Gao et al. [15], Cao et al. [7], and Zhang et al. [5].
The 10 phylogenetic results based on the five datasets and two analysis methods (Figure 8 and Figure 9; Figures S15–S22) indicate that mitochondrial genome sequences are informative of relationships within and among tribes of deltocephaline leafhoppers and within the tribe Chiasmini (bootstrap values, BS = 100; posterior probabilities, PP = 1.0). Within Chiasmini, relationships among the seven included genera (Aconurella, Doratura, Exitianus, Gurawa, Leofa, Nephotettix, and Zahniserius) are consistent among the 10 different topologies, with high support values (BS = 100, PP = 1.0) with one exception. In the PCG123-ML/BI and PCG123R-ML/BI trees, the topology is ((Zahniserius + Gurawa) + (Doratura + Aconurella)) + (Leofa + (Exitianus + Nephotettix)), while in the PCG12-ML/BI, PCG12R-ML/BI, and AA-ML/BI trees, the topology is ((Zahniserius + (Gurawa + (Doratura + Aconurella))) + (Leofa + (Exitianus + Nephotettix))). The latter topology has much higher support than the former and is more consistent with previous analyses based on combined mitochondrial and nuclear gene sequences (Zahniser and Dietrich [23]) and anchored hybrid nuclear gene data (Cao et al. [7]), suggesting that the phylogenetic relationships within Chiasmini based on the mitochondrial P12, PCG12R, and AA datasets are more reliable.
4. Conclusions
Comparative analysis of the complete mitogenomes of 16 species representing seven genera of the grass-specialist leafhopper tribe Chiasmini indicates a highly conserved overall genome structure and composition in this group, without any of the gene rearrangements reported for some other groups of deltocephaline leafhoppers. Overall, our phylogenetic results suggest that analyses of mitogenome sequence data provide good resolution of both deep and shallow branches within the phylogeny of Chiasmini and leafhoppers in general and yield results similar to analyses based on nuclear genes alone or combined nuclear and mitochondrial genes. Despite these results, the mitogenome sequences currently available represent only a tiny fraction of known species. More sequencing efforts are needed to increase the size of the available taxon sample and facilitate further exploration of the phylogeny of leafhoppers.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/insects15040253/s1, Figure S1: Predicted secondary structures of tRNA genes of Aconurella diplachnis; Figure S2: Predicted secondary structures of tRNA genes of Aconurella montana; Figure S3: Predicted secondary structures of tRNA genes of Aconurella prolixa; Figure S4: Predicted secondary structures of tRNA genes of Aconurella sibirica; Figure S5: Predicted secondary structures of tRNA genes of Exitianus nanus; Figure S6: Predicted secondary structures of tRNA genes of Doratura stylata; Figure S7: Predicted secondary structures of tRNA genes of Doratura homophyla; Figure S8: Predicted secondary structures of tRNA genes of Gurawa minorcephala; Figure S9: Predicted secondary structures of tRNA genes of Leofa pulchella; Figure S10: Predicted secondary structures of tRNA genes of Nephotettix malayanus; Figure S11: Predicted secondary structures of tRNA genes of Nephotettix nigropictus; Figure S12: Predicted secondary structures of tRNA genes of Nephotettix parvus; Figure S13: Predicted secondary structures of tRNA genes of Nephotettix virescens; Figure S14: Predicted secondary structures of tRNA genes of Zahniserius cylindricus; Figure S15: The phylogenetic relationships using the maximum likelihood (ML) analysis method based on the AA datasets. Numbers on each node correspond to the bootstrap values; Figure S16: The phylogenetic relationships using the maximum likelihood (ML) analysis method based on the PCG12 datasets. Numbers on each node correspond to the bootstrap values; Figure S17: The phylogenetic relationships using the Bayesian inference (BI) analysis method based on the PCG12 datasets. Numbers on each node correspond to the posterior probability (PP) values; Figure S18: The phylogenetic relationships using the maximum likelihood (ML) analysis method based on the PCG12R datasets. Numbers on each node correspond to the bootstrap values; Figure S19: The phylogenetic relationships using the Bayesian inference (BI) analysis method based on the PCG12R datasets. Numbers on each node correspond to the posterior probability (PP) values; Figure S20: The phylogenetic relationships using the maximum likelihood (ML) analysis method based on the PCG datasets. Numbers on each node correspond to the bootstrap values; Figure S21: The phylogenetic relationships using the maximum likelihood (ML) analysis method based on the PCGR datasets. Numbers on each node correspond to the bootstrap values; Figure S22: The phylogenetic relationships using the Bayesian inference (BI) analysis method based on the PCGR datasets. Numbers on each node correspond to the posterior probability (PP) values; Table S1: Best partitioning scheme and models for different datasets selected by PartitionFinder.
