Abstract
Background: IgG4-related hypertrophic pachymeningitis (IgG4-RHP) is an extremely rare central nervous system (CNS) autoimmune disorder, characterized by dural thickening, space-occupying effects, and neurological compression symptoms. It is frequently misdiagnosed as meningioma due to overlapping radiological features, leading to inappropriate management. This study aims to report a unique case of IgG4-RHP with skull destruction and subcutaneous mass formation, and summarize its diagnostic and therapeutic strategies through literature review. Methods: A 53-year-old male with a chronic subdural hematoma history was admitted for a progressive right frontal subcutaneous mass. Preoperative computed tomography (CT) and magnetic resonance imaging (MRI) were performed, followed by staged surgeries (subcutaneous biopsy and craniotomy with subtotal resection). Histopathological examinations (Hematoxylin and Eosin staining, IgG/IgG4 immunostaining) and serum IgG4 detection were conducted. The patient received postoperative prednisone acetate (60 mg/d) and 3-month follow-up. A literature search was also performed to analyze 34 previously reported IgG4-RHP cases. Results: Histopathology showed dense lymphoplasmacytic infiltration, storiform fibrosis, ≈40 IgG4+ plasma cells per high-power field (HPF), and an IgG4+/IgG+ ratio of ≈30%. Serum IgG4 was significantly elevated to 1521 μg/mL (normal < 1350 μg/mL), with marked reduction in residual lesions on follow-up MRI. Literature review revealed a 73.5% male predominance, mean age of 48.6 years, headache as the most common symptom (58.8%), and a 38.5% misdiagnosis rate. Glucocorticoids alone or combined with immunosuppressants achieved favorable outcomes in 96.0% of treated cases. Conclusions: Histopathological examination combined with serum IgG4 detection is the gold standard for IgG4-RHP diagnosis. Surgical resection relieves mass-occupying effects, while glucocorticoids are first-line therapy. Long-term follow-up is necessary for recurrence monitoring, and rituximab is effective for refractory cases. Awareness of atypical manifestations like skull destruction can reduce misdiagnosis and improve outcomes.
1. Introduction
IgG4-related hypertrophic pachymeningitis (IgG4-RHP) is an exceptionally rare autoimmune disorder of the central nervous system (CNS), characterized by space-occupying effects and neurologic compression symptoms stemming from focal or diffuse thickening of the dura mater and spinal meninges [1]. Striking IgG4+ plasma cell infiltrate (IgG4+/IgG+ ratio > 40% or >10 IgG4+ plasma cells per high-power field [HPF]) and storiform fibrosis are major pathological hallmarks of IgG4-RHP. Serum IgG4 can be elevated in some IgG4-RHP patients [2,3]. IgG4-RHP can be easily misdiagnosed as meningioma due to the similar radiological features [3]. An early recognition of the autoimmune nature of IgG4-RHP, which is distinctly different from tumors, can avoid excessive surgery and guide hormone therapy. IgG4-related disease (IgG4-RD) is often seen in the pancreas, lungs and salivary glands. A single manifestation in the dura mater can be extremely rare. Isolated IgG4-RHP is a rare subtype of IgG4-RD. We reported a case of IgG4-RHP confirmed by pathological examination, presenting with the triad of “a history of chronic subdural hematoma + skull invasion + subcutaneous soft tissue mass”, which has not been documented in previous literature. The case was initially misdiagnosed as atypical meningioma and later confirmed by pathological re-examination of the resected mass. Postoperatively, the patient was treated with prednisone acetate at a daily dose of 60 mg, and significant regression of residual lesions was observed after 3 months. This study aims to provide clinical experience for the identification, diagnosis, treatment and management of this rare disease, reduce the rate of misdiagnosis, and improve patient prognosis.
2. Case Presentation
2.1. Clinical Presentation
A 53-year-old male presented with a 1-year history of a progressively enlarged subcutaneous mass in the right frontal lobe. Two years earlier, he underwent burr hole evacuation for a chronic subdural hematoma in the right frontotemporal lobe. Neurological and systemic examinations were unremarkable. A family history of autoimmune disorders or similar conditions in family members was not documented.
2.2. Imaging
Head computed tomography (CT) scans (Philips Healthcare, Best, the Netherlands; software version R5.3) displayed a soft-tissue mass with a Hounsfield unit (HU) of 44, manifesting as a slightly high-density shadow in the right frontal lobe underneath the parietal bone, and compressed adjacent brain tissues (Figure 1A,B). Destruction of the right parietal bone was also seen on CT scans (Figure 1C,D). Nodular and strip-shaped thickening, uniform dural enhancement at the top of the right frontal, temporal and parietal lobes, and a spindle-shaped mass under the scalp with an abnormal cranial plate signal were found on preoperative contrast-enhanced magnetic resonance imaging (MRI) scans (Philips Healthcare, Best, the Netherlands; software version R6.1) (Figure 2A–D).
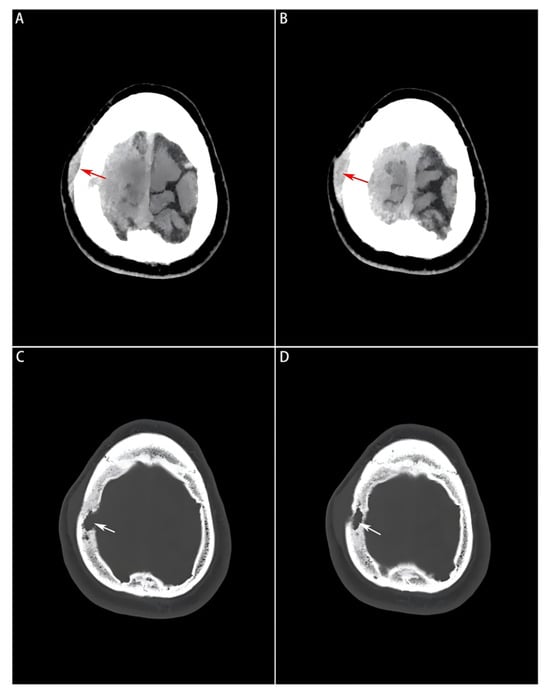
Figure 1.
