Abstract
We present a case of a patient in their 80s initially presenting with myelodysplastic syndromes (MDS). Chromosomal analysis showed an abnormal female karyotype with a complex karyotype. Metaphase FISH confirmed four copies of KMT2A (MLL) in 24.5% [49/200] and amplification of KMT2A (MLL) with more than four copies in 22% [44/200]. FISH also revealed the presence of MYC (8q24) on the long arm of chromosome 2 at 2q33 locus, two copies of BCR on each homolog 22, and two additional copies of BCR on a derivative chromosome 22. Flow cytometric analysis revealed a population of aberrant myeloid blasts (15–17%). Bone marrow analysis showed hypercellular marrow with a significant increase in myeloid blasts (~50%) and trilineage dysplasia. Eventually, these findings were consistent with a final diagnosis of acute myeloid leukemia non-M3 and a complex karyotype, correlating with cytogenetics, flow cytometry, molecular, and clinical findings. The patient’s clinical course was marked by a rapid deterioration, including recurrent arrhythmias, hypoxic respiratory failure, and septic shock. Given their poor clinical status and adverse-risk molecular profile, care was transported to hospice. The presence of KMT2A amplification is a rare event in AML and is present in ~1% of AML and MDS cases. MYC translocation, KMT2A (MLL) amplification, and 5q/20q losses suggest secondary therapy-related AML and categorize this case in the adverse risk prognosis under the ELN 2022 guidelines.
Keywords:
acute myeloid leukemia; myeloid; MLL; complex karyotype; MYC; FISH; secondary-related AML; adverse-risk 1. Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy characterized by uncontrolled clonal proliferation of immature myeloid precursors [1]. Meanwhile, myelodysplastic syndromes (MDS) are similar, involving ineffective hematopoiesis in bone marrow neoplasms [2]. The difference lies in a blast-count boundary; if the patient has less than 20% blasts, they are considered MDS, and if they have more than 20% blasts, they are considered AML [3]. However, this is not a hard absolute, and depending on the patient’s case, they can be classified as MDS or AML, regardless of being above or under a 20% blast count.
AML is characterized by clonal expansion of immature myeloid precursor cells in the bone marrow and peripheral blood, disrupting normal hematopoiesis [4]. It affects primarily patients >65 years with an overall survival (OS) of 32% (<10% in patients >60 years) [5,6,7]. Furthermore, AML comprises 15–20% of pediatric leukemia and 35% of adult leukemia along with numerous chromosomal abnormalities commonly reported, contributing to its poor prognosis [8]. MDS is sometimes referred to as preleukemia, given its ability to transform into leukemias such as AML. This is known as secondary AML, or AML-MR, which is much more complex than AML [9].
Although AML is most common de novo, development through AML-MR, prior cytotoxic therapy, or environmental exposures is also well documented. AML-MR typically involves complex cytogenetic abnormalities and is associated with an adverse prognosis under the ELN 2022 guidelines [10,11]. MYC is a well-studied oncogenic driver of cellular proliferation, differentiation inhibition, and other roles in myeloid malignancies. Rearrangements and amplifications of MYC are well studied in lymphoid malignancies but MYC translocations are exceedingly rare in AML and, when found, are associated with therapy-related myeloid neoplasms or relapses [12,13]. The most common KMT2A rearrangement is the reciprocal translocation, partnering with >130 genes and chimeric fusion proteins seen in acute lymphoid leukemia (ALL), MDS, and AML [14]. KMT2A is a common mutation in AML, found in 3–10% of adult AML, and found to be an aggressive and prognostically unfavorable leukemic marker. KMT2A-r disrupts epigenetic regulation through the formation of chimeric proteins that activate oncogenic signaling pathways such as HOXA and MEIS1 [14].
However, KMT2A amplification, in contrast to rearrangement, is exceedingly rare, occurring in approximately 1% of AML and MDS cases and carrying a particularly poor prognosis associated with low response to chemotherapy and extremely short survival [15]. The presence of KMT2A amplification in AML is seen more frequently in elderly patients and coexists with other high-risk abnormalities, including highly complex karyotypes, del5q, TP53 loss or mutation, and occasionally del7q. Furthermore, data have suggested that KMT2A amplification has a pathogenesis different from KMT2A-r, further complicating its role as a rare genetic abnormality [16]. Amplification of KMT2A in AML was first observed in 1999 as although extra copies of KMT2A were previously recorded in the literature, true gene amplification was not verified [17]. Then, in a 2015 study involving 21 patients with KMT2A amplification in AML/MDS (11 with AML-MR, six therapy-related AML, and four therapy-related MDS), again highly complex karyotypes, TP53 deletion and mutation, and del5q were frequently observed. Furthermore, all patients demonstrated resistance to therapy with a median OS of 1 month after KMT2A amplification detection [18].
