
PRMT1 Confers Resistance to Olaparib via Modulating MYC Signaling in Triple-Negative Breast Cancer

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Plasmids, Transfection, and Infection
2.3. MTT Assay
2.4. Colony Formation Assay
2.5. Western Blotting
2.6. Real-Time Polymerase Chain Reaction
2.7. Clinical and Molecular Data Acquisition
2.8. Statistical Analyses
3. Results
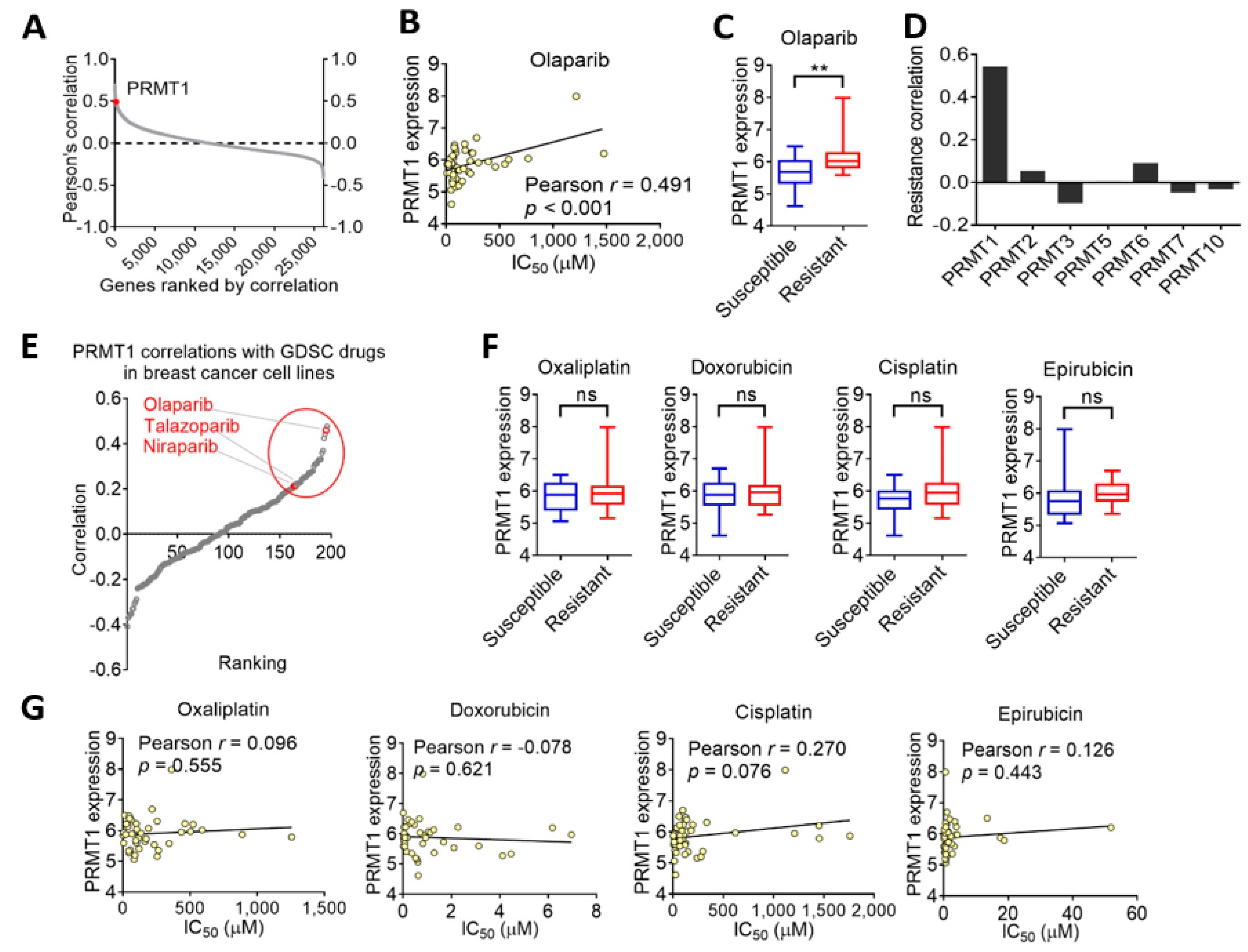
3.1. PRMT1 Expression Is Associated with Therapeutic Response to Olaparib in Breast Cancer Cells
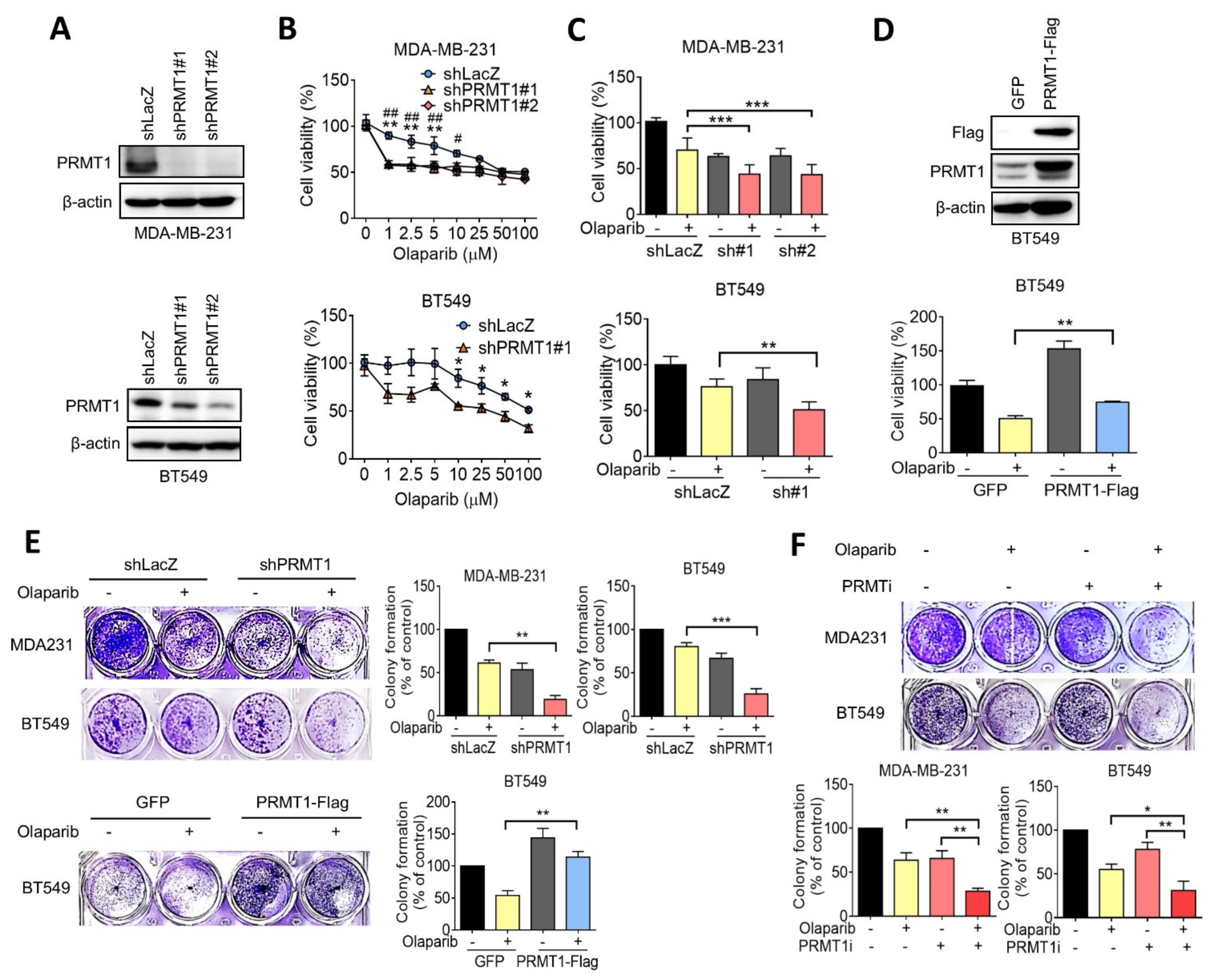
3.2. Inhibition of PRMT1 Increases Sensitivity to Olaparib in TNBC Cells
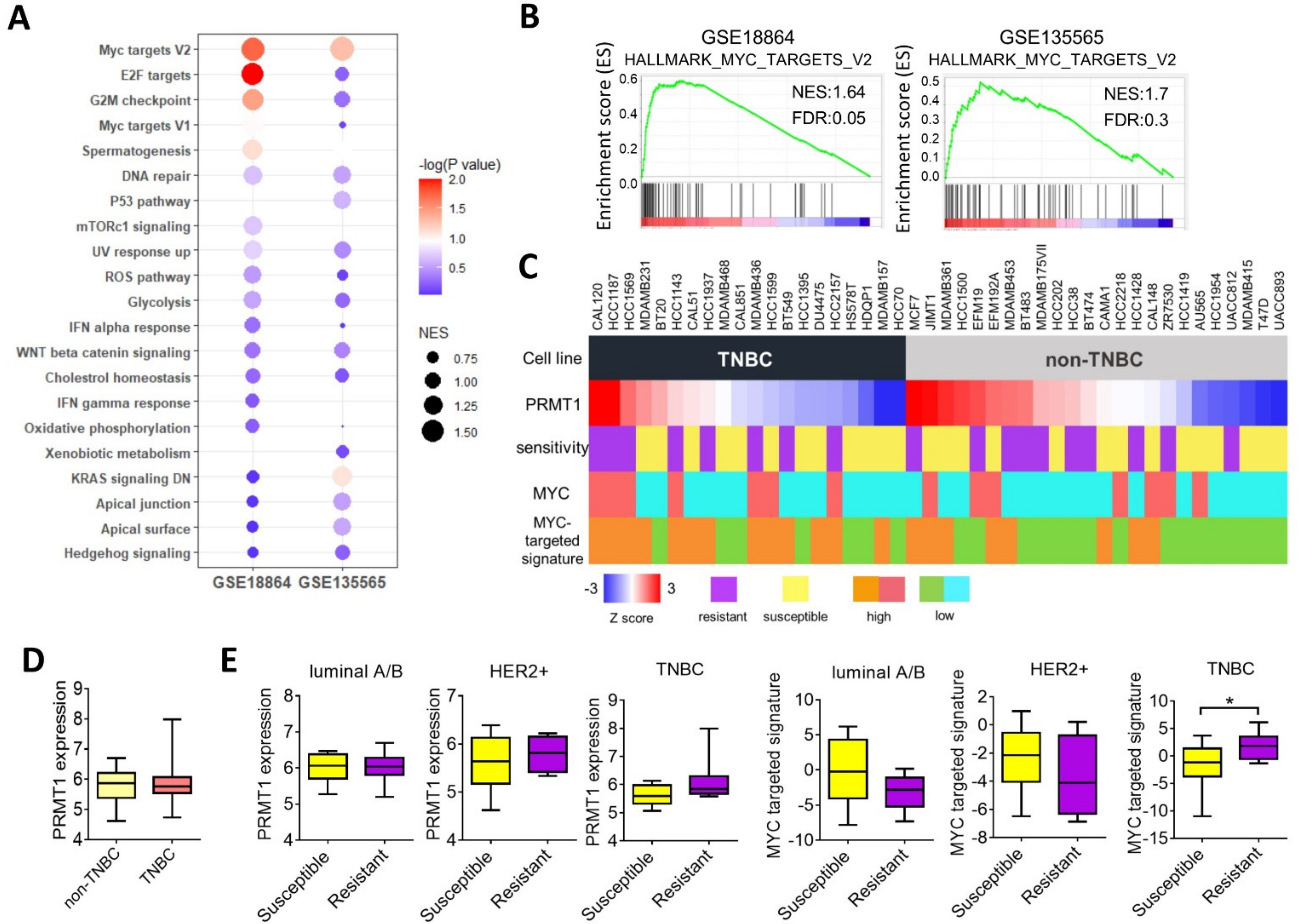
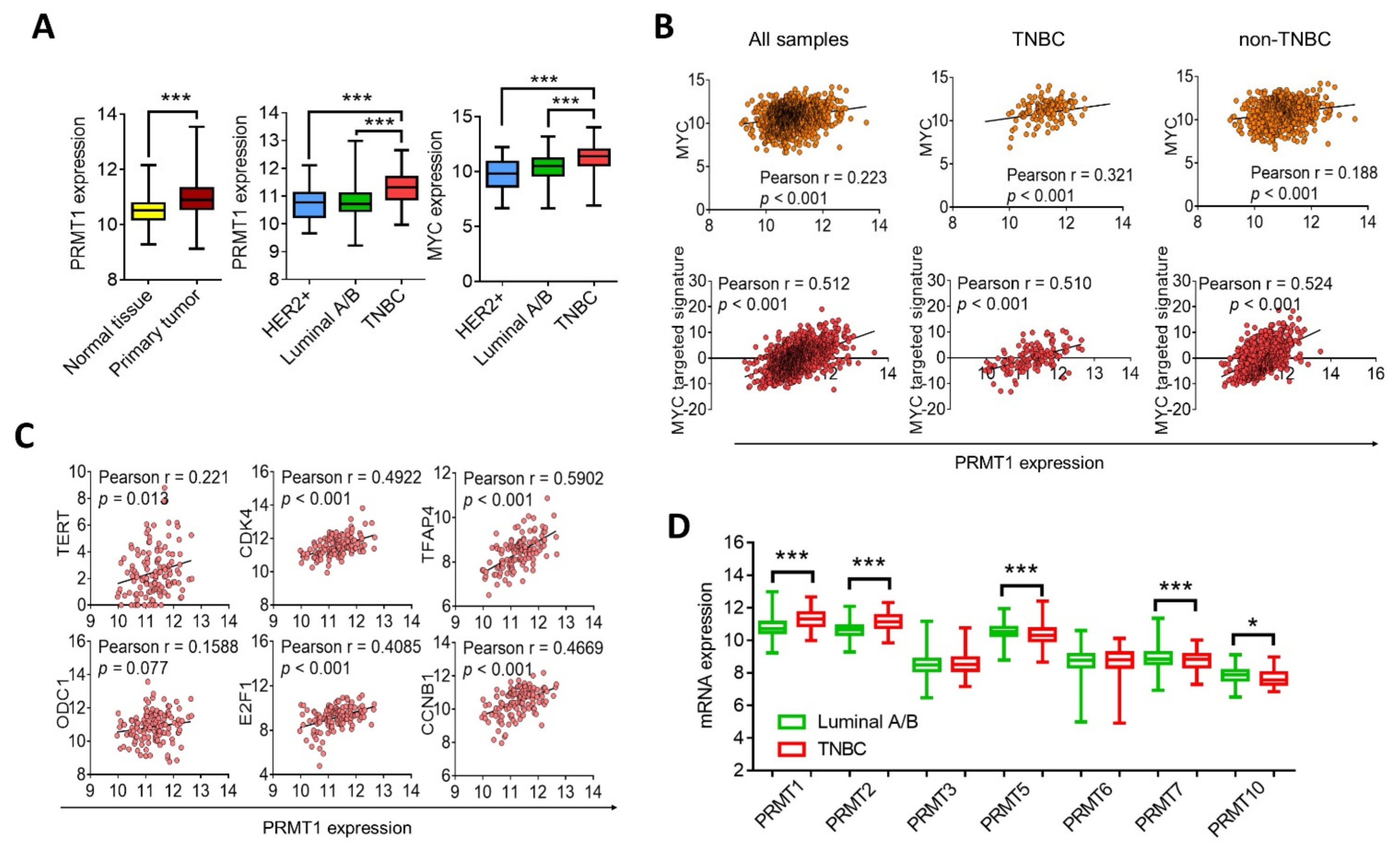
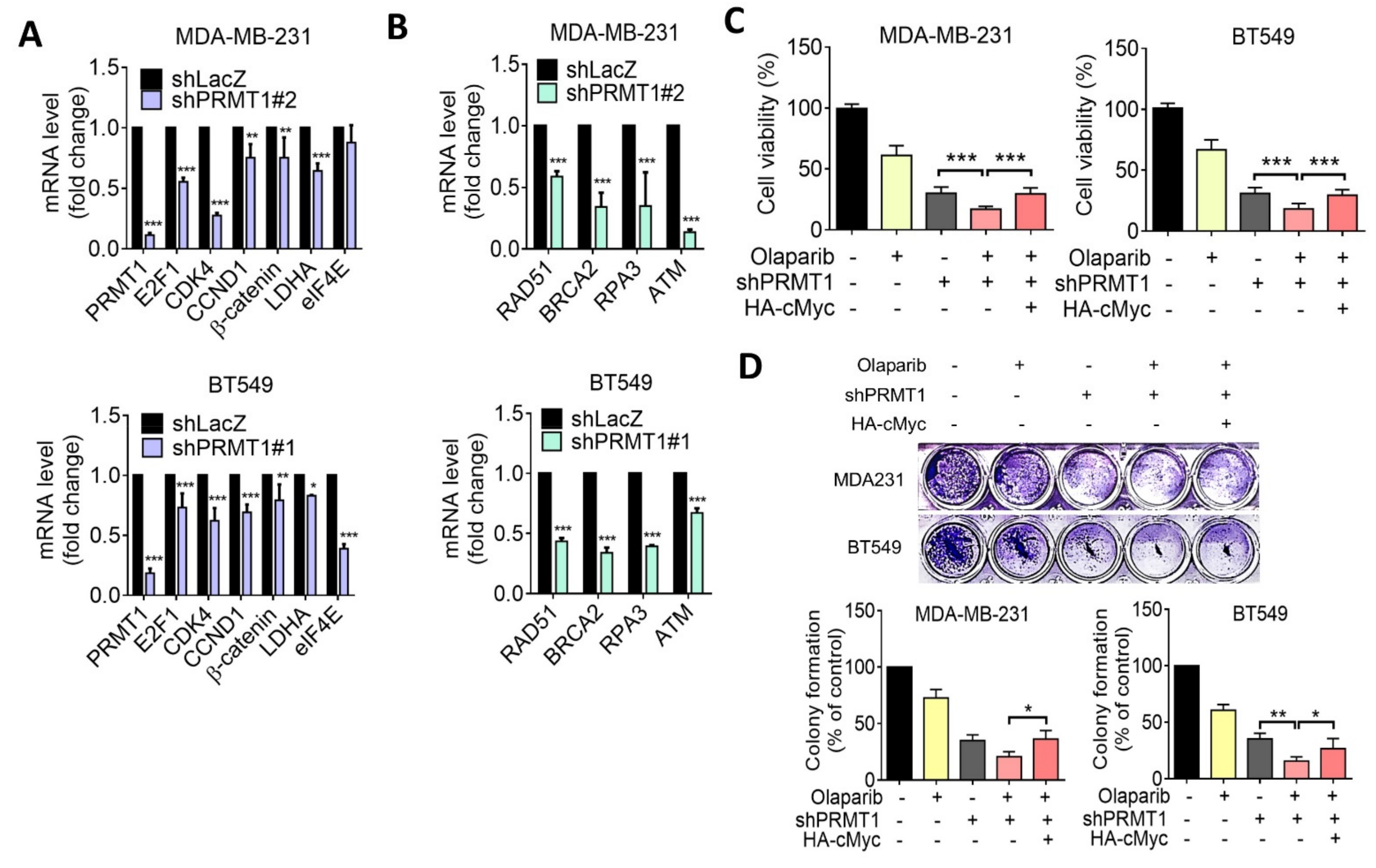
3.3. PRMT1 Expression Is Associated with MYC Signature
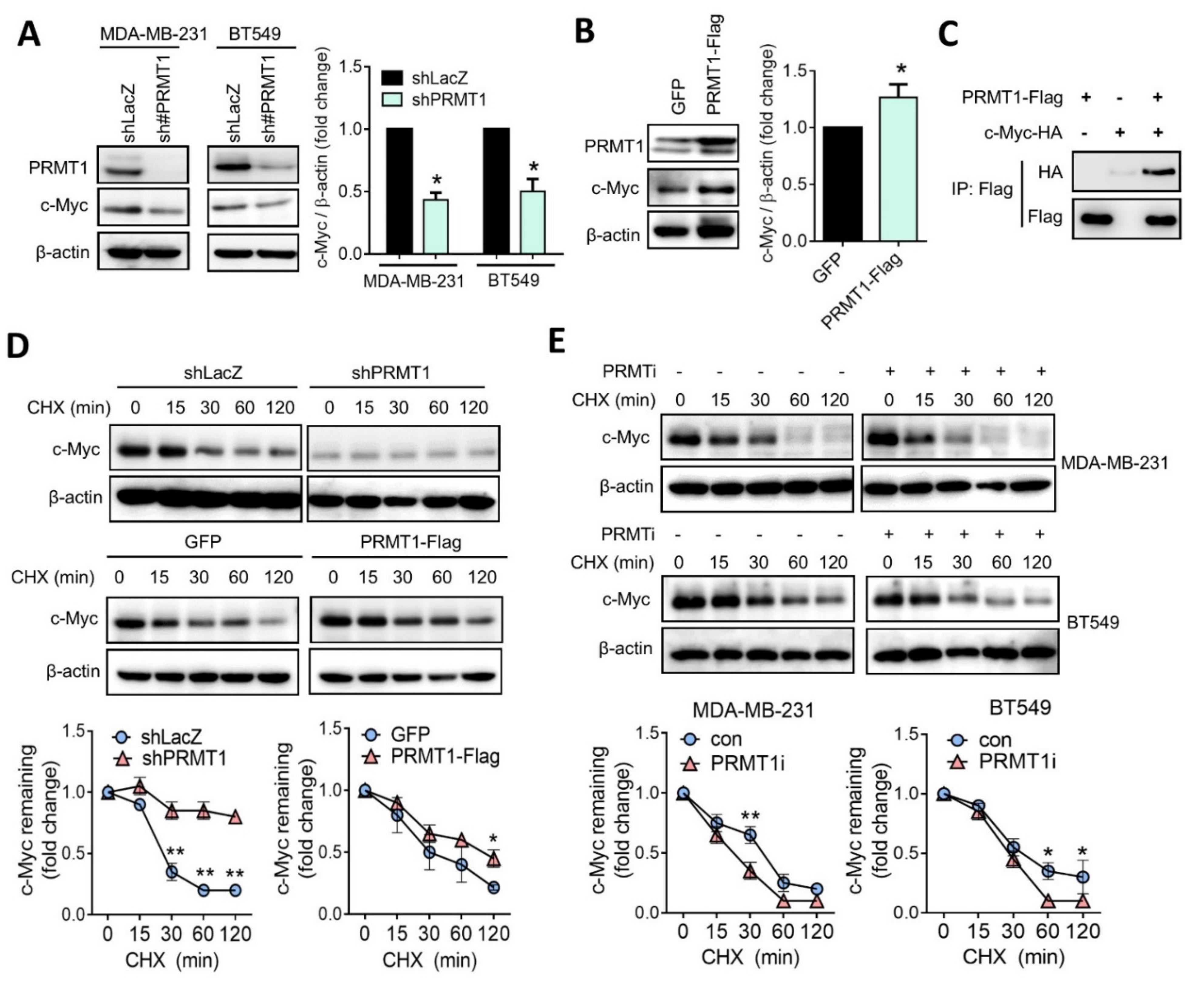
3.4. PRMT1 Regulates c-Myc Protein Stability
3.5. Suppression of PRMT1 Downregulates HR Gene Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baselga, J.; Norton, L. Focus on breast cancer. Cancer Cell 2002, 1, 319–322. [Google Scholar] [CrossRef]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef]