Preoperative head CT images of the soft tissue window and bone window. (A,B): A slightly high-density mass visualized on the soft tissue window, where the subcutaneous mass formed by the tumor lesion invading outside the bone window is indicated by a red arrow; (C,D): Bone erosion and defect visualized on the bone window image, and localized by a white arrow.
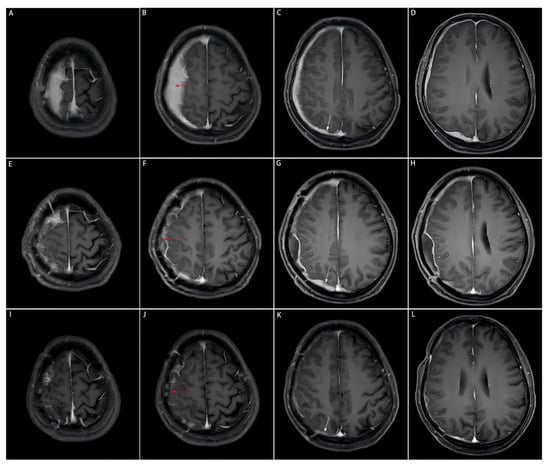
Figure 2.
Comparison of preoperative, postoperative, and post-medication enhanced-contrast MRI images. The area of the resected dural mass before and after the subtotal resection, and after medication on enhanced-contrast MRI images is indicated by the red arrow. The dural mass close to the cerebral falx on enhanced-contrast MRI images is indicated by the white arrow. This part is not surgically removed due to the tricky localization approach to the midline, but significantly shrinks after the three-month medication of prednisone acetate. (A–D): Enhanced-contrast MRI images preoperatively, (E–H): 2 months postoperatively, (I–L): 3 months of prednisone acetate.
2.3. Surgical Interventions
Staged surgeries were applied to the patient. First, the subcutaneous mass was biopsied for pathological examination, initially indicating a diagnosis of an atypical meningioma. A craniotomy was later performed, revealing a lesion invading two sites of the skull. The yellow-white, tough mass was located between the two layers of the dura mater without an invasion into the cerebral cortex, which was finally processed by a subtotal resection (Figure 3A,B). Contrast-enhanced MRI was performed during the follow-up period (Figure 2E–G).
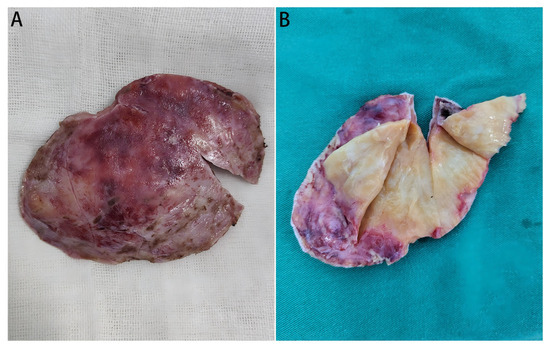
Figure 3.
Gross views of the resected mass. (A): The overall view of the resected tumor mass; (B): Gross view the mass (cut open), which is yellow-white and tough, with fine textured internal tissues.
2.4. Pathological Diagnosis
In pathological examination, Hematoxylin and Eosin (H&E) staining of the intracranial specimen showed fibrosis, inflammatory infiltration of lymphocytes and plasma cells (Figure 4A,B), and elevated number of IgG4+ plasma cells (≈40 IgG4+ plasma cells/HPF, IgG4+/IgG+ ratio ≈ 30%) (Figure 4C,D). Finally, a pathological diagnosis of IgG4-RHP was confirmed. H&E Staining and IgG/IgG4 Immunohistochemistry: Specimens were fixed in 4% formaldehyde, embedded in paraffin, and serially sectioned at a thickness of 4 μm. H&E staining was performed using a standard hematoxylin-eosin staining kit (Manufacturer: Thermo Fisher, Waltham, MA, USA; Catalog No.: NC166). For IgG immunohistochemistry, a rabbit anti-human IgG monoclonal antibody was used (Clone No.: EPR4421; Manufacturer: Abcam, Cambridge, UK; Catalog No.: ab109489; Dilution ratio: 1:500). For IgG4 immunohistochemistry, a rabbit anti-human IgG4 monoclonal antibody was employed (Clone No.: MRQ-44; Manufacturer: Cell Marque, Rocklin, CA, USA; Catalog No.: 276M-96; Dilution ratio: 1:300). The staining procedure followed the EnVision two-step method, with diaminobenzidine (DAB) as the chromogen and hematoxylin as the counterstain. Counting Criteria for IgG+ and IgG4+ Plasma Cells: Three positive cell-rich regions (hot spots) were selected under a high-power field (HPF, ×400) to count IgG+ and IgG4+ plasma cells. The assessment was conducted by two investigators in a double-blind manner, and the average value was used to calculate the IgG4+/IgG+ ratio.
