

Long-Read Sequencing and Structural Variant Detection: Unlocking the Hidden Genome in Rare Genetic Disorders

 ,
,  ,
,  , , ,
, , ,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Technological Landscape
2.1. PacBio HiFi Sequencing
2.2. Oxford Nanopore Sequencing
2.3. Fundamental Differences Between PacBio and ONT Sequencing
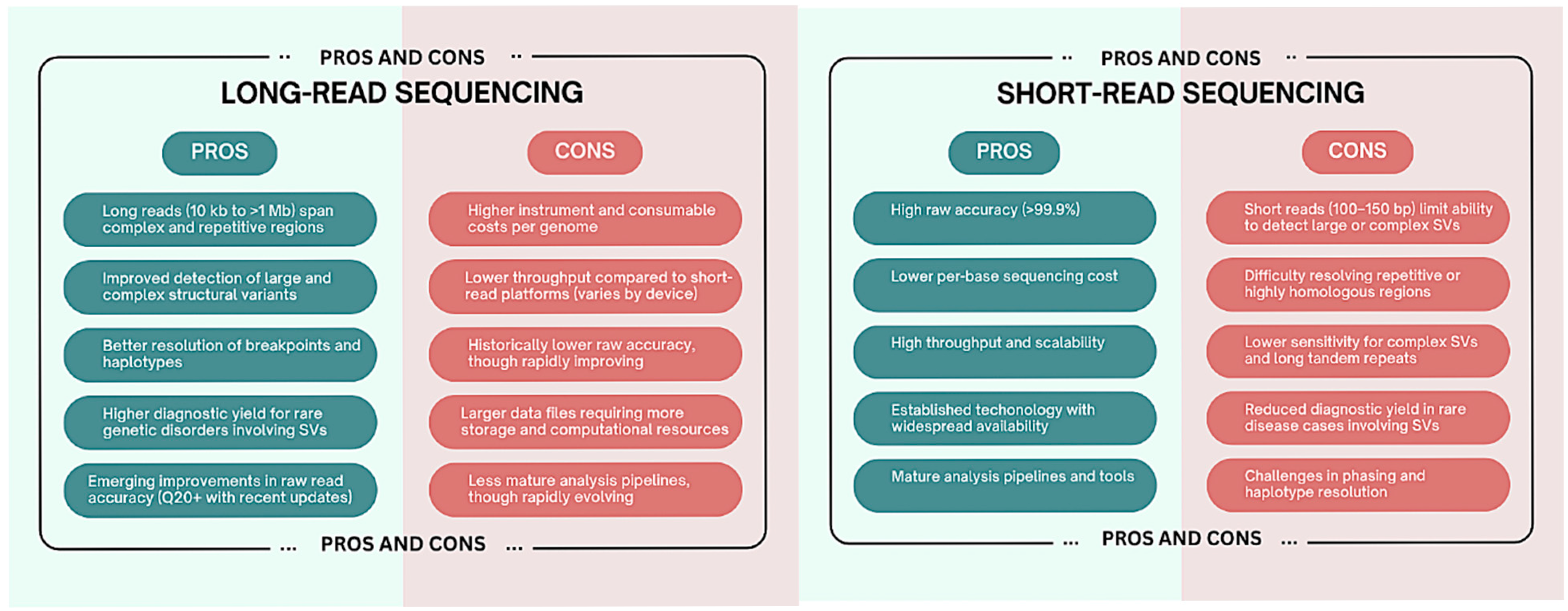
2.4. Comparative Summary
2.5. Benchmarking Performance for Structural Variant Detection
3. Structural Variant Detection
4. Applications in Rare Disease Diagnostics
5. Challenges and Limitations
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, C.E.; Singleton, K.S.; Wallin, M.; Faundez, V. Rare Genetic Diseases: Nature’s Experiments on Human Development. iScience 2020, 23, 101123. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Lee, D.; Hwang, A.; Kim, T.; Ryu, H.Y.; Choi, J. Rare disease genomics and precision medicine. Genom. Inf. 2024, 22, 28. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.A.; Nelson, E.T.; Koziura, M.; Cogan, J.D.; Hamid, R.; Phillips, J.A., 3rd. Lessons learned: Next-generation sequencing applied to undiagnosed genetic diseases. J. Clin. Investig. 2022, 132, e154942. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Del Gaudio, D.; Santani, A.; Scott, S.A. Applications of genome sequencing as a single platform for clinical constitutional genetic testing. Genet. Med. Open 2024, 2, 101840. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Roberts, R.; Mercer, T.R.; Xu, J.; Sedlazeck, F.J.; Tong, W. Towards accurate and reliable resolution of structural variants for clinical diagnosis. Genome Biol. 2022, 23, 68. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Ahn, J.W.; Bagnall, R.D.; Baralle, D.; Barton, S.; Campbell, C.; Downes, K.; Ellard, S.; Duff-Farrier, C.; FitzPatrick, D.R.; et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Bautista, R.; Larrosa, R.; Plata, O. Advancements in long-read genome sequencing technologies and algorithms. Genomics 2024, 116, 110842. [Google Scholar] [CrossRef] [PubMed]
- Jobanputra, V.; Andrews, P.; Felice, V.; Abhyankar, A.; Kozon, L.; Robinson, D.; London, F.; Hakker, I.; Wrzeszczynski, K.; Ronemus, M. Detection of Copy Number Variants by Short Multiply Aggregated Sequence Homologies. J. Mol. Diagn. 2020, 22, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Choo, Z.N.; Behr, J.M.; Deshpande, A.; Hadi, K.; Yao, X.; Tian, H.; Takai, K.; Zakusilo, G.; Rosiene, J.; Da Cruz Paula, A.; et al. Most large structural variants in cancer genomes can be detected without long reads. Nat. Genet. 2023, 55, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Oehler, J.B.; Wright, H.; Stark, Z.; Mallett, A.J.; Schmitz, U. The application of long-read sequencing in clinical settings. Hum. Genom. 2023, 17, 73. [Google Scholar] [CrossRef] [PubMed]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Keskus, A.G.; Wagner, J.; Izydorczyk, M.B.; Timp, W.; Sedlazeck, F.J.; Klein, A.P.; Zook, J.M.; Kolmogorov, M.; Schatz, M.C. Unraveling the hidden complexity of cancer through long-read sequencing. Genome Res. 2025, 35, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; He, J.; Li, M.; Peng, Y.; Jiang, H.; Zhao, J.; Li, Y.; Deng, F. Unlocking the Potential of Metagenomics with the PacBio High-Fidelity Sequencing Technology. Microorganisms 2024, 12, 2482. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Zhang, S.; Ren, P.; Liang, F.; Sun, Z.; Meng, G.; Tan, Y.; Li, X.; Lai, Q.; Han, L.; et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. Gigascience 2020, 9, giaa123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Soto-Serrano, A.; Li, W.; Panah, F.M.; Hui, Y.; Atienza, P.; Fomenkov, A.; Roberts, R.J.; Deptula, P.; Krych, L. Matching excellence: Oxford Nanopore Technologies’ rise to parity with Pacific Biosciences in genome reconstruction of non-model bacterium with high G+C content. Microb. Genom. 2024, 10, 001316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, H.; Jiang, M.; Hou, H.; Gao, Y.; Li, Y.; Wang, F.; Wang, J.; Peng, K.; Liu, Y.X. Nanopore sequencing: Flourishing in its teenage years. J. Genet. Genom. 2024, 51, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Ermini, L.; Driguez, P. The Application of Long-Read Sequencing to Cancer. Cancers 2024, 16, 1275. [Google Scholar] [CrossRef] [PubMed]
- Olson, N.D.; Wagner, J.; McDaniel, J.; Stephens, S.H.; Westreich, S.T.; Prasanna, A.G.; Johanson, E.; Boja, E.; Maier, E.J.; Serang, O.; et al. PrecisionFDA Truth Challenge V2: Calling variants from short and long reads in difficult-to-map regions. Cell Genom. 2022, 2, 100129. [Google Scholar] [CrossRef] [PubMed]
- Olivucci, G.; Iovino, E.; Innella, G.; Turchetti, D.; Pippucci, T.; Magini, P. Long read sequencing on its way to the routine diagnostics of genetic diseases. Front. Genet. 2024, 15, 1374860. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.K.; Yilmaz, K.C.; Acar, A. Benchmarking long-read structural variant calling tools and combinations for detecting somatic variants in cancer genomes. Sci. Rep. 2025, 15, 8707. [Google Scholar] [CrossRef] [PubMed]
- Udaondo, Z.; Sittikankaew, K.; Uengwetwanit, T.; Wongsurawat, T.; Sonthirod, C.; Jenjaroenpun, P.; Pootakham, W.; Karoonuthaisiri, N.; Nookaew, I. Comparative Analysis of PacBio and Oxford Nanopore Sequencing Technologies for Transcriptomic Landscape Identification of Penaeus monodon. Life 2021, 11, 862. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Liu, Y.H.; Zhou, X.M. VolcanoSV enables accurate and robust structural variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 2024, 15, 6956. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.E.; Dabernig-Heinz, J.; Lipp, M.; Cabal, A.; Simantzik, J.; Kohl, M.; Scheiber, M.; Lichtenegger, S.; Ehricht, R.; Leitner, E.; et al. Real-Time Nanopore Q20+ Sequencing Enables Extremely Fast and Accurate Core Genome MLST Typing and Democratizes Access to High-Resolution Bacterial Pathogen Surveillance. J. Clin. Microbiol. 2023, 61, e0163122. [Google Scholar] [CrossRef] [PubMed]
- Prodanov, T.; Bansal, V. Sensitive alignment using paralogous sequence variants improves long-read mapping and variant calling in segmental duplications. Nucleic Acids Res. 2020, 48, e114. [Google Scholar] [CrossRef] [PubMed]