- Won, K.A.; Spruck, C. Triple-negative breast cancer therapy: Current and future perspectives (Review). Int J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Yin, H.; Xiong, G.; Guo, S.; Xu, C.; Xu, R.; Guo, P.; Shu, D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019, 27, 1252–1261. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP Inhibitors in Ovarian Cancer. Recent Pat. Anticancer Drug Discov. 2018, 13, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yu, X.; Li, S.; Tian, Y.; Liu, C. New Perspectives for Resistance to PARP Inhibitors in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Therapeutic applications of PARP inhibitors: Anticancer therapy and beyond. Mol. Asp. Med. 2013, 34, 1217–1256. [Google Scholar] [CrossRef]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Qualmann, B.; Kessels, M.M. The Role of Protein Arginine Methylation as Post-Translational Modification on Actin Cytoskeletal Components in Neuronal Structure and Function. Cells 2021, 10, 1079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Jiang, Y.B.; Zhong, C.Q.; Ma, N.; Zhang, E.B.; Zhang, F.; Li, J.J.; Deng, Y.Z.; Wang, K.; Xie, D.; et al. PRMT1 Promoted HCC Growth and Metastasis In Vitro and In Vivo via Activating the STAT3 Signalling Pathway. Cell Physiol. Biochem. 2018, 47, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.H.; Hubbell-Engler, B.; Adelmant, G.; Huang, J.; Joyce, C.E.; Vazquez, F.; Weir, B.A.; Montgomery, P.; Tsherniak, A.; Giacomelli, A.O.; et al. PRMT1-Mediated Translation Regulation Is a Crucial Vulnerability of Cancer. Cancer Res. 2017, 77, 4613–4625. [Google Scholar] [CrossRef]
- Malbeteau, L.; Poulard, C.; Languilaire, C.; Mikaelian, I.; Flamant, F.; Le Romancer, M.; Corbo, L. PRMT1 Is Critical for the Transcriptional Activity and the Stability of the Progesterone Receptor. iScience 2020, 23, 101236. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Ge, J.; Liu, M.; Pang, X.; Liu, J.; Luo, C.; Xu, Y.; Zhao, Q. The methyltransferase PRMT1 regulates γ-globin translation. J. Biol. Chem. 2021, 296, 100417. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Hua, Z.-Y.; Hansen, J.N.; He, M.; Dai, S.-K.; Choi, Y.; Fulton, M.D.; Lloyd, S.M.; Szemes, M.; Sen, J.; Ding, H.-F.; et al. PRMT1 promotes neuroblastoma cell survival through ATF5. Oncogenesis 2020, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Jhe, Y.L.; Kim, J.; Lim, J.Y.; Lee, J.E.; Shin, M.K.; Cheong, J.H. FoxM1-dependent and fatty acid oxidation-mediated ROS modulation is a cell-intrinsic drug resistance mechanism in cancer stem-like cells. Redox Biol. 2020, 36, 101589. [Google Scholar] [CrossRef]