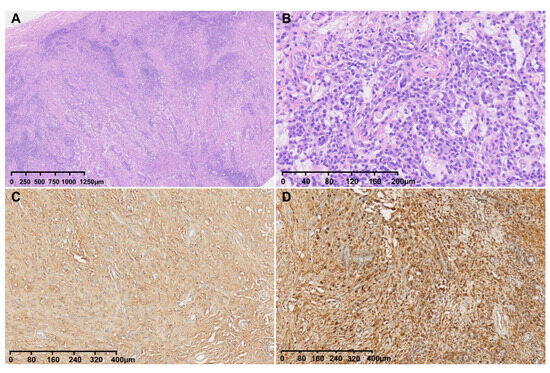
Figure 4.
H&E and IgG/IgG4 staining of tumor sections. (A): H&E staining of tumors under low magnification; (B): H&E staining of the mass showing an abundant infiltration of plasma cells; (C): IgG staining of the mass; (D): IgG4 staining of the mass (about 40 IgG4+ plasma cells/HPF in the hot spots, and 30% of the IgG4+/IgG+ ratio).
2.5. Laboratory Findings
Serum IgG and IgG4 testing was not timely performed due to an initial diagnosis of meningioma. After the second surgery of subtotal resection, an elevated serum IgG4 level (1521 μg/mL;36 μg/mL < normal range < 1350 μg/mL), but normal total IgG level (17,200 μg/mL; 860 μg/mL < normal range < 1740 μg/mL) were detected in the patient.
2.6. Treatment and Follow-Up
The patient was postoperatively medicated with prednisone acetate (60 mg/d), calcium supplements and proton pump inhibitors to prevent complications. The follow-up imaging at 3 months postoperatively showed a significant reduction in diffuse meningeal thickening and enhancement, suggesting a sensitive response of IgG4-RHP to glucocorticoid therapy (Figure 2I–L).
3. Literature Review on 34 Cases of IgG4-RHP
We searched PubMed and Google Scholar for literature using the keywords “IgG4”, “hypertrophic pachymeningitis”, and “intracranial lesions”. Cases without pathological confirmation and those involving only intraspinal lesions were excluded (Please refer to the Supplementary Materials: File S1: Inclusion and Exclusion Criteria Analysis), and a total of 34 patient case reports were ultimately selected (as shown in Table 1).

Table 1.
Clinical, Pathological, Therapeutic and Outcome Characteristics of 34 Cases.
A multidimensional characterization analysis was performed on these 34 cases. The patient cohort predominantly consisted of individuals ranging from young to elderly (mean age: 48.6 years, range: 15–86 years), including 25 males (73.5%) and 9 females (26.5%). The main clinical manifestations were as follows: headache in 20 cases (58.8%), seizures in 7 cases (20.6%), visual impairment in 5 cases (14.7%), hearing loss in 5 cases (14.7%), limb sensory disturbance in 4 cases (11.8%), facial sensory disturbance in 3 cases (8.8%), nausea in 3 cases (8.8%), ataxia in 2 cases (5.9%), dysarthria in 2 cases (5.9%), dizziness in 2 cases (5.9%), limb weakness in 1 case 2.9%), scalp mass in 1 case (2.9%), syncope in 1 case 2.9%) and facial mass in 1 case (2.9%). Two patients presented with chronic subdural hematoma on the affected side, and one patient developed superior sagittal sinus venous thrombosis at the central site of the lesion. Among all enrolled cases, only 13 were correctly diagnosed at initial presentation, 8 case reports did not mention the initial diagnosis, and 13 were misdiagnosed initially (misdiagnosis rate: 38.2%). The main misdiagnoses included meningioma, metastatic tumor, tuberculous meningoencephalitis, and meningitis. These findings indicate that IgG4-RHP is associated with a high misdiagnosis rate. Among all enrolled cases, 11 underwent subtotal resection, 19 were subjected to meningeal biopsy, and the surgical approach was not mentioned in 4 cases (as shown in Table 2).
Table 2.
Supplementary Clinical and Surgical Details.
All 34 enrolled cases were confirmed by pathological examination (including cases verified by re-examination), among which 6 patients (17.6%) were misdiagnosed in the initial pathological assessment. Dense lymphoplasmacytic infiltration and storiform fibrosis were identified in all cases (34/34). Obliterative phlebitis was detected in 8 cases, not detected in 9 cases, and not mentioned in 17 case reports. Among the cases, 27 (79.4%) met the criterion of >10 IgG4+ plasma cells per high-power field (HPF), while this indicator was not reported in 7 cases. Twenty-four cases (70.6%) satisfied the standard of an IgG4+/IgG+ plasma cell ratio exceeding 40%, 5 cases did not meet this standard, and 5 cases lacked relevant information (as shown in Table 2).
Among the 34 cases included in this study, the intracranial distribution of lesion sites exhibited distinct anatomical clustering characteristics, with some cases involving multiple sites. In terms of lesion location classification, supratentorial lesions were the most common, accounting for 50.0% (17 cases), mainly including unilateral/bilateral supratentorial regions and lesions involving both the supratentorium and skull base. The skull base/cavernous sinus region was the second most frequent site, involving 14 cases (41.2%), covering the clival dura mater, middle/posterior skull base, cavernous sinus, and anterior skull base. Posterior fossa lesions were observed in 8 cases (23.5%), primarily including lesions of the posterior fossa dura mater and those involving both the posterior fossa and tentorium cerebelli. Skull invasion was relatively rare, occurring in only 5 cases (14.7%), all presenting as supratentorial lesions combined with skull invasion. Mixed supratentorial and infratentorial lesions were the rarest, with only 1 case (2.9%), involving simultaneous bilateral supratentorial and infratentorial dural involvement (as shown in Table 3). One case invaded the retina, and one case invaded the maxillary sinus (as shown in Table 2).
Table 3.
Statistics of intracranial disease locations.