- Conlin, L.K.; Aref-Eshghi, E.; McEldrew, D.A.; Luo, M.; Rajagopalan, R. Long-read sequencing for molecular diagnostics in constitutional genetic disorders. Hum. Mutat. 2022, 43, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Eisfeldt, J.; Ek, M.; Nordenskjold, M.; Lindstrand, A. Toward clinical long-read genome sequencing for rare diseases. Nat. Genet. 2025, 57, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Damian, A.; Nunez-Moreno, G.; Jubin, C.; Tamayo, A.; de Alba, M.R.; Villaverde, C.; Fund, C.; Delepine, M.; Leduc, A.; Deleuze, J.F.; et al. Long-read genome sequencing identifies cryptic structural variants in congenital aniridia cases. Hum. Genom. 2023, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Kaplun, L.; Krautz-Peterson, G.; Neerman, N.; Schindler, Y.; Dehan, E.; Huettner, C.S.; Baumgartner, B.K.; Stanley, C.; Kaplun, A. ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges. Int. J. Mol. Sci. 2025, 26, 2725. [Google Scholar] [CrossRef] [PubMed]
- Bronner, I.F.; Dawson, E.; Park, N.; Piepenburg, O.; Quail, M.A. Evaluation of controls, quality control assays, and protocol optimisations for PacBio HiFi sequencing on diverse and challenging samples. Front. Genet. 2024, 15, 1505839. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Jasielczuk, I.; Szmatola, T.; Sawicki, S.; Semik-Gurgul, E.; Dlugosz, B.; Bugno-Poniewierska, M. Application of Nanopore Sequencing for High Throughput Genotyping in Horses. Animals 2023, 13, 2227. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.S.; Urban, A.E.; Mills, R.E. Structural variation in the sequencing era. Nat. Rev. Genet. 2020, 21, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.J.; Chiang, C.; Hall, I.M. Structural variants are a major source of gene expression differences in humans and often affect multiple nearby genes. Genome Res. 2021, 31, 2249–2257. [Google Scholar] [CrossRef] [PubMed]
- D’Haene, E.; Vergult, S. Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genet. Med. 2021, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Spies, N.; Weng, Z.; Bishara, A.; McDaniel, J.; Catoe, D.; Zook, J.M.; Salit, M.; West, R.B.; Batzoglou, S.; Sidow, A. Genome-wide reconstruction of complex structural variants using read clouds. Nat. Methods 2017, 14, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, S.; Bartalucci, N.; Vannucchi, A.M. Resolving complex structural variants via nanopore sequencing. Front. Genet. 2023, 14, 1213917. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; Van Broeckhoven, C. Newest Methods for Detecting Structural Variations. Trends Biotechnol. 2019, 37, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Tvedte, E.S.; Gasser, M.; Sparklin, B.C.; Michalski, J.; Hjelmen, C.E.; Johnston, J.S.; Zhao, X.; Bromley, R.; Tallon, L.J.; Sadzewicz, L.; et al. Comparison of long-read sequencing technologies in interrogating bacteria and fly genomes. G3 (Bethesda) 2021, 11, jkab083. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.; Paulin, L.F.; Grochowski, C.M.; Horner, D.W.; Mahmoud, M.; Behera, S.; Kalef-Ezra, E.; Gandhi, M.; Hong, K.; Pehlivan, D.; et al. Detection of mosaic and population-level structural variants with Sniffles2. Nat. Biotechnol. 2024, 42, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Sindi, S.S.; Onal, S.; Peng, L.C.; Wu, H.T.; Raphael, B.J. An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol. 2012, 13, R22. [Google Scholar] [CrossRef] [PubMed]