- Choucair, A.; Pham, T.H.; Omarjee, S.; Jacquemetton, J.; Kassem, L.; Trédan, O.; Rambaud, J.; Marangoni, E.; Corbo, L.; Treilleux, I.; et al. The arginine methyltransferase PRMT1 regulates IGF-1 signaling in breast cancer. Oncogene 2019, 38, 4015–4027. [Google Scholar] [CrossRef]
- Montenegro, M.; González Guerrero, R.; Campo, L.; Piñero, A.; Cabezas-Herrera, J.; Rodríguez-López, J. PRMT1-dependent methylation of BRCA1 contributes to the epigenetic defense of breast cancer cells against ionizing radiation. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Liu, L.M.; Sun, W.Z.; Fan, X.Z.; Xu, Y.L.; Cheng, M.B.; Zhang, Y. Methylation of C/EBPα by PRMT1 Inhibits Its Tumor-Suppressive Function in Breast Cancer. Cancer Res. 2019, 79, 2865–2877. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Kuo, T.C.; Chou, C.M.; Hsu, W.J.; Lee, W.C.; Dai, J.Z.; Wu, S.M.; Lin, C.W. Upregulation of CD109 Promotes the Epithelial-to-Mesenchymal Transition and Stemness Properties of Lung Adenocarcinomas via Activation of the Hippo-YAP Signaling. Cells 2020, 10. [Google Scholar] [CrossRef]