In this report, we present a case of AML with a KMT2A amplification, MYC translocation, and a complex karyotype. We will provide a summary of the case presentation, diagnostic workup, outcome, and insight into the management of the patient. We will conclude with a thorough discussion of the abnormalities discovered in this report and similar cases involving KMT2A amplifications and MYC rearrangements. Although KMT2A amplifications, found in 1% of AML and MDS, and MYC translocations are two notably rare events in their own sense, they are exceedingly rare in co-occurrence with very few reports in the literature, highlighting the importance of this case [16].
2. Case Presentation
A woman in her 80s with a history of coronary artery disease status post ST-elevation myocardial infarction and coronary artery bypass grafting, hypertension, hyperlipidemia, and a complicated postoperative sternal wound infection presented for evaluation of tachycardia. She was noted to have atrial fibrillation with rapid ventricular response and dyspnea on exertion following prolonged travel. Workup for pulmonary embolism was negative, and she was admitted for cardiac monitoring. Review of prior laboratory data showed normal blood counts before cardiac surgery.
Her postoperative course following coronary artery bypass grafting was complicated by recurrent wound infections and bacteremia requiring multiple surgical debridements and prolonged antibiotic therapy over the ensuing weeks. During this postoperative period, new-onset cytopenias were first noted. On admission, laboratory evaluation demonstrated pancytopenia with a white blood cell count of 2.56 × 109/L (ANC 1.26 × 109/L), hemoglobin 6.8 g/dL (requiring transfusion of one unit of packed red blood cells), and platelet count of 57 × 109/L. Hematology was consulted for the evaluation of pancytopenia. Initial workup revealed no evidence of hemolysis, nutritional deficiency, or monoclonal gammopathy (vitamin B12 in the 400s pg/mL, folate 14 ng/mL, and iron studies were consistent with inflammation, with an elevated ferritin of 420 ng/mL). The cytopenias were initially attributed to chronic inflammation and recent infection.
Given that the patient demonstrated clinical stability and slow improvement in blood counts, management options, including observation versus bone marrow biopsy, were discussed, with a shared decision to pursue close monitoring and supportive care. However, after approximately two months of persistent pancytopenia without hematologic recovery, a bone marrow aspiration and core biopsy were performed.
A complete blood count (CBC) obtained in July of 2025 demonstrated a white blood cell count (WBC) of 3.00 k/µL, hemoglobin of 9.2 g/dL, hematocrit of 29.5%, mean corpuscular volume of 91.6 fL, and a platelet count of 83 k/µL. Evaluation of bone marrow was performed, including aspiration, core biopsy, touch preparation, and clot section. The aspirate and touch-prep are markedly hypercellular and demonstrate trilineage arrest with an increase in blasts. Histological analysis demonstrated that blasts are medium to large in size with moderate cytoplasm and absent cytoplasmic granules and no Auer rods. Dysplastic changes were present in all three hematopoietic lineages, with the myeloid lineage showing neutrophils with hypogranular forms as well as abnormally lobate forms. The erythroid lineage displayed megablastoid maturation, and megakaryocytes showed hypolobated nuclei. A 200-cell bone marrow aspirate differential showed markedly increased blasts and increased nucleated red blood cells along with reduced mature myeloid forms (Table 1).

Table 1.
Bone marrow aspirate differential (200-cell count).
Flow cytometric analysis revealed a population of aberrant myeloid blasts (about 15–17% of total events). The cells express CD45 (dim+), CD34 (+), CD117 (+), CD33 (dim+), CD13 (+), CD123 (weak+), HLA-DR (+), CD38 (variable negative to dim positive), CD7 (small subset, variable dim+), CD4 (−), CD11b (−), CD15 (−), CD11c (weak/−), CD135 (+), and MPO (+). The marrow was markedly hypercellular (80–90%) with clusters of myeloblasts and mild marrow fibrosis (grade 0–1). Iron staining revealed increased ring sideroblasts.