Regarding serum IgG4 levels among the 34 enrolled cases, 11 cases had normal levels (<1350 μg/mL), 11 cases had elevated levels (>1350 μg/mL), and 12 case reports indicated that serum IgG4 levels were not measured.
In terms of treatment, among the 34 enrolled cases, 19 underwent meningeal biopsy, 11 underwent subtotal tumor resection, and the surgical approach was not specified in 4 case reports. Among the 19 patients who underwent meningeal biopsy: 13 achieved favorable outcomes, 5 had no mentioned outcome data, and 1 showed poor response. Among the 11 patients who underwent subtotal tumor resection: 7 obtained favorable outcomes, and 4 had no mentioned outcome data (as shown in Table 1 and Table 2). Postoperatively, 25 cases received pharmacotherapy with the following regimens: 11 cases treated with glucocorticoids alone, 9 cases with glucocorticoids combined with rituximab, 2 cases with glucocorticoids combined with azathioprine, 2 case with glucocorticoids combined with methotrexate, and 1 case with glucocorticoids combined with paclitaxel liposome. Three cases did not receive pharmacotherapy due to poor physical status, and 6 case reports did not mention relevant medication details. Among the 25 patients who received pharmacotherapy, only 1 showed no response to treatment, while the remaining patients achieved significant improvements in both clinical symptoms and imaging findings (as shown in Table 1).
4. Discussion
4.1. Epidemiology and Clinical Symptoms
IgG4-RHP is an extremely rare autoimmune disorder of the central nervous system (CNS), first reported in 2009 [26], with an incidence of less than 1 per 100,000 population, based on a nationwide survey of hypertrophic pachymeningitis in Japan [27,28]. The incidence is higher in males than in females, with a male-to-female ratio of approximately 2:1, and the mean age at onset is around 52 years. IgG4-RHP is rarely associated with systemic diseases, and more than half of the cases present as isolated lesions [29].
A retrospective study of 33 cases by Lucy X. Lu et al. demonstrated that 22 cases (67%) had headache, 11 cases (33%) were complicated with cranial nerve palsy, 7 cases (21%) had visual abnormalities (diplopia or decreased visual acuity), 5 cases (15%) had limb weakness, 4 cases (12%) were accompanied by limb numbness, 3 cases (9%) had sensorineural hearing loss, and 2 cases (6%) experienced seizures [30]. These results are highly consistent with the statistical findings of the present study. Headache may be caused by dural inflammation or increased intracranial pressure. Cranial nerve palsy and other neurological deficit symptoms are mostly attributed to the compression of brain tissue and cranial nerves by the thickened dura mater, or secondary injuries and complications resulting from vascular compression [14].
4.2. Imaging Characteristics
The imaging manifestations of IgG4-RHP are crucial for initial diagnosis. On computed tomography (CT) and magnetic resonance (MR) imaging, it can present as band-like or nodular dural thickening, or mass-like protrusions [30], which are easily confused with Rosai-Dorfman disease, vasculitis, tuberculous meningitis, atypical meningioma, central nervous system leukemia, and other conditions [1]. A positron emission tomography (PET)-CT scan is useful for excluding the involvement of other tissues or organs. Extracranial manifestations can be detected in at least 40% of IgG4-RD patients. Therefore, a thorough systematic examination is necessary to rule out comorbidities such as asymptomatic cirrhosis, pancreatitis, nephritis, aneurysms, and arteritis. Overall, regular imaging during follow-up is preferred for the continuous monitoring of IgG4-RHP [31].
Matias et al. [1] summarized the MRI features of 32 patients with IgG4-RHP and found that “diffuse dural thickening + homogeneous enhancement” is the most typical manifestation, with a specificity of 83% but a sensitivity of only 67%, suggesting that imaging alone is insufficient for a definitive diagnosis. Skull involvement is a rare manifestation of IgG4-RHP. Most reported cases in the literature are associated with skull hyperostosis at the site of lesion involvement [8], while the case reported in this study presents with skull destruction and defects caused by lesion invasion, with proliferative dural tissue extending subcutaneously through the bone defect area. Patients are often misdiagnosed with malignant tumors due to skull erosion or defects observed on CT scans. In addition, cystic lesions [32], tumor-like masses [14], and other special manifestations have been reported, further increasing the difficulty of clinical diagnosis. The present case is characterized by “a history of chronic subdural hematoma + skull destruction + subcutaneous soft tissue mass”, a combination that has not been documented in previous studies. This unique clinical presentation enriches the phenotypic spectrum of IgG4-RHP.
4.3. Diagnostic Methods
Meningeal biopsy is the gold standard for diagnosing IgG4-RHP after excluding infectious, neoplastic, and other inflammatory pathologies [16]. Since IgG4-RHP is a rare subtype of IgG4-RD, the pathological diagnostic criteria for IgG4-RHP are identical to those for IgG4-RD.The classic three key histopathological features required for the diagnosis of IgG4-RD, known as the IgG4-RD triad, consist of dense lymphoplasmacytic infiltration, storiform fibrosis (radially arranged fibroblasts/myofibroblasts), and obliterative phlebitis (inflammatory venous occlusion). The diagnosis of IgG4-RD can be confirmed if two or more key histopathological features are present, accompanied by a marked increase in the number of IgG4-positive plasma cells (IgG4+/IgG+ ratio > 40% or >10 IgG4-positive plasma cells per high-power field [HPF, ×400]), especially for meningeal lesions [30]. The case reported herein fits this scenario. The pathological findings fulfilled the criteria of dense lymphoplasmacytic infiltration and storiform fibrosis. Although the IgG4+/IgG+ ratio was approximately 30% (<40%), the number of IgG4-positive plasma cells was elevated (≈40 IgG4+ plasma cells per high-power field [HPF, ×400]). Typical fibrosis or phlebitis may be absent in lymph nodes, lungs, and salivary glands [1]. It should be noted that small biopsy specimens may hinder the detection of obliterative phlebitis or an adequate number of IgG4-positive plasma cells, so a comprehensive assessment combining multi-site biopsy and clinical findings is required.