Author Contributions
Conceptualization, Y.D. and C.H.D.; methodology, B.S., B.X., K.W., H.N. and Y.D.; software, B.S., B.X., K.W., M.A.H. and M.Y.; formal analysis, B.S. and M.A.H.; investigation, B.S., M.A.H. and Y.D.; resources, Y.D.; data curation, B.S., K.W., M.Y. and Y.D.; writing—original draft preparation, B.S., M.A.H., B.X., M.Y. and H.N.; writing—review and editing, B.S., M.A.H., B.X., H.N., M.Y., C.H.D. and Y.D.; supervision, Y.D.; project administration, Y.D.; funding acquisition, Y.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Anhui Provincial Colleges and Universities Natural Science Foundation (2022AH050868), the National Natural Science Foundation of China (31000968), and the Innovation and Entrepreneurship Training Program for College Students (X202210364113).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Acknowledgments
We express our sincere thanks to J.R. Schrock, Emporia State University, USA, for commenting on an earlier draft of this paper and to anonymous referees and the subject editor for constructive criticism.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Abascal, F.; Posada, D.; Knight, R.D.; Zardoya, R. Parallel evolution of the genetic code in arthropod mitochondrial genomes. PLoS Biol. 2006, 4, e127. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Wilson, A.C.; Cann, R.L.; Carr, S.M.; George, M.; Gyllensten, U.B.; Helm-Bychowski, K.M.; Higuchi, R.B.; Palumbi, S.R.; Prager, E.M.; Sage, R.D.; et al. Mitochondrial DNA and two perspectives on evolutionary genetics. Biol. J. Linn. Soc. 1985, 26, 375–400. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Gao, Y.; Xiong, J.L.; Dietrich, C.H.; Duan, Y.N. Phylogenetic analyses of the leafhopper tribe Chiasmini and species delimitation of the genus Exitianus Ball (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini) in China based on molecular data. Eur. J. Taxon. 2024, 921, 276–297. [Google Scholar]
- Zahniser, J.N.; Dietrich, C.H. A review of the tribes of Deltocephalinae (Hemiptera: Auchenorrhyncha: Cicadellidae). Eur. J. Taxon. 2013, 45, 1–211. [Google Scholar] [CrossRef]
- Cao, Y.H.; Dietrich, C.H.; Zahniser, J.N.; Dmitriev, D.A. Dense sampling of taxa and characters improves phylogenetic resolution among deltocephaline leafhoppers (Hemiptera: Cicadellidae: Deltocephalinae). Syst. Entomol. 2022, 47, 430–444. [Google Scholar] [CrossRef]
- Duan, Y.N.; Zhang, Y.L.; Webb, M.D. Review of the grassland leafhopper subgenus Leofa (Prasutagus) Distant (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini). Zootaxa 2009, 1972, 35–43. [Google Scholar] [CrossRef]
- Duan, Y.N.; Zhang, Y.L. A taxonomic review of the grassland leafhopper genera Gurawa Distant and Chiasmus Mulsant & Rey (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini) from China with description of a new species. Zootaxa 2012, 3537, 41–52. [Google Scholar]
- Duan, Y.N.; Zhang, Y.L. Redescription of the grassland leafhopper genus Doraturopsis Lindberg (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini) with description of a new genus and species from China. Zootaxa 2012, 3177, 24–32. [Google Scholar] [CrossRef]
- Duan, Y.N.; Zhang, Y.L. New records of the grassland leafhopper genus Doratura Sahlberg (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini) from China. Zootaxa 2012, 3175, 54–62. [Google Scholar] [CrossRef]
- Duan, Y.N.; Zhang, Y.L. A taxonomic review of the grassland leafhopper genus Aconurella Ribaut (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini) from China with the description of two new species. Zootaxa 2012, 3397, 28–44. [Google Scholar] [CrossRef]