The patient was initially planned for hypomethylating agent–based therapy but experienced rapid clinical deterioration, including recurrent atrial arrhythmias, hypoxic respiratory failure, neutropenic fever, pneumonia, and septic shock requiring intensive care. Care was transitioned to a comfort-focused approach, and the patient was discharged to hospice.
3. Materials and Methods
3.1. Conventional Cytogenetics
Chromosome analysis was performed using conventional cytogenetics protocols, including trypsin-Giemsa (G-band) banding of twenty metaphase spreads, eleven from a 24 h bone marrow culture and nine from a 72 h culture at a resolution of 400 chromosome bands. Results were annotated according to the International System for Human Cytogenomic Nomenclature 2024 standards [19]
3.2. Molecular Cytogenetics
Fluorescence in situ hybridization (FISH) was performed using the following probes: XL KMT2A BA (11q23.3) break-apart, XL MECOM (3q26), XL t(15;17) Dual Fusion (15q24, 17q21.1-q21.2) (MetaSystems, Medford, MA, USA), LSI 5q EGR1/D5S23, D5S721 (5q31, 5p15.2), LSI D7S486/CEP7 (7q31/7p11.1-q11.1 [D7Z1]), LSI RUNX1/RUNX1T1 (8q21.3, 21q22), LSI MYC DC BAR (8q24), LSI BCR/ABL1 DC DF (9q34, 22q11.2) (Abbott, Des Plaines, IL, USA), LSI CBFB (16q22) DC BAR, LSI CEP8 (D8Z2), LSI BCL2 (18q21) BAR, PDGFRA (4q12) BA (Empire Genomics, Depew, NY, USA), and del(20q) Deletion (20q12, 20q13.1) (CytoCell, Cambridge, UK).
This case report was conducted in accordance with institutional ethical standards and the Declaration of Helsinki. Due to the patient’s death during the same hospitalization and the absence of identifiable next of kin, written informed consent specific to publication could not be obtained. However, a general consent for treatment and use of de-identified clinical information for educational and research purposes had been signed at the time of hospital admission. All identifying information has been removed to protect patient privacy.
4. Results
4.1. Flow Cytometry and Blast Assessment
The immunophenotypic profile suggests a clonal population of immature myeloid blasts showing abnormal maturation. Altogether, the biopsy findings provide important information not fully realized within the initial flow cytometry and aspirate differential. Flow cytometry identified 15–17% aberrant myeloid blasts, and the aspirate differential showed a diagnostically significant blast percentage, while the performed core biopsy and CD34/CD117 immunostains demonstrated a markedly higher blast burden of approximately 40–50%. The blast burden of approximately 40–50% on core biopsy was estimated based on morphologic assessment of representative sections along with CD34 and CD117 immunohistochemical staining. Given the discrepancy between modalities, the highest blast percentage identified on core biopsy was used for the final disease classification, as the aspirate and flow cytometric assessments were felt to underestimate blast burden due to sampling limitations.
4.2. Conventional Cytogenetics
Conventional cytogenetics utilizing the 20 Giemsa-banded metaphase spreads demonstrated an abnormal female karyotype with a highly complex pattern of structural and numerical abnormalities (Figure 1B). The composite karyotype was described as 45~48,XX,add(2)(q33),add(4)(q13),add(5)(q22),add(8)(q24),add(10)(q22),add(16) (p13.3), 18,−20,add(22)(q13),+add(22)(q13),+1~5 mar[cp20]ish. The findings were consistent with a complex karyotype characterized by multiple unbalanced rearrangements.
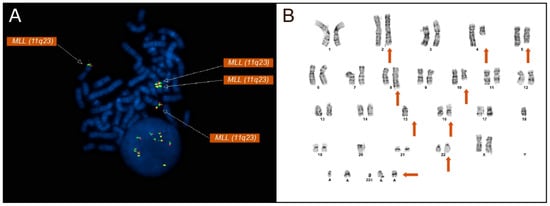
Figure 1.