It is crucial to emphasize that elevated serum IgG4 concentration is not a necessary condition for the diagnosis of isolated IgG4-RHP. Unlike systemic IgG4-RD, due to the relatively low blood circulation in the dural tissue, patients with IgG4-RHP involving only the dura mater may have normal peripheral serum IgG4 concentrations, making them highly susceptible to misdiagnosis as meningitis or meningioma [22]. Therefore, elevated IgG4 levels in cerebrospinal fluid (CSF) are more specific for diagnosis. However, based on the cases analyzed in this study, due to limited clinical awareness of this disease and the invasive nature of lumbar puncture, there are few case reports on CSF IgG4 testing in clinical practice [33]. In addition, CSF examination often shows leukocytosis (WBC > 5/mm3) and elevated protein levels (Protein > 40 mg/dL), but these indicators lack specificity [34].
4.4. Differential Diagnosis
4.4.1. Meningioma
Meningioma is the most common benign tumor of the central nervous system, accounting for approximately 15% of all intracranial tumors [35]. Its imaging features are similar to those of IgG4-RHP, with both presenting as dural-based masses accompanied by significant enhancement. However, meningiomas are usually well-circumscribed, localized round or oval masses, and calcification can be observed in 20–30% of cases. Pathologically, meningiomas are composed of circumferentially arranged meningothelial cells, without IgG4+ plasma cell infiltration or storiform fibrosis [36]. In the present case, CT showed a slightly hyperdense mass, and MRI revealed dural enhancement. The initial biopsy misdiagnosed it as atypical meningioma. Final pathological examination did not identify meningothelial cell proliferation or psammoma bodies, but showed extensive IgG4+ plasma cell infiltration, which is the key basis for differentiating this disease from meningioma.
4.4.2. Metastatic Tumor
Intracranial dural metastatic tumors are lesions formed by the hematogenous spread, direct extension, or CSF dissemination of malignant tumors to the dura mater. Their magnetic resonance (MRI) imaging features are often focal nodular or massive masses with obvious heterogeneous enhancement and ill-defined boundaries. Lesions are mostly located in the cerebral convexity and parasagittal region, and are often accompanied by erosion or destruction of adjacent skull bones. Clinically, most patients have a history of malignant tumors, and symptoms (such as headache, vomiting, seizures, or focal neurological deficits) progress rapidly. CSF cytological examination may detect tumor cells, but a definitive diagnosis relies on pathological biopsy, which shows atypical tumor cells under microscopy. This disease is insensitive to glucocorticoid therapy or only has a transient response, and treatment requires comprehensive therapies such as radiotherapy and chemotherapy targeting the primary tumor [37].
4.4.3. Tuberculous Meningitis
Tuberculous pachymeningitis is an infectious disease, usually with acute or subacute onset, accompanied by toxic symptoms such as fever and night sweats. Most patients have a history of pulmonary tuberculosis or tuberculosis exposure. The key differentiating point between the two lies in CSF examination: patients with tuberculous pachymeningitis show typical “three highs and one low” changes in CSF (increased pressure, elevated protein, increased cell count, decreased glucose, and decreased chloride). Acid-fast bacilli can be detected in smears or cultures, and PCR testing for Mycobacterium tuberculosis is positive [38]. In contrast, IgG4-RHP is an autoimmune disease with a chronic course, mainly presenting with intractable headache, and often complicated with systemic multi-organ involvement. Significantly elevated serum IgG4 levels are one of the core features of IgG4-RHP, and the disease responds well to glucocorticoid therapy. A definitive diagnosis ultimately depends on pathological biopsy.
4.5. Treatment and Prognosis
For cases with intracranial mass effect caused by dural thickening, subtotal tumor resection is often performed to relieve pressure and obtain specimens for pathological examination; for cases without intracranial mass effect, minimally invasive puncture and meningeal biopsy can be performed to confirm the diagnosis. However, the local cerebral tissue compression symptoms in patients who underwent subtotal tumor resection were all significantly relieved after the surgery. After pathological confirmation, most cases are treated with glucocorticoid monotherapy or combined with immunosuppressive therapy, and all achieve favorable prognoses. GCs are the mainstay of treatment for inducing and maintaining remission in patients with IgG4-RD. In the case reported in this study, the patient was administered oral prednisone acetate 60 mg once daily after meals. Concurrently, oral calcium supplements and proton pump inhibitors were prescribed to mitigate the adverse effects of glucocorticoids. Literature reports indicate that lesions in some cases can be completely resolved after pharmacotherapy, but even with glucocorticoid maintenance therapy, the recurrence rate remains relatively high [39]. Data from a study by Kenichi Takano et al. [40] showed that after glucocorticoid treatment and maintenance, the clinical remission rate of patients was 73.8%, the annual recurrence rate was 11.5%, and 50% of cases relapsed within 7 years after initial treatment. If recurrence occurs, it is necessary to increase the dose of glucocorticoids or switch to other immunosuppressive agents (such as rituximab) [38]. Rituximab is an alternative for severe, recurrent, or glucocorticoid-dependent IgG4-RD [28]. Emerging evidence supports the high efficacy of intrathecal rituximab in treating refractory IgG4-RD, with immunological and imaging improvements observed in patients with a poor response to intravenous administration [41].