- Tafazoli, A.; Hemmati, M.; Rafigh, M.; Alimardani, M.; Khaghani, F.; Korostynski, M.; Karnes, J.H. Leveraging long-read sequencing technologies for pharmacogenomic testing: Applications, analytical strategies, challenges, and future perspectives. Front. Genet. 2025, 16, 1435416. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xie, Z.; Li, M. Comprehensive and deep evaluation of structural variation detection pipelines with third-generation sequencing data. Genome Biol. 2024, 25, 188. [Google Scholar] [CrossRef] [PubMed]
- Meleshko, D.; Yang, R.; Maharjan, S.; Danko, D.C.; Korobeynikov, A.; Hajirasouliha, I. Blackbird: Structural variant detection using synthetic and low-coverage long-reads. bioRxiv 2024, 5, vbaf151. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Wang, A.; Au, K.F. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 2019, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Hu, H.; Jiang, Z.; Cao, S.; Wang, G.; Zhao, Y.; Jiang, T. SVHunter: Long-read-based structural variation detection through the transformer model. Brief. Bioinform. 2025, 26, bbaf203. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, E.; Bellin, M.; Di Resta, C. Advance in Genomics of Rare Genetic Diseases. Biomolecules 2023, 13, 1441. [Google Scholar] [CrossRef] [PubMed]
- Nisar, H.; Wajid, B.; Shahid, S.; Anwar, F.; Wajid, I.; Khatoon, A.; Sattar, M.U.; Sadaf, S. Whole-genome sequencing as a first-tier diagnostic framework for rare genetic diseases. Exp. Biol. Med. 2021, 246, 2610–2617. [Google Scholar] [CrossRef] [PubMed]
- Pitsava, G.; Hawley, M.; Auriga, L.; de Dios, I.; Ko, A.; Marmolejos, S.; Almalvez, M.; Chen, I.; Scozzaro, K.; Zhao, J.; et al. Genome sequencing reveals the impact of non-canonical exon inclusions in rare genetic disease. medRxiv 2024. [Google Scholar] [CrossRef]
- Wojcik, M.H.; Lemire, G.; Berger, E.; Zaki, M.S.; Wissmann, M.; Win, W.; White, S.M.; Weisburd, B.; Wieczorek, D.; Waddell, L.B.; et al. Genome Sequencing for Diagnosing Rare Diseases. N. Engl. J. Med. 2024, 390, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Rudaks, L.I.; Stevanovski, I.; Yeow, D.; Reis, A.L.M.; Chintalaphani, S.R.; Cheong, P.L.; Gamaarachchi, H.; Worgan, L.; Ahmad, K.; Hayes, M.; et al. Targeted Long-Read Sequencing as a Single Assay Improves the Diagnosis of Spastic-Ataxia Disorders. Ann. Clin. Transl. Neurol. 2025, 12, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Doss, R.M.; Lopez-Ignacio, S.; Dischler, A.; Hiatt, L.; Dashnow, H.; Breuss, M.W.; Dias, C.M. Mosaicism in Short Tandem Repeat Disorders: A Clinical Perspective. Genes 2025, 16, 216. [Google Scholar] [CrossRef] [PubMed]