(A) Metaphase FISH using a KMT2A (MLL, 11q23) break-apart probe demonstrating multiple KMT2A signals, including additional amplified KMT2A material on chromosome 16p13. The presence of multiple KMT2A signals indicates gene copy number amplification rather than a balanced chromosomal rearrangement. All KMT2A signals on the metaphase FISH are denoted by arrows. (B) The patient’s G-banded karyotype displays a complex karyotype including multiple structural and numeric abnormalities (orange arrows denote derivative chromosomes displaying abnormalities). A complex karyotype is defined by the presence of multiple structural and numerical chromosomal abnormalities. Additional material involving chromosomes 2, 5, 8, 10, 16, and 22, as well as marker chromosomes, is indicated.
4.3. Molecular Cytogenetics
FISH analysis was performed on 200 nuclei utilizing a panel of probes targeting recurrent AML and MDS-associated abnormalities. Most notably, FISH analysis with the KMT2A (MLL) break-apart probe revealed an increased copy number of KMT2A signals. Four copies of KMT2A were found in 24.5% [49/200] of nuclei, and more than four copies were found in 22% [44/200], displaying an amplification of KMT2A not commonly observed in AML and MDS cases (Figure 1A). FISH analysis confirmed the presence of an amplification of KMT2A rather than the more commonly associated rearrangements seen in AML and MDS patients.
Additionally, the FISH results were consistent with one copy of EGR1 in 83.5% [167/200] of nuclei. Similarly to KMT2A, four copies of BCR were found in 88% [176/200] of the examined nuclei. Additionally, one copy of BCL2, PTPRT, and MYBL2 was found in 85.5% [171/200], 81% [162/200], and 81% [162/200], respectively.
Metaphase FISH showed the relocation of MYC signals to the long arm of chromosome 2 at 2q33 (Figure 2), along with loss of 5q31 on one homolog of chromosome 5 (Figure 1B). Metaphase FISH also revealed two additional KMT2A signals localized to chromosome 16p13, indicating insertion of amplified KMT2A material at this locus (Figure 1A). Similarly, two copies of BCR were identified on each normal chromosome 22, with two additional BCR signals present on a derivative chromosome 22, consistent with the BCR copy number gain. Additionally molecular testing identified a TP53 mutation.
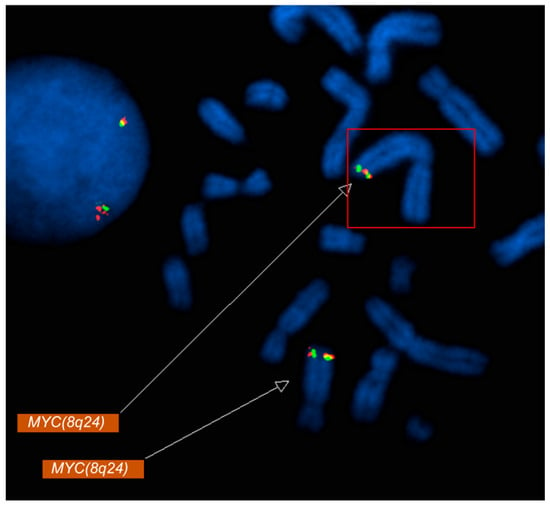
Figure 2.
Metaphase FISH analysis using a MYC (8q24) probe demonstrates relocation of MYC signals to the long arm of chromosome 2 (2q33). Fluorescent MYC signals (red/green) are visualized on metaphase chromosomes (DAPI counterstain in blue). The boxed region and upper arrow highlight the aberrant MYC signal on chromosome 2q33, consistent with a MYC rearrangement/translocation, while the lower arrow indicates the MYC signal at the native 8q24 locus.
Deletion probes targeting 5q and 20q displayed a loss of material consistent with a report of del(5q) and del(20q). No evidence of t(15;17)(q24;q21), RUNX1::RUNX1T1, CBFB rearrangement, MECOM rearrangement, PDGFRA rearrangement, or BCL2 rearrangement was identified.
5. Discussion
KMT2A is located on chromosome 11q23 and catalyzes the methylation of lysine residues on histone tails, particularly histone H3 typically at lysine 4. Unlike other epigenetic enzymes like histone acetyltransferases (HATs), KMTs modify just one or two lysines per histone [20,21]. It is primarily the SET domain at the C-terminus of KMTs that is responsible for methylation of H3 at lysine 4 (H3K4). In previous studies, KMT2A has been shown to be part of a macro protein complex that functions to promote stability of KMT2A and regulate transcriptional activation of HOX genes [21,22]. KMT2A amplifications, in contrast to translocations, are defined as focal copy number amplifications that result in increases in gene copy number of the KMT2A gene [23]. Furthermore, mutations involving balanced rearrangements of KMT2A (KMT2A-r) have been commonly observed in AML, appearing in 3–10% of AML patients, while KMT2A amplifications are much rarer in AML [14,16].