5. Limitations
This study has several limitations that should be acknowledged. First, as a single case report combined with a literature review, the generalizability of conclusions regarding treatment strategies and prognosis is inherently limited. Large-scale prospective studies are therefore warranted to validate the observed findings. Second, the 3-month follow-up duration is relatively short given the known high relapse risk of IgG4-related diseases (with an annual recurrence rate of 11.5% as previously reported). Longer-term clinical and imaging follow-up (≥1 year) is essential to evaluate the sustained efficacy of glucocorticoid therapy and closely monitor for potential recurrence. Third, preoperative serum IgG4 measurement was not performed, which restricts the assessment of its value in early diagnostic decision-making and may have contributed to the initial misdiagnosis. Fourth, cerebrospinal fluid (CSF) IgG4 analysis—an indicator with higher specificity for isolated intracranial IgG4-RHP—was not conducted in this case, reflecting the gap between ideal diagnostic standards and real-world clinical practice. Finally, the literature review presented herein is descriptive rather than quantitative (e.g., meta-analysis). Future studies could employ systematic review and meta-analysis methodologies to further clarify the correlations between pathological thresholds, treatment regimens, and clinical outcomes.
6. Conclusions
Histopathological examination remains the gold standard for the definitive diagnosis of IgG4-RHP, as imaging findings and serum IgG4 levels may be nonspecific or overlapping with other intracranial lesions. Surgical intervention, whether subtotal resection or meningeal biopsy, not only provides critical tissue samples for pathological confirmation but also effectively alleviates the mass-occupying effect caused by dural thickening, relieving neurological compression symptoms. Glucocorticoids are the first-line therapeutic option for IgG4-RHP, demonstrating significant efficacy in reducing lesions and improving clinical outcomes, as evidenced by the notable regression of residual lesions in the reported case and most literature-reviewed cases. Given the relatively high recurrence rate of IgG4-RHP, long-term clinical and imaging follow-up is indispensable to timely detect recurrence and adjust treatment strategies. For refractory cases unresponsive to glucocorticoids or those with recurrence, rituximab serves as a reliable alternative therapeutic agent. Additionally, enhancing clinical awareness of atypical manifestations of IgG4-RHP (such as skull destruction and subcutaneous mass formation) and emphasizing the combination of histopathological features, serum IgG4 detection, and clinical context can effectively reduce misdiagnosis rates and optimize patient management.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/diagnostics16050682/s1, File S1: Inclusion and Exclusion Criteria Analysis.
Author Contributions
Conceptualization, X.-M.L., L.-J.Y. and J.-L.W.; methodology, X.-M.L. and J.-L.W.; investigation, X.-M.L., L.J. and X.-L.S.; data curation, X.-M.L. and X.-L.S.; writing—original draft preparation, X.-M.L. and L.-J.Y.; writing—review and editing, J.-L.W.; supervision, X.-M.L., L.-J.Y. and J.-L.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Medical Science Research Project of Hebei (Grant No. 20250526).
Institutional Review Board Statement
Ethical review and approval were waived for this study in accordance with Chinese law, as this is a case report that only requires data anonymization and informed consent (no experimental research was involved, so ethics committee approval is not mandatory for publication).
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request, as ethical restrictions prevent public deposition of individual patient data.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| IgG4 | immunoglobulin G subclass 4 |
| IgG | immunoglobulin G |
| CNS | central nervous system |
| CT | computed tomography |
| MRI | magnetic resonance imaging |
| HPF | high-power field |
| IgG4-RD | IgG4-related disease |
| IgG4-RHP | IgG4-related hypertrophic pachymeningitis |
| PET | positron emission tomography |
| HP | hypertrophic pachymeningitis |
| CSF | cerebrospinal fluid |
| GCs | Glucocorticoid |
| RTX | Rituximab |
| MTX | Methotrexate |
| AZA | Azathioprine |
| PL | Paclitaxel Liposome |
| LPI | Lymphoplasmacytic infiltration |
| SF | Storiform fibrosis |
| OP | Obliterative phlebitis |
| HU | Hounsfield unit |
References
- Matias, T.B.; Cordeiro, R.A.; Duarte, J.A.; de Jarry, V.M.; Appenzeller, S.; Villarinho, L.; Reis, F. Immune-Mediated Hypertrophic Pachymeningitis and its Mimickers: Magnetic Resonance Imaging Findings. Acad. Radiol. 2023, 30, 2696–2706. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, J. Imaging features, clinicopathological analysis and diagnostic strategy of IgG4-related hypertrophic pachymeningitis. Ann. Palliat. Med. 2020, 9, 2551–2558. [Google Scholar] [CrossRef]