- Duan, Y.N.; Zhang, Y.L. Review of the grassland leafhopper genus Exitianus Ball (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini) from China. ZooKeys 2013, 333, 31–43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duan, Y.N.; Zhang, Y.L. Review of the grassland leafhopper genus Nephotettix Matsumura (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini) from the Chinese mainland. Zootaxa 2014, 3755, 201–229. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Y.L.; Dietrich, C.H.; Duan, Y.N. Phylogenetic analyses and species delimitation of Nephotettix Matsumura (Hemiptera: Cicadellidae: Deltocephalinae: Chiasmini) in China based on molecular data. Zool. Anz. 2021, 293, 202–214. [Google Scholar] [CrossRef]
- Carloni, E.; Virla, E.; Paradell, S.; Carpane, P.; Nome, C.; Laguna, I.; Giménez Pecci, M.P. Exitianus obscurinervis (Hemiptera: Cicadellidae), a new experimental vector of Spiroplasma kunkelii. J. Econom. Entomol. 2011, 104, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Salehi, M.; Izadpanah, K.; Siampour, M.; Taghizadeh, M. Molecular characterization, and transmission of Bermuda grass white leaf phytoplasma in Iran. J. Plant Pathol. 2009, 91, 655–661. [Google Scholar]
- Zahniser, J.N. Seven new species and new distributions of Old World Chiasmini (Hemiptera: Cicadellidae: Deltocephalinae), with a redescription, key to genera, and species checklist for the tribe. Zootaxa 2008, 1808, 1–32. [Google Scholar] [CrossRef]
- Zahniser, J.N. An enigmatic new leafhopper genus, Protochiasmus (Hemiptera: Cicadellidae: Deltocephalinae), from Brazil. Dtsch. Entomol. Z. 2010, 57, 271–274. [Google Scholar] [CrossRef]
- Zahniser, J.N. New generic synonymies and combinations in Chiasmini (Cicadellidae: Deltocephalinae). Int. J. Trop. Insect Sci. 2012, 32, 173–176. [Google Scholar] [CrossRef]
- Zahniser, J.N.; Dietrich, C.H. Phylogeny of the leafhopper subfamily Deltocephalinae (Insecta: Auchenorrhyncha: Cicadellidae) and related subfamilies based on morphology. Syst. Biodivers. 2008, 6, 1–24. [Google Scholar] [CrossRef]
- Zahniser, J.N.; Dietrich, C.H. Phylogeny of the leafhopper subfamily Deltocephalinae (Hemiptera: Cicadellidae) based on molecular and morphological data with a revised family-group classification. Syst. Entomol. 2010, 35, 489–511. [Google Scholar] [CrossRef]
- Zahniser, J.N.; Dietrich, C.H. Phylogeny, evolution, and historical biogeography of the grassland leafhopper tribe Chiasmini (Hemiptera: Cicadellidae: Deltocephalinae). Zool. J. Linn. Soc. 2015, 175, 473–495. [Google Scholar] [CrossRef]
- Du, Y.M.; Dietrich, C.H.; Dai, W. Complete mitochondrial genome of Macrosteles quadrimaculatus (Matsumura) (Hemiptera: Cicadellidae: Deltocephalinae) with a shared tRNA rearrangement and its phylogenetic implications. Int. J. Biol. Macromol. 2019, 122, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Cai, W.Z.; Li, H. Insufficient power of mitogenomic data in resolving the auchenorrhynchan monophyly. Zool. J. Linn. Soc. 2018, 183, 776–790. [Google Scholar] [CrossRef]
- Wu, K.Q.; Yan, M.H.; Zhang, Y.L.; Dietrich, C.H.; Duan, Y.N. The complete mitochondrial genome of Aconurella prolixa (Lethierry 1885) (Deltocephalinae: Chiasmini). Mitochondrial DNA Part B 2022, 7, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.A.; Tan, Z.X.; Shen, R.R.; Xing, J.C. Comparative mitochondrial genome analysis of three leafhopper species of the genus Abrus Dai & Zhang (Hemiptera: Cicadellidae: Deltocephalinae) from China with phylogenetic implication. BMC Genomics 2023, 24, 714. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313319. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE-visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinformatics 2014, 15, 294. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; IQ-TREE, R.L. 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Liu, J.; Bu, C.P.; Wipfler, B.; Liang, A.P. Comparative analysis of the mitochondrial genomes of Callitettixini spittlebugs (Hemiptera: Cercopidae) confirms the overall high evolutionary speed of the AT-Rich region but reveals the presence of short conservative elements at the tribal level. PLoS ONE 2014, 9, e109140. [Google Scholar] [CrossRef] [PubMed]