MYC is a proto-oncogene located at chromosome 8q24, playing roles in cell-cycle progression, apoptosis, and cellular transformation [24,25]. As an oncogene, MYC is one of the most frequently mutated events in cancer, including one of the most frequently amplified regions in AML [25]. In recent studies, MYC has shown to regulate downstream genes responsible for apoptosis and differentiation of AML cells as well as being overexpressed/required for myeloid leukemias triggered by FLT3-ITD and PML-RARA and other fusion oncoproteins [26]. In de novo AML patients, overexpression of MYC has been associated with inferior survival outcomes with more limited clinical data on MYC rearrangements in AML patients though some studies have revealed chemotherapy resistance [25,26].
This patient’s case highlights many unique features of a complex karyotype present with atypical abnormalities such as KMT2A amplifications and translocations of MYC in AML. As a patient in her 80s, this falls under the observation that KMT2A amplifications, if seen, are typically present in elderly patients with AML or MDS [16]. This patient’s prior history of major surgery and coronary heart disease, recurring infections, and subsequent prolonged antibiotic therapy all contribute to the comorbidities that may have affected the eventual prognosis of this patient.
Bone marrow evaluations helped reveal a significantly hypercellular marrow with trilineage dysplasia and a large increase in myeloid blasts, demonstrating progression to AML rather than MDS and establishing a final diagnosis of acute myeloid leukemia (non-M3). The presence of ring sideroblasts and mild marrow fibrosis further illustrates the degree of marrow dysregulation in this patient.
The differences in blast percentages reported across flow cytometry (15–17%), bone marrow aspirate differential (26%) and core biopsy (40–50%), illustrate an important clinical discordance. This is a common discrepancy in AML, reflecting sampling variability and differences in methodology, and is important to recognize. These differences are critical as relying on a single assessment may underestimate disease significance or delay diagnosis and risk stratification, even misclassifying a patient (AML v. MDS). Integrating all morphological and immunophenotypic data is a necessity in ensuring an accurate evaluation of blast burden and guiding appropriate decision making.
Cytogenetic and molecular findings in this patient also demonstrate the aggressive nature of her disease. KMT2A amplification co-occurred with a MYC translocation to chromosome 2q33, TP53 was found to be mutated, and there were marked losses of 5q and 20q. While BCR copy number gains were also observed, they are deemed to likely be less clinically relevant than the MYC and KMT2A abnormalities. MYC translocations combined with amplification of KMT2A in an AML patient are an exceptionally rare occurrence. Coupled with the complex karyotype of the patient and the TP53 mutation found present, these indicators effectively foreshadowed an extremely poor prognosis, especially when the age and history of the patient are considered. These findings correlate closely with the patient’s rapid clinical deterioration, including recurrent atrial arrhythmias, hypoxic respiratory failure, neutropenic fever, and ultimately the transition to hospice care.
In 2022, karyotyping/FISH data on 42 AML/MDS patients and next-generation sequencing (NGS) and prognostic data on a patient subset was studied, all with KMT2A amplification [27]. With a median age at diagnosis of 70 years, the median survival was 45 days and a complex karyotype was found in 100% (n = 38) of cases. 5q and 17p deletions were the most common findings followed by del7q and gain of chromosome 8 with TP53 mutations, the most commonly found in NGS [27]. As seen in previous studies, AML/MDS with KMT2A amplification was associated with poor prognosis and specific cytogenetic abnormalities including complex karyotypes, del5q, and TP53 mutations. MYC translocations are also seen to carry a significantly adverse prognosis, as in one study involving t(3;8)(q26.2;q24) patients (8 with t-AML, 3 with t-MDS), a common MYC translocation, 18 out of 20 patients died with a median survival of ~6 months [28].