- Kaplun, L.; Krautz-Peterson, G.; Neerman, N.; Stanley, C.; Hussey, S.; Folwick, M.; McGarry, A.; Weiss, S.; Kaplun, A. ONT long-read WGS for variant discovery and orthogonal confirmation of short read WGS derived genetic variants in clinical genetic testing. Front. Genet. 2023, 14, 1145285. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Marques-Bonet, T.; Sharp, A.J.; Mefford, H.C. The genetics of microdeletion and microduplication syndromes: An update. Annu. Rev. Genom. Hum. Genet. 2014, 15, 215–244. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, S.H.; Spielmann, M.; Mundlos, S.; Veltman, J.A.; Gilissen, C. Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum. Mutat. 2015, 36, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Brennan, N.F.; Trachana, K.; Katsandres, S.; Bodamer, O.; Belmont, J.; Veenstra, D.L.; Peng, S. A meta-analysis of diagnostic yield and clinical utility of genome and exome sequencing in pediatric rare and undiagnosed genetic diseases. Genet. Med. 2025, 27, 101398. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, I.; Aschbacher, R.; Pagani, E. Current Uses and Future Perspectives of Genomic Technologies in Clinical Microbiology. Antibiotics 2023, 12, 1580. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Ou, S.; Jenike, K.M.; Schatz, M.C. Approaching complete genomes, transcriptomes and epi-omes with accurate long-read sequencing. Nat. Methods 2023, 20, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.V.; Goodwin, S.; McCombie, W.R. Leveraging the power of long reads for targeted sequencing. Genome Res. 2024, 34, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Scarano, C.; Veneruso, I.; De Simone, R.R.; Di Bonito, G.; Secondino, A.; D’Argenio, V. The Third-Generation Sequencing Challenge: Novel Insights for the Omic Sciences. Biomolecules 2024, 14, 568. [Google Scholar] [CrossRef] [PubMed]
- Mastrorosa, F.K.; Miller, D.E.; Eichler, E.E. Applications of long-read sequencing to Mendelian genetics. Genome Med. 2023, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Handler, H.P.; Victorsen, A.; Flaten, Z.; Ellison, A.; Knutson, T.P.; Munro, S.A.; Martinez, R.J.; Billington, C.J.; Laffin, J.J.; et al. Validation of a comprehensive long-read sequencing platform for broad clinical genetic diagnosis. Front. Genet. 2025, 16, 1499456. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.M.; Prybutok, V. Balancing Privacy and Progress: A Review of Privacy Challenges, Systemic Oversight, and Patient Perceptions in AI-Driven Healthcare. Appl. Sci. 2024, 14, 675. [Google Scholar] [CrossRef]
- Austin-Tse, C.A.; Jobanputra, V.; Perry, D.L.; Bick, D.; Taft, R.J.; Venner, E.; Gibbs, R.A.; Young, T.; Barnett, S.; Belmont, J.W.; et al. Best practices for the interpretation and reporting of clinical whole genome sequencing. NPJ Genom. Med. 2022, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Sharo, A.G.; Hu, Z.; Sunyaev, S.R.; Brenner, S.E. StrVCTVRE: A supervised learning method to predict the pathogenicity of human genome structural variants. Am. J. Hum. Genet. 2022, 109, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Zawar, A.; Manoj, G.; Nair, P.P.; Deshpande, P.; Suravajhala, R.; Suravajhala, P. Variants of uncertain significance: At the crux of diagnostic odyssey. Gene 2025, 962, 149587. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [PubMed]