- Buechler, S.A.; Gokmen-Polar, Y.; Badve, S.S. RespondR signature to predict potential alternative therapies for taxane resistant triple-negative breast cancer patients. J. Clin. Oncol. 2016, 34, 1078. [Google Scholar] [CrossRef]
- Wang, S.; Zou, X.; Chen, Y.; Cho, W.C.; Zhou, X. Effect of N6-Methyladenosine Regulators on Progression and Prognosis of Triple-Negative Breast Cancer. Front. Genet. 2021, 11. [Google Scholar] [CrossRef]
- Lai, Y.-W.; Hsu, W.-J.; Lee, W.-Y.; Chen, C.-H.; Tsai, Y.-H.; Dai, J.-Z.; Yang, C.-C.; Lin, C.-W. Prognostic Value of a Glycolytic Signature and Its Regulation by Y-Box-Binding Protein 1 in Triple-Negative Breast Cancer. Cells 2021, 10, 1890. [Google Scholar] [CrossRef] [PubMed]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef]
- Luoto, K.R.; Meng, A.X.; Wasylishen, A.R.; Zhao, H.; Coackley, C.L.; Penn, L.Z.; Bristow, R.G. Tumor cell kill by c-MYC depletion: Role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res. 2010, 70, 8748–8759. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Bryant, H.E.; Schultz, N. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 2005, 4, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed]