Gray et al. in 2023 then demonstrated that histone H3 lysine 9 mono- and di-methylation (H3K9me1/2) balance at the KMT2A locus can regulate these amplifications and rearrangements [29]. This balance, through crosstalk between the depletion of lysine demethylase KDM3B that increases H3K9me1/2 levels and methyltransferase G9a/EHMT2, increases amplification of KMT2A. It was seen that depleting CFTC, a transcription factor, at the KMT2A locus through KDM3B depletion was also enough to generate these genetic aberrations [29].
This case demonstrated the challenges of managing a high-risk elderly and frail patient diagnosed with AML. Despite initial stability and careful monitoring, the patient’s disease progressed rapidly, limiting the feasibility of hypomethylating agent therapy. Along with the patient’s presentation of a complex karyotype, elderly age, and features suggestive of secondary or therapy-related AML, the selection of FISH targets was driven by their relevance in AML and MDS. KMT2A amplification, though rare, is generally associated with patients with similar molecular profiles. Evaluating KMT2A among other prognostic markers such as a TP53 mutation provides a more comprehensive risk stratification. Furthermore, compared to previously reported cases of KMT2A amplified AML, our case is distinguished by the concurrent presence of a MYC translocation. Although KMT2A amplification carries an adverse outcome, typically along a complex karyotype and TP53 mutations, a MYC translocation is exceptionally rare. This co-occurrence is rarely seen in the literature, underscoring the unique molecular profile of this patient.
The correlation seen between the patients’ cytogenetic and molecular abnormalities, their aggressive clinical course, and poor prognosis demonstrates the importance of early diagnostic evaluations and risk stratification in other similar patients. Integration of flow cytometry, bone marrow morphology, conventional cytogenetics, and FISH/molecular testing provided a detailed understanding of the patient’s high-risk disease biology, which helps to inform clinical decision-making and counseling in future cases with similarly rare abnormalities.
Although the current prognosis of AML with KMT2A aberrations is poor, there are emerging therapeutic strategies that may hold promise. For example, menin inhibitors that disrupt the interaction between menin and KMT2A, and suppress aberrant HOX/MEIS1 expression, have shown promising preclinical applications [14]. Revumenib and Ziftomenib, among other menin inhibitors, are being evaluated in phase I/II studies and demonstrate reduction in leukemic blasts in patient cohorts.
6. Conclusions
In conclusion, KMT2A amplifications occur in just 1% of AML/MDS cases and are functionally distinct from KMT2A rearrangements. Furthermore, MYC rearrangements are similarly rare in AML and also confer a poor prognosis. Although there are limited cases in the literature of these reports, KMT2A amplifications in AML have consistently been observed alongside complex karyotypes, multiple chromosomal abnormalities, and poor survival outcomes. Along with the characteristic low median OS and high resistance to therapy, the unique co-occurrence of a MYC rearrangement with KMT2A amplification is a rare case that illustrates the importance of KMT2A amplifications as a prognostic and therapeutic marker for AML.
7. Limitations
This report is limited by its single-case design; thus the findings may not be generalizable to a larger population of KMT2A-amplified AML patients. Given the rarity of this molecular profile, it can be difficult to directly compare results to similar patients. Longitudinal molecular monitoring was not performed, limiting assessment of clonal evolution over time. Additionally, the patient’s rapid clinical decline prevented further evaluation of treatment response or resistance patterns. Finally, given the highly complex karyotype, it is also difficult to determine the individual contribution of each cytogenetic marker to the overall course and prognosis.
Author Contributions
Conceptualization, C.A.T., C.W., F.H. and J.C.; methodology, J.C. and F.H.; software, J.C. and F.H.; validation, F.H., J.C. and C.A.T.; formal analysis, J.C.; investigation, F.H. and C.W.; resources, C.A.T.; data curation, J.C. and F.H.; writing—original draft preparation, C.W.; writing—review and editing, J.C. and F.H.; visualization, F.H.; supervision, C.A.T.; project administration, C.A.T.; funding acquisition, C.A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Single-patient case reports do not require Institutional Review Board approval, as they are typically not classified as research under regulations in the U.S. The work was conducted in accordance with the Declaration of Helsinki.