- Dawood, M.; Heavner, B.; Wheeler, M.M.; Ungar, R.A.; LoTempio, J.; Wiel, L.; Berger, S.; Bernstein, J.A.; Chong, J.X.; Delot, E.C.; et al. GREGoR: Accelerating Genomics for Rare Diseases. arXiv 2024, arXiv:2412.14338. [Google Scholar] [CrossRef]
- Henglin, M.; Ghareghani, M.; Harvey, W.T.; Porubsky, D.; Koren, S.; Eichler, E.E.; Ebert, P.; Marschall, T. Graphasing: Phasing diploid genome assembly graphs with single-cell strand sequencing. Genome Biol. 2024, 25, 265. [Google Scholar] [CrossRef] [PubMed]
- Showpnil, I.A.; Gonzalez, M.E.H.; Ramadesikan, S.; Marhabaie, M.; Daley, A.; Dublin-Ryan, L.; Pastore, M.T.; Gurusamy, U.; Hunter, J.M.; Stone, B.S.; et al. Long-read genome sequencing resolves complex genomic rearrangements in rare genetic syndromes. NPJ Genom. Med. 2024, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Hook, P.W.; Timp, W. Beyond assembly: The increasing flexibility of single-molecule sequencing technology. Nat. Rev. Genet. 2023, 24, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.M.; Yeung, M.H.Y.; Wong, A.N.N.; Tsang, H.F.; Yu, A.C.S.; Yim, A.K.Y.; Wong, S.C.C. Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells 2023, 12, 493. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.C.; Solomon, B.; Mun, T.; Iyer, S.; Langmead, B. Reference flow: Reducing reference bias using multiple population genomes. Genome Biol. 2021, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Meredith, M.; Daida, K.; Jerez, P.A.; Negi, S.; Malik, L.; Genner, R.M.; Moller, A.; Zheng, X.; Gibson, S.B.; et al. Long-read sequencing of hundreds of diverse brains provides insight into the impact of structural variation on gene expression and DNA methylation. bioRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.M.; Ali, B.R.; Al-Jasmi, F.; Sinnott, R.O.; Al Dhaheri, N.; Mohamad, M.S. A review of genetic variant databases and machine learning tools for predicting the pathogenicity of breast cancer. Brief. Bioinform. 2023, 25, bbad479. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, J.A.; Gibson, S.B.; Damaraju, N.; Zalusky, M.P.G.; Hoekzema, K.; Twesigomwe, D.; Yang, L.; Snead, A.A.; Richmond, P.A.; De Coster, W.; et al. High-coverage nanopore sequencing of samples from the 1000 Genomes Project to build a comprehensive catalog of human genetic variation. Genome Res. 2024, 34, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Emerson, J.J.; Macdonald, S.J.; Long, A.D. Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits. Nat. Commun. 2019, 10, 4872. [Google Scholar] [CrossRef] [PubMed]
- Brlek, P.; Bulic, L.; Bracic, M.; Projic, P.; Skaro, V.; Shah, N.; Shah, P.; Primorac, D. Implementing Whole Genome Sequencing (WGS) in Clinical Practice: Advantages, Challenges, and Future Perspectives. Cells 2024, 13, 504. [Google Scholar] [CrossRef] [PubMed]
- Negi, S.; Stenton, S.L.; Berger, S.I.; Canigiula, P.; McNulty, B.; Violich, I.; Gardner, J.; Hillaker, T.; O’Rourke, S.M.; O’Leary, M.C.; et al. Advancing long-read nanopore genome assembly and accurate variant calling for rare disease detection. Am. J. Hum. Genet. 2025, 112, 428–449. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Feature | PacBio HiFi | Oxford Nanopore (ONT) |