- Su, T.J.; He, B.; Li, K.; Liang, A.P. Comparative analysis of the mitochondrial genomes of oriental spittlebug trible Cosmoscartini: Insights into the relationships among closely related taxa. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.Y.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.Z.; Sota, T.; Li, H. Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Liang, A.P.; Gao, J.; Zhao, X. Characterization of the complete mitochondrial genome of the treehopper Darthula hardwickii (Hemiptera: Aetalionidae). Mitochondrial DNA Part A 2016, 27, 3291–3292. [Google Scholar] [CrossRef]
- Wang, X.Y.; Wang, J.J.; Dai, R.H. Mitogenomics of five Olidiana leafhoppers (Hemiptera: Cicadellidae: Coelidiinae) and their phylogenetic implications. Peerj 2021, 9, e11086. [Google Scholar] [CrossRef]
- Wang, J.J.; Li, H.; Dai, R.H. Complete mitochondrial genome of Taharana fasciana (Insecta, Hemiptera: Cicadellidae) and comparison with other Cicadellidae insects. Genetica 2017, 145, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Xing, J.C. Complete mitochondrial genome of Abrus expansivus (Hemiptera: Cicadellidae: Deltocephalinae) from China. Mitochondrial DNA Part B 2019, 4, 197–198. [Google Scholar] [CrossRef]
- Song, N.; Cai, W.Z.; Li, H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci. Rep. 2017, 7, 10429. [Google Scholar] [CrossRef]
- Yu, P.F.; Wang, M.X.; Cui, L.; Chen, X.X.; Han, B.Y. The complete mitochondrial genome of Tambocerus sp. (Hemiptera: Cicadellidae). Mitochondrial DNA Part A 2015, 28, 133–134. [Google Scholar] [CrossRef]
- Yang, W.J.; Gao, Y.R.; Li, C.; Song, Y.H. The complete mitochondrial genome of Chlorotettix nigromaculatus (Hemiptera: Cicadellidae: Deltocephalinae) with phylogenetic consideration. Mitochondrial DNA Part B 2019, 4, 624–625. [Google Scholar] [CrossRef]
- Du, Y.M.; Zhang, C.N.; Dietrich, C.H.; Zhang, Y.L.; Dai, W. Characterization of the complete mitochondrial genomes of Maiestas dorsalis and Japananus hyalinus (Hemiptera: Cicadellidae) and comparison with other Membracoidea. Sci. Rep. 2017, 7, 14197. [Google Scholar] [CrossRef]
- Wu, Y.F.; Dai, R.H.; Zhan, H.P.; Qu, L. Complete mitochondrial genome of Drabescoides nuchalis (Hemiptera: Cicadellidae). Mitochondrial DNA Part A 2016, 27, 3626–3627. [Google Scholar] [CrossRef]
- Xu, D.L.; Yu, T.H.; Zhang, Y.L. Characterization of the complete mitochondrial genome of Drabescus ineffectus and Roxasellana stellata (Hemiptera: Cicadellidae: Deltocephalinae: Drabescini) and their phylogenetic implications. Insects 2020, 11, 534. [Google Scholar] [CrossRef]
- Luo, H.; Wang, Y.; Di, X.C.; Shan, L.C.Y.; Wang, S.S. The complete mitochondrial genome of Fieberiella septentrionalis (Hemiptera: Cicadellidae: Deltocephalinae). Mitochondrial DNA Part B 2021, 6, 1426–1428. [Google Scholar] [CrossRef]
- Mao, M.; Yang, X.S.; Bennett, G. The complete mitochondrial genome of Macrosteles quadrilineatus (Hemiptera: Cicadellidae). Mitochondrial DNA Part B 2017, 2, 173–175. [Google Scholar] [CrossRef]
- Yang, H.Y.; Dai, R.H. Complete mitochondrial genome of Mukaria splendida Distant (Hemiptera: Cicadellidae: Deltocephalinae: Mukariini) and phylogenetic analysis. Mitochondrial DNA Part B 2021, 6, 622–623. [Google Scholar] [CrossRef]
- Xing, J.C.; Wang, J.J. Complete mitochondrial genome of Paralaevicephalus gracilipenis (Hemiptera: Cicadellidae: Deltocephalinae) from China. Mitochondrial DNA Part B 2019, 4, 1372–1373. [Google Scholar] [CrossRef]
- Xu, T.L.; Dai, R.H. Complete mitochondrial genome of Penthimia melanocephala Motschulsky, 1863 (Hemiptera: Cicadellidae: Deltocephalinae). Mitochondrial DNA Part B 2021, 6, 568–569. [Google Scholar] [CrossRef]