- Dong, L.L.; Sheikh, I.S.; Huang, A.H.; Wu, X.H.; Chen, E.G.; Ying, K.J. Immunoglobulin G4-related disease: Case report and literature review. Immunol. Res. 2021, 69, 415–421. [Google Scholar] [CrossRef]
- Wallace, Z.S.; Carruthers, M.N.; Khosroshahi, A.; Carruthers, R.; Shinagare, S.; Stemmer-Rachamimov, A.; Deshpande, V.; Stone, J.H. IgG4-related disease and hypertrophic pachymeningitis. Medicine 2013, 92, 206–216. [Google Scholar] [CrossRef]
- Lin, C.K.; Lai, D.M. IgG4-related intracranial hypertrophic pachymeningitis with skull hyperostosis: A case report. BMC Surg. 2013, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Utsuki, S.; Kijima, C.; Fujii, K.; Miyakawa, S.; Iizuka, T.; Hara, A. Investigation of IgG4-positive cell infiltration in biopsy specimens from cases of hypertrophic pachymeningitis. Clin. Neuropathol. 2013, 32, 84–90. [Google Scholar] [CrossRef]
- Rodríguez-Castro, E.; Fernández-Lebrero, A.; López-Dequidt, I.A.; Rodríguez-Osorio, X.; López-González, F.J.; Suárez-Peñaranda, J.M.; Arias, M. Hypertrophic pachymeningitis secondary to IgG4-related disease: Case report and review of the literature. Rev. Neurol. 2015, 61, 308–312. [Google Scholar]
- Nambirajan, A.; Chand Sharma, M.; Garg, K.; Sriram, S.; Boorgula, M.T.; Suri, V. Large dural-based mass with bony hyperostosis in a 16-year-old male: IgG4-related disease mimicking lymphoplasmacyte-rich meningioma. Childs Nerv. Syst. 2019, 35, 1423–1427. [Google Scholar] [CrossRef]
- Levraut, M.; Cohen, M.; Bresch, S.; Yokoyama, R.; Kawasaki, H.; Tamura, N.; Yamamoto, T. Immunoglobulin G4-related hypertrophic pachymeningitis: A case-oriented review. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e568. [Google Scholar] [CrossRef]
- Ota, K.; Nakazato, Y.; Okuda, R.; Yokoyama, R.; Kawasaki, H.; Tamura, N.; Yamamoto, T. Polycystic subdural hygroma associated with immunoglobulin G4-related intracranial hypertrophic pachymeningitis: A case report. BMC Neurol. 2020, 20, 228. [Google Scholar] [CrossRef]
- Suisa, H.; Soustiel, J.F.; Grober, Y. IgG4-related pachymeningitis masquerading as foramen magnum meningioma: Illustrative case. J. Neurosurg. Case Lessons 2021, 2, CASE21398. [Google Scholar] [CrossRef] [PubMed]
- Yamamuro, S.; Negishi, H.; Shijo, K.; Yoshino, A. Treatment-responsive case of focal clivus IgG4-related hypertrophic pachymeningitis mimicking meningioma; case report. Acta Neurol. Belg. 2021, 121, 1395–1397. [Google Scholar] [CrossRef]
- Woo, P.Y.M.; Ng, B.C.F.; Wong, J.H.M.; Ng, O.K.S.; Chan, T.S.K.; Kwok, N.-F.; Chan, K.-Y. The protean manifestations of central nervous system IgG4-related hypertrophic pachymeningitis: A report of two cases. Chin. Neurosurg. J. 2021, 7, 13. [Google Scholar] [CrossRef]
- Esmaeilzadeh, M.; Dadak, M.; Atallah, O.; Möhn, N.; Skripuletz, T.; Hartmann, C.; Banan, R.; Krauss, J.K. IgG4-related hypertrophic pachymeningitis with tumor-like intracranial and intracerebral lesions. Acta Neurochir. 2022, 164, 2781–2787. [Google Scholar] [CrossRef]
- Yu, Y.; Lv, L.; Yin, S.L.; Chen, C.; Jiang, S.; Zhou, P.-Z. Clivus-involved immunoglobulin G4 related hypertrophic pachymeningitis mimicking meningioma: A case report. World J. Clin. Cases. 2022, 10, 6269–6276. [Google Scholar] [CrossRef]
- Paira, S.; Reibaldi, A.; Froullet, C. Hypertrophic pachymeningitis due to IgG4-related disease (RD-IgG4): A case report. Reumatol. Clin. 2023, 19, 338–344. [Google Scholar] [CrossRef]
- Lichtblau, N.; Aliaga-Arias, J.; Kalaitzoglou, D.; Bodi, I.; Ashkan, K.; Bhangoo, R.; Vergani, F.; Joe, D.; Stanton, B.; Galloway, J.; et al. IgG4-related hypertrophic pachymeningitis with chronic subdural haematoma. Pract. Neurol. 2023, 23, 441–445. [Google Scholar] [CrossRef]
- Gautier, F.; Neumann, L.; Adle-Biassete, H.; Rubenstein, E.; Bernat, A.-L.; Chimon, A.; Mouly, S.; Sène, D.; Comarmond, C. Pachymeningitis associated with IgG4-related disease and ANCA positivity: Case report and review of the literature. Autoimmunity Rev. 2023, 22, 103285. [Google Scholar] [CrossRef]
- Suezumi, K.; Uehara, T.; Taira, A.; Akamatsu, N.; Tanaka, T.; Hayashi, Y.; Komuta, M.; Shiomi, T.; Murai, H. IgG4-related pachyleptomeningitis with inflammatory pseudotumor. eNeurologicalSci 2024, 34, 100490. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.; Yee, G.T.; Seo, J.; Seo, M.R.; Baek, H.J.; Choi, H.-J. Immunoglobulin G4-related hypertrophic pachymeningitis with an isolated scalp mass mimicking a brain tumor: A case report and literature review. J. Rheum. Dis. 2024, 31, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Chen, F. A case report of Immunoglobulin-G4-related hypertrophic sclerosing pachymeningitis. Radiol. Case Rep. 2024, 19, 1758–1763. [Google Scholar] [CrossRef]