|---|---|---|
| Read Length | 10–25 kb (HiFi reads) | Up to >1 Mb (typical reads 20–100 kb) |
| Accuracy | >99.9% (HiFi consensus) | ~98–99.5% (Q20+ with recent improvements) |
| Throughput | Moderate–High (up to ~160 Gb/run Sequel IIe) | High (varies by device; PromethION > Tb) |
| Instrument Cost | High (Sequel IIe system) | Lower (MinION, GridION, scalable options) |
| Consumable Cost | Higher per Gb | Lower per Gb |
| Notable Strengths | Exceptional accuracy, suited to clinical applications | Ultra-long reads, portability, real-time analysis |
| Metric | PacBio HiFi | ONT (Q20+/R10.4.1) |
|---|---|---|
| SV Calling F1 Score | >95% | 85–90% (improving with basecaller upgrades) |
| Typical Assembly N50 | 20–30 Mb (HiFi reads) | >50 Mb (ultra-long reads) |
| Pathogenic SV Detection | High precision, fewer false positive | Higher sensitivity for large/repetitive SVs |
| Per-genome Cost (USD) | USD 1000–1500 | USD 400–800 (varies by scale and platform) |
| Diagnostic Yield Gain (rare disease cases) | +10–15% vs. short-read WGS | Similar, with strengths in large SVs and TRs |
| Variant Type | Disease Group | Representative Diseases | Example of Long-Read Utility |
|---|---|---|---|
| Repeat Expansion | Neurological and Neuromuscular |
|
|
| Deep Intronic Insertion | Neurodevelopmental Disorders |
|
|
| Large Deletions/Duplications | Syndromic Disorders |
|
|
| Mobile Element Insertions | Neurological Disorders |
|
|
| Complex Structural Rearrangements | Developmental Disorders |
|
|
| Mosaic Structural Variants | Somatic Mosaic Disorders |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moustakli, E.; Christopoulos, P.; Potiris, A.; Zikopoulos, A.; Mavrogianni, D.; Karampas, G.; Kathopoulis, N.; Anagnostaki, I.; Domali, E.; Tzallas, A.T.; et al. Long-Read Sequencing and Structural Variant Detection: Unlocking the Hidden Genome in Rare Genetic Disorders. Diagnostics 2025, 15, 1803. https://doi.org/10.3390/diagnostics15141803
Moustakli E, Christopoulos P, Potiris A, Zikopoulos A, Mavrogianni D, Karampas G, Kathopoulis N, Anagnostaki I, Domali E, Tzallas AT, et al. Long-Read Sequencing and Structural Variant Detection: Unlocking the Hidden Genome in Rare Genetic Disorders. Diagnostics. 2025; 15(14):1803. https://doi.org/10.3390/diagnostics15141803
Chicago/Turabian StyleMoustakli, Efthalia, Panagiotis Christopoulos, Anastasios Potiris, Athanasios Zikopoulos, Despoina Mavrogianni, Grigorios Karampas, Nikolaos Kathopoulis, Ismini Anagnostaki, Ekaterini Domali, Alexandros T. Tzallas, and et al. 2025. "Long-Read Sequencing and Structural Variant Detection: Unlocking the Hidden Genome in Rare Genetic Disorders" Diagnostics 15, no. 14: 1803. https://doi.org/10.3390/diagnostics15141803
APA StyleMoustakli, E., Christopoulos, P., Potiris, A., Zikopoulos, A., Mavrogianni, D., Karampas, G., Kathopoulis, N., Anagnostaki, I., Domali, E., Tzallas, A. T., Drakakis, P., & Stavros, S. (2025). Long-Read Sequencing and Structural Variant Detection: Unlocking the Hidden Genome in Rare Genetic Disorders. Diagnostics, 15(14), 1803. https://doi.org/10.3390/diagnostics15141803