Informed Consent Statement
As informed consent was not possible with the patient dying the same week of admission as well as a lack of contactable family members, the patient profile was edited to remove all identifiable features. Furthermore, all mentions of age of the patient were edited to “80s” to protect patient privacy.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chen, J.; Hassan, F.; Tirado, C.A. A Mutational Landscape in Acute Myeloid Leukemia: Overview and Prognostic Impacts. Diagnostics 2025, 15, 2537. [Google Scholar] [CrossRef]
- Vakiti, A.; Reynolds, S.B.; Mewawalla, P. Cancer, Acute Myeloid Leukemia (AML, Erythroid Leukemia, Myelodysplasia-Related Leukemia, BCR-ABL Chronic Leukemia); StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Chaudhuri, D.; Kokab, I.K.; Roba, A.S.; Akhil, A.; Ferguson, A.A.; Aujala, I.K.; Baraa, A.; Sai, D.G.; Hamid, P. Secondary Acute Myeloid Leukemia in Myelodysplastic Syndrome Patients Aged over 60 Years. Curēus 2023, 15, e40124. [Google Scholar] [CrossRef]
- Shimony, S.; Stahl, M.; Stone, R.M. Acute Myeloid Leukemia: 2025 Update on Diagnosis, Risk-Stratification, and Management. Am. J. Hematol. 2025, 100, 860–891. [Google Scholar] [CrossRef]
- Juliusson, G.; Antunovic, P.; Derolf, Å.; Lehmann, S.; Möllgård, L.; Stockelberg, D.; Tidefelt, U.; Wahlin, A.; Höglund, M. Age and Acute Myeloid Leukemia: Real World Data on Decision to Treat and Outcomes from the Swedish Acute Leukemia Registry. Blood 2009, 113, 4179–4187. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Acute Myeloid Leukemia—Cancer Stat Facts; National Cancer Institute: Bethesda, MD, USA, 2024. [Google Scholar]
- Sasaki, K.; Ravandi, F.; Kadia, T.M.; DiNardo, C.D.; Short, N.J.; Borthakur, G.; Jabbour, E.; Kantarjian, H.M. De Novo Acute Myeloid Leukemia: A Population-Based Study of Outcome in the United States Based on the Surveillance, Epidemiology, and End Results (SEER) Database, 1980 to 2017. Cancer 2021, 127, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Makkar, H.; Majhi, R.K.; Goel, H.; Gupta, A.K.; Chopra, A.; Tanwar, P.; Seth, R. Acute Myeloid Leukemia: Novel Mutations and Their Clinical Implications. Am. J. Blood Res. 2023, 13, 12–27. [Google Scholar]
- Green, S.D.; Wang, E.S. How I Treat: Secondary Acute Myeloid Leukemia. Blood J. 2024, 145, 1260–1272. [Google Scholar] [CrossRef]
- Capelli, D.; Menotti, D.; Fiorentini, A.; Saraceni, F.; Olivieri, A. Secondary Acute Myeloid Leukemia: Pathogenesis and Treatment. Leukemia 2022, 145, 111–127. [Google Scholar]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood 2022, 140, 1260–1272. [Google Scholar] [CrossRef]
- Murgas, K.A.; Materum, P., III; Li, L.Z.; Rocha, J.; Schuster, M.; Ahmed, T.; Tirado, C.A. A Relapsed AML Case Featuring MYC and MECOM Rearrangements. Diagnostics 2025, 15, 2410. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.D.; Albajar, M.; Gomez-Casares, M.T.; Batlle, A.; León, J. MYC Oncogene in Myeloid Neoplasias. Clin. Transl. Oncol. 2012, 15, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Zehtabcheh, S.; Soleimani Samarkhazan, H.; Asadi, M.; Zabihi, M.; Parkhideh, S.; Mohammadi, M.H. Insights into KMT2A Rearrangements in Acute Myeloid Leukemia: From Molecular Characteristics to Targeted Therapies. Biomark. Res. 2025, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Ittel, A.; Vey, N.; Mozziconacci, M. Multiple Cytogenetics Forms of KMT2A Amplification and Jumping Translocation in a Case of Myelodysplastic Syndrome. eJHaem 2021, 2, 905–906. [Google Scholar] [CrossRef]