- Chou, M.L.; Chen, H.C.; Tai, S.H.; Huang, C.W. Seronegative Immunoglobulin G4-related hypertrophic pachymeningitis misdiagnosed as meningitis and meningioma. Kaohsiung J. Med. Sci. 2024, 40, 506–508. [Google Scholar] [CrossRef]
- da Silva, G.E.; Medeiros, U.L.; Meira, A.T.; Braz, A.S. A case report: Hypertrophic pachymeningitis and IgG4-related disease. Int. J. Rheum. Dis. 2024, 27, e15410. [Google Scholar] [CrossRef]
- Yang, L.; Kaninia, S.; Urankar, K.; Brown, W.; Szewczyk-Krolikowski, K.; Rice, C.M. Cerebral venous sinus thrombosis complicating IgG4-related hypertrophic pachymeningitis. Pract. Neurol. 2025, pn-2025-004845. [Google Scholar] [CrossRef]
- Khanna, S.; Yadav, S. Balakrishnan CIntracranial IgG4-related Disease: Insights from Two Cases. J. Assoc. Physicians India 2025, 73, 54–56. [Google Scholar] [CrossRef]
- Chan, S.K.; Cheuk, W.; Chan, K.T.; Chan, J.K. IgG4-related sclerosing pachymeningitis: A previously unrecognized form of central nervous system involvement in IgG4-related sclerosing disease. Am. J. Surg. Pathol. 2009, 33, 1249–1252. [Google Scholar] [CrossRef]
- Yonekawa, T.; Murai, H.; Utsuki, S.; Matsushita, T.; Masaki, K.; Isobe, N.; Yamasaki, R.; Yoshida, M.; Kusunoki, S.; Sakata, K.; et al. A nationwide survey of hypertrophic pachymeningitis in Japan. J. Neurol. Neurosurg. Psychiatry 2014, 85, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Saitakis, G.; Chwalisz, B.K. The neurology of IGG4-related disease. J. Neurol. Sci. 2021, 424, 117420. [Google Scholar] [CrossRef] [PubMed]
- Umehara, H.; Okazaki, K.; Nakamura, T.; Satoh-Nakamura, T.; Nakajima, A.; Kawano, M.; Mimori, T.; Chiba, T. Current approach to the diagnosis of IgG4-related disease—Combination of comprehensive diagnostic and organ-specific criteria. Mod. Rheumatol. 2017, 27, 381–391. [Google Scholar] [CrossRef]
- Lu, L.X.; Della-Torre, E.; Stone, J.H.; Clark, S.W. IgG4-related hypertrophic pachymeningitis: Clinical features, diagnostic criteria, and treatment. JAMA Neurol. 2014, 71, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Elmaci, I.; Altinoz, M.A.; Akdemir, G.; Sari, R.; Baskan, O.; Ozpinar, A.; Hacker, E.; Sav, A. Neurosurgical and neuro-immunological management of IgG4-related hypertrophic sclerosing pachymeningitis. Clin. Neurol. Neurosurg. 2021, 200, 106342. [Google Scholar] [CrossRef]
- Araújo, D.A.B.S.; Ribeiro, R.M.; Lima, P.L.G.S.B.; de Queiroz, D.C.; Pitombeira, M.S.; Martins, B.; Coimbra, P.P.A.; Nogueira, C.D.; Braga-Neto, P.; Silva, G.D.; et al. Spinal cord compression by cystic IgG4-related spinal pachymeningitis mimicking neurocysticercosis: A case report. BMC Neurol. 2024, 24, 318. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Wan, Q.; Li, L. Roles of IgG4 and IgG4/IgG ratio to IgG4-related disease in patients with elevated serum IgG4 level. Clin. Rheumatol. 2023, 42, 793–800. [Google Scholar] [CrossRef]
- Terrim, S.; Mahler, J.V.; Filho, F.V.M.; Lucato, L.T.; Giardini, H.M.; Adoni, T.; Silva, G.D. Clinical Presentation, Investigation Findings, and Outcomes of IgG4-Related Pachymeningitis: A Systematic Review. JAMA Neurol. 2025, 82, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Soni, N.; Ora, M.; Bathla, G.; Szekeres, D.; Desai, A.; Pillai, J.J.; Agarwal, A. Meningioma: Molecular Updates from the 2021 World Health Organization Classification of CNS Tumors and Imaging Correlates. AJNR Am. J. Neuroradiol. 2025, 46, 240–250. [Google Scholar] [CrossRef]
- Alsharif, N.; Alkhathami, A.; Almalki, A.S.; Elkholy, S.S. Atypical Imaging Features of an Incidentally Discovered Intracranial Meningioma: A Case Report. Cureus 2024, 16, e64503. [Google Scholar] [CrossRef]
- Wu, H.; Beylerli, O.; Gareev, I.; Beilerli, A.; Ilyasova, T.; Talybov, R.; Sufianov, A.; Guo, X. Are there reliable multiparametric MRI criteria for differential diagnosis between intracranial meningiomas and solitary intracranial dural metastases? Oncol. Lett. 2023, 26, 350. [Google Scholar] [CrossRef]
- Ahlawat, S.; Chaudhary, R.; Dangi, M.; Bala, K.; Singh, M.; Chhillar, A.K. Advances in tuberculous meningitis diagnosis. Expert. Rev. Mol. Diagn. 2020, 20, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Zen, Y.; Pillai, S.; Stone, J.H. IgG4-related disease. Lancet 2015, 385, 1460–1471. [Google Scholar] [CrossRef]
- Takano, K.; Yamamoto, M.; Takahashi, H.; Himi, T. Recent advances in knowledge regarding the head and neck manifestations of IgG4-related disease. Auris Nasus Larynx 2017, 44, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Balaban, D.T.; Hutto, S.K.; Panzarini, B.P.; O’SHea, A.; Varma, A.; Jones, P.S.; Chwalisz, B.K.; Stone, J.H.; Venna, N. Treatment of IgG4-related disease-associated hypertrophic pachymeningitis with intrathecal rituximab: A case report. Front. Neurol. 2023, 14, 1189778. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.