- Musiani, D.; Giambruno, R.; Massignani, E.; Ippolito, M.R.; Maniaci, M.; Jammula, S.; Manganaro, D.; Cuomo, A.; Nicosia, L.; Pasini, D.; et al. PRMT1 Is Recruited via DNA-PK to Chromatin Where It Sustains the Senescence-Associated Secretory Phenotype in Response to Cisplatin. Cell Rep. 2020, 30, 1208–1222. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, Q.; Li, C.; Wang, X.; Jiang, L.; Huang, L.; Wang, C.; Chen, H. PRMT1 regulates the tumour-initiating properties of esophageal squamous cell carcinoma through histone H4 arginine methylation coupled with transcriptional activation. Cell Death Dis. 2019, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jia, Z.; Xie, D.; Zhao, T.; Tan, Z.; Zhang, S.; Kong, F.; Wei, D.; Xie, K. Methylation of HSP70 Orchestrates Its Binding to and Stabilization of BCL2 mRNA and Renders Pancreatic Cancer Cells Resistant to Therapeutics. Cancer Res. 2020, 80, 4500. [Google Scholar] [CrossRef]
- Liao, H.W.; Hsu, J.M.; Xia, W.; Wang, H.L.; Wang, Y.N.; Chang, W.C.; Arold, S.T.; Chou, C.K.; Tsou, P.H.; Yamaguchi, H.; et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J. Clin. Investig. 2015, 125, 4529–4543. [Google Scholar] [CrossRef]
- Li, Z.; Owonikoko, T.K.; Sun, S.-Y.; Ramalingam, S.S.; Doetsch, P.W.; Xiao, Z.-Q.; Khuri, F.R.; Curran, W.J.; Deng, X. c-Myc suppression of DNA double-strand break repair. Neoplasia 2012, 14, 1190–1202. [Google Scholar] [CrossRef]