- López de la Osa María, J.; Juárez-Salcedo Luis, M.; Ortiz, J.; Benzo, G.; Cannata, J.; Loscertales, J.; Dalia, S.; Alegre, A.; Arranz, E. The Role of KMT2A Gene Amplification in Acute Myeloid Leukemia, Serie of Cases and Literature Review. Int. J. Clin. Stud. Med. Case Rep. 2025, 49, 1–4. [Google Scholar]
- Hervé, A.-L.; Godon, C.; Li, J.-Y.; Axelle, D.; Marie-Paule, M.; Pascaline, T.; Jean-Luc, H.; Bataille, R. Amplification of the 11q23 Region in Acute Myeloid Leukemia. Genes Chromosomes Cancer 1999, 26, 166–170. [Google Scholar]
- Tang, G.; DiNardo, C.; Zhang, L.; Farhad, R.; Khoury, J.D.; Huh, Y.O.; Tariq, M.; Medeiros, L.J.; Wang, S.A.; Bueso-Ramos, C.E. MLL Gene Amplification in Acute Myeloid Leukemia and Myelodysplastic Syndromes Is Associated with Characteristic Clinicopathological Findings and TP53 Gene Mutation. Hum. Pathol. 2015, 46, 65–73. [Google Scholar] [CrossRef]
- Hastings, R.J.; Moore, S.; Chia, N. ISCN 2024: An International System for Human Cytogenomic Nomenclature (2024). Cytogenet. Genome Res. 2024, 164, 1–224. [Google Scholar] [CrossRef]
- Castiglioni, S.; Di Fede, E.; Bernardelli, C.; Lettieri, A.; Parodi, C.; Grazioli, P.; Colombo, E.A.; Ancona, S.; Milani, D.; Ottaviano, E.; et al. KMT2A: Umbrella Gene for Multiple Diseases. Genes 2022, 13, 514. [Google Scholar] [CrossRef]
- Milan, T.; Celton, M.; Lagacé, K.; Roques, É.; Safa-Tahar-Henni, S.; Bresson, E.; Bergeron, A.; Hebert, J.; Meshinchi, S.; Cellot, S.; et al. Epigenetic Changes in Human Model KMT2A Leukemias Highlight Early Events during Leukemogenesis. Haematologica 2020, 107, 86–99. [Google Scholar] [CrossRef]
- Slany, R.K. The Molecular Biology of Mixed Lineage Leukemia. Haematologica 2009, 94, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xing, S.; Zhang, H.; Mao, X.; Xiao, M.; Wang, Y. FISH Improves Risk Stratification in Acute Leukemia by Identifying KMT2A Abnormal Copy Number and Rearrangements. Sci. Rep. 2022, 12, 9585. [Google Scholar] [CrossRef]
- NCBI. MYC MYC Proto-Oncogene, BHLH Transcription Factor [Homo Sapiens (Human)]—Gene; NCBI: Bethesda, MD, USA, 2026. [Google Scholar]
- Boddu, P.; Pemmaraju, N.; Tang, G. MYC Rearrangements in Myeloid Neoplasms. Blood 2021, 130, 5077. [Google Scholar]
- Yun, S.; Sharma, R.; Chan, O.; Vincelette, N.D.; Sallman, D.A.; Sweet, K.; Padron, E.; Rami, K.; Lancet, J.E.; Abraham, I.; et al. Prognostic Significance of MYC Oncoprotein Expression on Survival Outcome in Patients with Acute Myeloid Leukemia with Myelodysplasia Related Changes (AML-MRC). Leuk. Res. 2019, 84, 106194. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.; Zhao, J.; Andersen, E.; Hong, B. 62. Characterization of Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML) with KMT2A Amplification. Cancer Genet. 2024, 286–287, S20. [Google Scholar] [CrossRef]
- Tang, G.; Hu, S.; Wang, S.A.; Xie, W.; Lin, P.; Xu, J.; Gokce, T.; Zhao, M.; Gu, J.; Doty, M.; et al. T(3;8)(Q26.2;Q24) Often Leads to MECOM/MYC Rearrangement and Is Commonly Associated with Therapy-Related Myeloid Neoplasms And/or Disease Progression. J. Mol. Diagn. 2019, 21, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Gray, Z.H.; Chakraborty, D.; Duttweiler, R.R.; Alekbaeva, G.D.; Murphy, S.E.; Chetal, K.; Ji, F.; Ferman, B.I.; Honer, M.A.; Wang, Z.; et al. Epigenetic Balance Ensures Mechanistic Control of MLL Amplification and Rearrangement. Cell 2023, 186, 4528–4545.e18. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.