- Xu, T.L.; Dai, R.H. Complete mitogenome of Reticuluma hamata (Hemiptera: Cicadellidae: Deltocephalinae) from China. Mitochondrial DNA Part B 2020, 5, 1437–1438. [Google Scholar] [CrossRef]
- Du, Y.M.; Dai, W.; Dietrich, C.H. Mitochondrial genomic variation and phylogenetic relationships of three groups in the genus Scaphoideus (Hemiptera: Cicadellidae: Deltocephalinae). Sci. Rep. 2017, 7, 16908. [Google Scholar] [CrossRef][Green Version]
- Wang, J.J.; Yang, M.F.; Dai, R.H.; Li, H.; Wang, X.Y. Characterization and phylogenetic implications of the complete mitochondrial genome of Idiocerinae (Hemiptera: Cicadellidae). Int. J. Biol. Macromol. 2018, 120, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.H.; Wang, J.J.; Yang, M.F. The complete mitochondrial genome of the leafhopper Idioscopus clypealis (Hemiptera: Cicadellidae: Idiocerinae). Mitochondrial DNA Part B 2018, 3, 32–33. [Google Scholar]
- Yuan, Z.W.; Yang, X.; Li, C.; Song, Y.H. The complete mitochondrial genome of the leafhopper Evacanthus acuminatus (Hemiptera: Cicadellidae: Evacanthinae). Mitochondrial DNA Part B 2019, 4, 3866–3867. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.M.; Liang, Z.L.; Dietrich, C.H.; Dai, W. Comparative analysis of mitochondrial genomes of Nirvanini and Evacanthini (Hemiptera: Cicadellidae) reveals an explicit evolutionary relationship. Genomics 2021, 113, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Huang, W.J.; Zhang, Y.L. The complete mitochondrial genome of four Hylicinae (Hemiptera: Cicadellidae): Structural features and phylogenetic implications. Insects 2020, 11, 869. [Google Scholar] [CrossRef]
- Wang, J.J.; Wu, Y.F.; Dai, R.H.; Yang, M.F. Comparative mitogenomes of six species in the subfamily Iassinae (Hemiptera: Cicadellidae) and phylogenetic analysis. Int. J. Biol. Macromol. 2020, 149, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Li, D.F.; Li, H.; Yang, M.F.; Dai, R.H. Structural and phylogenetic implications of the complete mitochondrial genome of Ledra auditura. Sci. Rep. 2019, 9, 15746. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, Y.L. Characterization of two complete mitochondrial genomes of Ledrinae (Hemiptera: Cicadellidae) and phylogenetic analysis. Insects 2020, 11, 609. [Google Scholar] [CrossRef]
- Wang, J.J.; Dai, R.H.; Li, H.; Zhan, H.P. Characterization of the complete mitochondrial genome of Japanagallia spinosa and Durgades nigropicta (Hemiptera: Cicadellidae: Megophthalminae). Biochem. Syst. Ecol. 2017, 74, 33–41. [Google Scholar] [CrossRef]
- He, H.L.; Yang, M.F. Characterization of the leafhopper mitogenome of Mileewa alara (Hemiptera: Cicadellidae: Mileewinae) and its phylogenetic analysis. Mitochondrial DNA Part B 2021, 6, 1265–1266. [Google Scholar] [CrossRef]
- Yu, T.H.; Zhang, Y.L. Two complete mitochondrial genomes of Mileewinae (Hemiptera: Cicadellidae) and a phylogenetic analysis. Insects 2021, 12, 668. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Yuan, Z.W.; Li, C.; Song, Y.H. Complete mitochondrial genome sequence of Empoascanara dwalata (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 2260–2261. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Chen, X.X.; Li, C.; Song, Y.H. The complete mitochondrial genome of Empoascanara sipra (Hemiptera: Cicadellidae: Typhlocybinae) with phylogenetic consideration. Mitochondrial DNA Part B 2020, 5, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liang, A.P. Complete DNA sequence of the mitochondrial genome of the treehopper Leptobelus gazella (Membracoidea: Hemiptera). Mitochondrial DNA Part A 2016, 27, 3318–3319. [Google Scholar] [CrossRef]
- Mao, M.; Yang, X.H.; Bennett, G. The complete mitochondrial genome of Entylia carinata (Hemiptera: Membracidae). Mitochondrial DNA Part B 2016, 1, 662–663. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).