- Yi, J.; Liu, C.; Tao, Z.; Wang, M.; Jia, Y.; Sang, X.; Shen, L.; Xue, Y.; Jiang, K.; Luo, F.; et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine 2019, 43, 225–237. [Google Scholar] [CrossRef]
- Campaner, S.; Amati, B. Two sides of the Myc-induced DNA damage response: From tumor suppression to tumor maintenance. Cell Div. 2012, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Ning, J.-F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.K.; Mahapatra, S.; Kesherwani, V.; Kling, M.J.; Shukla, M.; Ray, S.; Kanchan, R.; Perumal, N.; McGuire, T.R.; Sharp, J.G.; et al. Role of protein arginine methyltransferase 5 in group 3 (MYC-driven) Medulloblastoma. BMC Cancer 2019, 19, 1056. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Salvatori, L.; Nanni, S.; Iwamoto-Stohl, L.K.; Valente, S.; Mai, A.; Scagnoli, F.; Fontanella, R.A.; Totta, P.; Nasi, S.; et al. The Protein Arginine Methyltransferases 1 and 5 affect Myc properties in glioblastoma stem cells. Sci. Rep. 2019, 9, 15925. [Google Scholar] [CrossRef]
- Tikhanovich, I.; Zhao, J.; Bridges, B.; Kumer, S.; Roberts, B.; Weinman, S.A. Arginine methylation regulates c-Myc-dependent transcription by altering promoter recruitment of the acetyltransferase p300. J. Biol. Chem. 2017, 292, 13333–13344. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, W.-J.; Chen, C.-H.; Chang, Y.-C.; Cheng, C.-H.; TsaI, Y.-H.; Lin, C.-W. PRMT1 Confers Resistance to Olaparib via Modulating MYC Signaling in Triple-Negative Breast Cancer. J. Pers. Med. 2021, 11, 1009. https://doi.org/10.3390/jpm11101009
Hsu W-J, Chen C-H, Chang Y-C, Cheng C-H, TsaI Y-H, Lin C-W. PRMT1 Confers Resistance to Olaparib via Modulating MYC Signaling in Triple-Negative Breast Cancer. Journal of Personalized Medicine. 2021; 11(10):1009. https://doi.org/10.3390/jpm11101009
Chicago/Turabian StyleHsu, Wen-Jing, Cheng-Hsun Chen, Yu-Chu Chang, Chia-Hsiung Cheng, Ying-Huei TsaI, and Cheng-Wei Lin. 2021. "PRMT1 Confers Resistance to Olaparib via Modulating MYC Signaling in Triple-Negative Breast Cancer" Journal of Personalized Medicine 11, no. 10: 1009. https://doi.org/10.3390/jpm11101009
APA StyleHsu, W.-J., Chen, C.-H., Chang, Y.-C., Cheng, C.-H., TsaI, Y.-H., & Lin, C.-W. (2021). PRMT1 Confers Resistance to Olaparib via Modulating MYC Signaling in Triple-Negative Breast Cancer. Journal of Personalized Medicine, 11(10), 1009. https://doi.org/10.3390/jpm11101009